DOI:
10.1039/C0MD00228C
(Concise Article)
Med. Chem. Commun., 2011,
2, 261-269
Conformational effects on potency of thioimidazoles and dihydrothiazolines
Received
19th November 2010
, Accepted 5th January 2011
First published on 27th January 2011
Abstract
We investigated the effect of steric restriction and hindrance on inhibitors directed against the biological activity of p38α MAP kinase. The structural reduction of the early lead compound SKF86002 into three structural classes of optimized SKF86002-like inhibitors produced a strong effect on inhibitory potency. This effect was investigated by conformational analysis and docking experiments.
1 Introduction
The inhibition of interleukin-1 beta (IL-1β) production by the anti-inflammatory compound SKF86002 was first reported in 1988,1 but it was not until 1994 that the molecular target of this class of pyridinyl imidazole compounds was identified as p38α MAPK. Originally developed from the early lead compound SKF860022 (Fig. 1), the first generation p38α MAPK inhibitors were structurally related to the prototypical pyridinyl imidazole SB203580 (Fig. 1). The central pharmacophore consists of a vicinal pyridine/fluorophenyl system and, to date, several inhibitors fulfilling this concept have been advanced into clinical trials.3
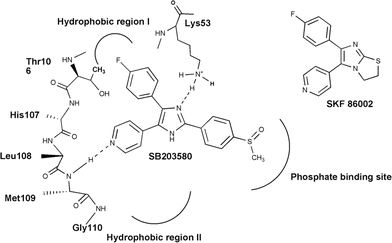 |
| | Fig. 1 Binding mode of SB203580. | |
Crystallographic data have demonstrated that SB203580 binds in the ATP binding site of p38α.4 Specifically, the pyridine ring nitrogen accepts a hydrogen bond from the backbone NH of Met109 in the hinge region. Vicinal to this interaction site, the 4-fluorophenyl moiety occupies the “hydrophobic pocket I,” conferring to the vicinal pyridine/fluorophenyl inhibitors selectivity for MAPK over other PKs. Previous reports in the literature have shown that N-substitution (N-1) on the imidazole core leads to decreased inhibitory potencies,5 but further studies to investigate this effect are missing. We considered that the loss of inhibitory potency caused by N-substitution at the imidazole moiety could be due to conformational changes that interfered with the interaction of the inhibitor with the p38α binding site. To investigate the negative effects of the imidazoleN-substitution, we synthesized three different inhibitor classes based on the core structure SKF86002 as well as their modified analogues. We determined IC50 values for the new inhibitors against p38α, and analyzed the results in the context of additional studies involving conformational analysis and docking experiments.
We initiated the aforementioned modifications by structural reductions of SKF86002 (Scheme 1). In an effort to clarify which structural features had the greatest importance for inhibition of p38α MAP kinase, we used selective reduction of the structure of the parent compound to clarify the negative effects of N-substitution. Since we assumed that conformational effects were a possible explanation, we chose the core structures 1–3 (Scheme 1) as the basis of the generated substance library. Sulfanylimidazoles without N-substitution (3) are highly potent structures.5Reduction of the dihydrothiazoline to N-methyl-S-methyl imidazole 2 leads to a loss of steric restriction but to a gain in steric hindrance by repulsion between the methyl moieties. In a further structural reduction of the S-methylimidazole 3, we neutralized this effect by N-demethylation, leading to a loss of the mentioned steric hindrance. Analogues of the core structures 1–3 were designed by known optimization methods.5 Modification at C-2 of the pyridine moiety by amine- or acyl-substitution should lead to further interactions with the hinge region, as a result of bidentate hydrogen bonding.5 The presence of lipophilic residues could strongly increase potency by the enhanced hydrophobic interaction with the hydrophobic region II of the kinase. The presence of acyl-residues has been shown to improve metabolic stability, due to obstruction of N-demethylation.6
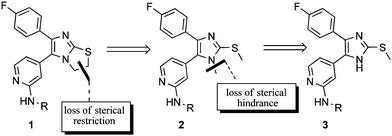 |
| | Scheme 1 Structural reduction of the SKF86002 core structure. | |
2 Results and discussion
We synthesized a panel of highly potent acylated inhibitors of the three above mentioned core structures 1–3. The syntheses followed known literature procedures.5,6,7,8 Synthesis of 1,2,4,5-tetrasubstituted imidazoles is shown in Scheme 2. The fluorination of 1-(2-aminopyridin-4-yl)-2-(4-fluorophenyl)ethanone to 4 was carried out with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundsodium nitrite in dry COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen fluoride/COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpyridine. The alpha-oximination was easily performed in COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundglacial acetic acid with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundsodium nitrite, yielding 5. Subsequent ring closure with the appropriate triazinanes afforded the N-oxides 6 in good yields which were then sulfurated to yield imidazole-2-thiones 7. In the case of R = hydroxymethyl, the imidazole-2-thione 7 was mesylated, resulting in a subsequent ring closure yielding dihydrothiazoline 8. Alternative imidazole-2-thiones 7 were alkylated with alkylhalogenides yielding compound 11. Nucleophilic aromatic substitution of 8, 11, or 18 with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNH3 resulted in 9, 12, or 19, respectively. The amides 10, 13, and 20 were obtained by amide coupling of the corresponding activated carbonic acid (CDI activation) and the amino-substituted compounds 9, 12, or 19. Synthesis of 2,4,5-trisubstituted imidazoles (Scheme 3) was performed starting from COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundethanone 14via the intermediates 15 and 16 and pursuing a four step nitrosation, reduction, and cyclization strategy.
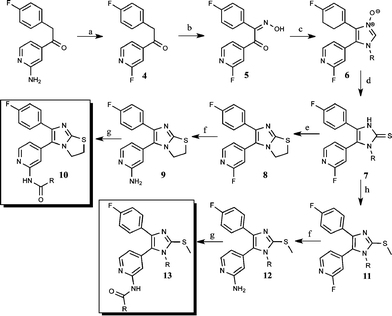 |
| | Scheme 2 Synthetic pathway to 1,2,4,5-tetrasubstituted imidazoles and 5,6-disubstituted dihydrothiazolines: a) NaNO2, 70% COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundHF-pyridine, −15 to −10 °C, then rt; b) NaNO2, glacial AcOH, rt; c) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH, (CH2NR)2, reflux; d) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compound2,2,4,4-tetramethylcyclobutane-1,3-dithione, CH2Cl2, rt; e) CH3O2SCl, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundpyridine, 50 °C f) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNH3, reactor; h) R-X, K2CO3, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundMeOH, rt. | |
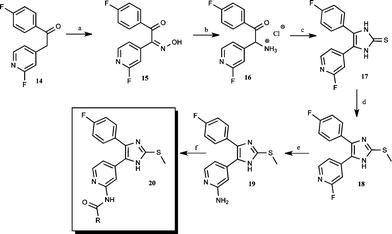 |
| | Scheme 3 Synthetic pathway to 2,4,5-trisubstituted imidazoles: a) NaNO2, glacial AcOH, rt; b) H2, Pd/C 10%, 1 atm, 2-propanolic COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundhydrogen chloride, rt; c) DMF, KSCN, reflux; d) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundiodomethane, Na2CO3, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundEtOH, rt; e) COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundNH3, reactor f) RCOOH, CDI, NMP, rt, then reflux. | |
The inhibitory potency of the synthesized compounds was determined in an in vitro p38α MAP kinase assay,9 using SB203580 as the reference compound. The inhibitors exhibited IC50-values in the range of 498 nM to 9 nM (Table 1). We also calculated the more reproducible grading factors (gf) (due to the variabilities of IC50-values of the performed inter-assays) that are the ratios of IC50 SB203580/IC50 compound. To directly compare the activity profiles, we constructed a diagram that shows the relationship of the grading factors (Fig. 2).
Table 1
IC50 values and gradient factors (gf: IC50 SB203580/IC50 compound) of compounds 1a–h, 2a–h and 3a–h
| R (compound) |
p38α
IC50, μMa of 1 |
p38α
IC50, μMa of 2 |
p38α
IC50, μMa of 3 |
|
Mean ± SEM of three experiments.
|

|
0.083 ± 0.0179 |
0.337 ± 0.030 |
0.019 ± 0.0020 |
| gf: 0.89 (ref: 0.074 ± 0.006) |
gf: 0.18 (ref: 0.062 ± 0.008) |
gf: 1.36 (ref: 0.026 ± 0.003) |
|
0.046 ± 0.0026 |
0.133 ± 0.034 |
0.009 ± 0.0004 |
| gf: 1.93 (ref: 0.089 ± 0.012) |
gf: 0.24 (ref: 0.033 ± 0.003) |
gf: 3.55 (ref: 0.032 ± 0.001) |

|
0.046 ± 0.0036 |
0.498 ± 0.1390 |
0.022 ± 0.0037 |
| gf: 0.97 (ref: 0.045 ± 0.025) |
gf: 0.05 (ref: 0.027 ± 0.005) |
gf: 1.23 (ref: 0.027 ± 0.005) |

|
0.140 ± 0.0097 |
0.173 ± 0.1011 |
0.013 ± 0.005 |
| gf: 0.63 (ref: 0.089 ± 0.012) |
gf: 0.35 (ref: 0.062 ± 0.008) |
gf: 1.46(ref: 0.019 ± 0.003) |

|
0.029 ± 0.0017 |
0.411 ± 0.0818 |
0.014 ± 0.002 |
| gf: 0.96 (ref: 0.028 ± 0.040) |
gf: 0.10 (ref: 0.041 ± 0.004) |
gf: 1.93 (ref: 0.027 ± 0.002) |
|
0.020 ± 0.0042 |
0.186 ± 0.0667 |
0.020 ± 0.008 |
| gf: 1.40 (ref: 0.028 ± 0.040) |
gf: 0.22 (ref: 0.041 ± 0.004) |
gf: 1.85 (ref: 0.037 ± 0.006) |
|
0.115 ± 0.00523 |
0.430 ± 0.1026 |
0.053 ± 0.017 |
| gf: 0.77 (ref: 0.089 ± 0.012) |
gf: 0.10 (ref: 0.041 ± 0.004) |
gf: 0.70 (ref: 0.037 ± 0.004) |

|
0.036 ± 0.006 |
0.396 ± 0.0389 |
0.044 ± 0.0066 |
| gf: 1.44 (ref: 0.052 ± 0.005) |
gf: 0.16 (ref: 0.062 ± 0.008) |
gf: 0.61 (ref: 0.027 ± 0.005) |
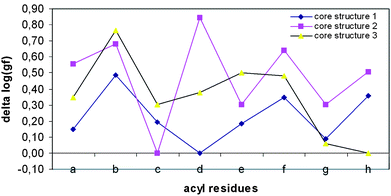 |
| | Fig. 2 Comparison of the grading factors (IC50 SB203580/IC50 compound) of inhibitors 1a–h-3a–h. | |
The hydrophobic interactions with the hydrophobic region II (Fig. 1) of the residues 1b, 1f and 1h (Table 1) show the highest effect on potency in the case of core structure 1 (Scheme 1). The poorest IC50 values were observed with the residues 1a, 1c, 1d, 1e and 1g. A general strongly decreased potency was observed for core structure 2. Structures of core compound 2 showing the highest gf values are the acyl analogues 2d. The most potent structures are 3b, 3e and 3f, and within this series 3a, 3c, 3g and 3h showed the lowest activity. Furthermore, similar activity profiles of gf values were observed for 1a–h and 3a–h, whereas the activity profiles for 2a–h differed markedly. This outcome suggests that the trends in the activity profiles depend more on the core structure and that the inhibitory properties conferred by the substituents are not interchangeable with different core structures. Clearly, the gain or loss of inhibitory potency with respect to the presence of the same acyl substituents is not constant for the three core structures, even though the hydrophobic interactions of the acyl substituent with the hydrophobic region II of the kinase should be the same.
The superiority of the core structures 1 and 3 as inhibitors with respect to the inhibitors of class 2 (Fig. 2) can be explained by the conformation of core structures 1–3. Structures 1a–h are characterized by a fixed conformation, which is neutralized in structures 2a–h and 3a–h by structural reduction to methylsulfanyl imidazoles. The loss of sterical restriction in 2 is compounded by decreased potencies, whereas N-demethylation in 3 resulted in increased potencies. A crystal structure of 1b, one of the compounds with a low IC50 value, was obtained (Fig. 3a).10 We measured the angle between the S–Me and N–Me group of this dihydrothiazoline compound, and this angle fell within the ideal range of N–Me, S–Me conformations, which could be adopted by the inhibitors with core structures 2 and 3. The two planes of the dihydrothiazoline are defined by selecting atoms N-4, C-7a, and N-7 for the first plane and C-7a, S, and S–CH2 for the second. The angle between the two planes is 13.20° (Fig. 3a).
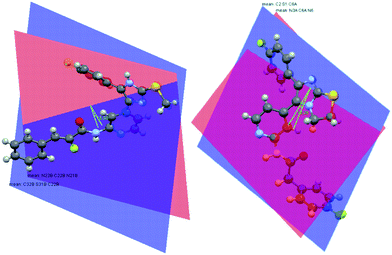 |
| | Fig. 3 Calculated11 angles between the imidazole plane and S-CH3/2 of the crystal structures of 1b (a) and analogue structures of 3 (b). | |
A crystal structure of an analogue structure of 3a–h (Fig. 3b) was published by our group.12 We measured the angle between the imidazole plane (N-1, C-2, N-2) and the S–Me plane (C-2, S, CH3) as being 16.00°. This conformation may differ from the real conformation in solution due to the loss of rigidity, but, interestingly, the difference in the angles conforms with the conformation of the thiazoline system within the small range of 2.8°. These studies indicate that the conformations of core structures 1 and 3 are similar. Structures 3a–h show, in contrast to 1a–h, improved or equal potencies in our panel of synthesized inhibitors. One obvious difference is the flexible S–Me group of 3. Due to the cyclization of the thiazoline group, we would expect different C-2/7a, S, CH2/3 angles between 1 and 3. These angles are 88.7° for the thiazoline and 99.3° for the trisubstituted imidazole, a difference of 10.6° (Fig. 4). In contrast to 1 and 2, another obvious difference is the NH function of 3, which serves as an additional hydrogen bond donor. This additional donor character is clearly shown in the crystal structure of the methylsulfanyl imidazole.
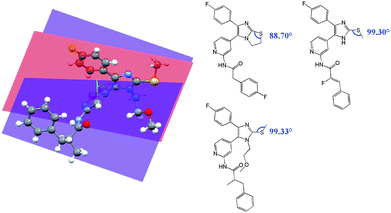 |
| | Fig. 4 Calculated11 angles between the N1–C2–N2 and the C2–S–CH3 plane of the crystal structure of structural analogues of 2 and dihedral angles of the C2/7a–S–CH3/2. | |
We next considered why structures 1a–h and 3a–h showed generally better activity profiles than structures 2a–h. The loss of the NH donor function due to the N-methylation cannot explain the lower activity since this functionality was present in the potent inhibitors 1a–h. The changes in the dihedral angles of 2 with respect to 1 can be also excluded since this feature was present in the highly potent inhibitors 3a–h. We then considered the steric hindrance of the methyl groups to be the possible culprit in the loss of activity. Through the repulsion of the methyl groups, a new conformation is favored, which differs from SKF86002-inhibitor 1 by the orientation of the S–Me group. A related crystal structure to 2a–h has also been published by our group.13 The orientation of the S–Me group in this crystal structure supports the presumed orientation of the S–Me group. Of course, the crystal structure does not show the real conformation in solution, but it is an indication of the steric intramolecular parameters of tetrasubstituted imidazoles. The angle between N1–C2–N2 plane and the C2–S–Me plane is 155.75° (Fig. 4).
To quantify this effect, we performed a conformational analysis (Fig. 5).14 The energetic dependence on the orientation of the S–methyl group in relation to the N–methyl group is shown in Fig. 6.
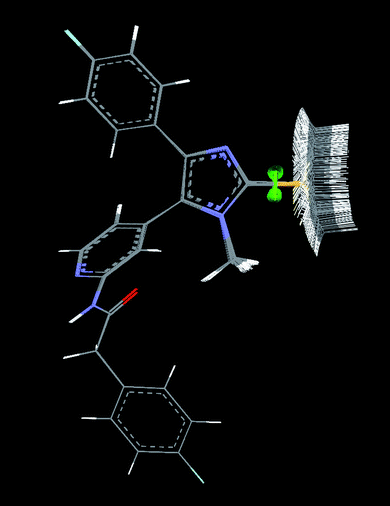 |
| | Fig. 5 Orientations of S-Me to N–Me (conformation method: Boltzman Jump; energy threshold: 20 kcal mol−1; max. conformations: 250). | |
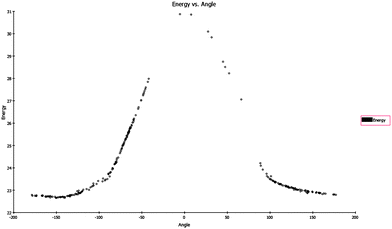 |
| | Fig. 6 Energy of the orientations of the S–Me to the imidazole plane. | |
In the case of the core structure 3, the orientation of the S–methyl moiety is energetically degenerated, therefore the nearly synperiplanar conformation of the fixed SKF86002 core structure can be adopted (Fig. 7). In contrast to this orientation, the favored anti-periplanar orientation of the S–methyl in core structure 2 is positioned in the opposite direction. The energetic difference of 2 to the assumed favored orientation is nearly 8 kcal mol−1. Thus, for binding to the p38α binding pocket, the unfavorable orientation of 1 and 3 has to be essential.
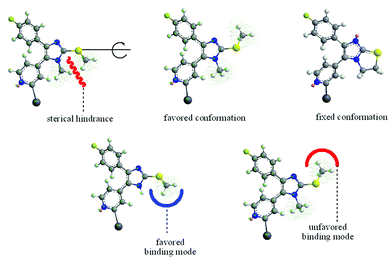 |
| | Fig. 7 Orientation of the S-methyl group in compounds 2 (left/middle top, right below), 1 (right top) and 3 (left below). | |
Docking studies conducted with docking tools from Schroedinger15 are shown in Fig. 8.
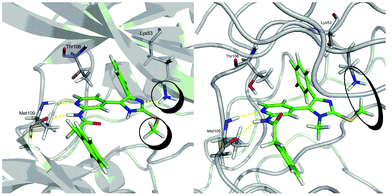 |
| | Fig. 8 Docking results for compounds 3c (left) (GScore: −12.46) and 2c (right) (GScore: −8.30). For these results the X-ray structure 1YWR from the PDB database was used for 3c and 1OUK for 2c. | |
A possible explanation for the decreased activities of 2 is the hydrophobic repulsion of the flexible Lys53 moiety by the restricted flexibility in conformations of the S–Me moiety. The highly flexible Lys53 can interact with the imidazole N-2 via a hydrogen bond. The calculated energetically favored antiperiplanar conformation of 2 induces a hydrophobic repulsion of the flexible Lys53 away from the S–methyl moiety. Therefore, the required proximity for N–H interactions is not achieved, resulting in a loss of inhibitory potency for the 2 core series. With respect to the core structures 1, this repulsion is avoided by the rigidity inherent in the inhibitor design. The advantageous nature of core structure 3 is the energetic degeneration of conformations of the S–Me moiety. The repulsions of the kinase Lys53 or the S–Me moiety are competing forces with respect to the synperiplanar conformation of the inhibitor. In the case of core structure 2, the maximum energetic difference of nearly 8 kcal mol−1 is sufficient for the repulsion of Lys53. In the case of core structure 3, Lys53 forces the S–Me group to adopt the synperiplanar orientation due the energetic degeneration. These results demonstrate the strong energetic contribution of Lys53 to the binding constant of these inhibitors at the p38α binding pocket.
3 Conclusions
In summary, we have shown the strong conformational effects on inhibitor potency caused by steric hindrance of the S–methyl group in the well-known class of pyridinyl thioimidazoles. The steric effects were demonstrated by conformational analysis and docking experiments. By a loss of rigidity, the induced steric hindrance then favors a new orientation which is in opposition to the rigid structures; the loss of rigidity dramatically decreases potency of these compounds. By further structural reduction, the loss of steric hindrance increased inhibitory potency up to 2 fold compared to the comparable dihydrothiazoline structures. Furthermore, we synthesized highly potent inhibitiors with up to 3.5 fold increased potency compared to the well known lead structure SB203580 with IC50 values in the range of 9–20 nM.
4 Experimental
4.1 Molecular modeling
All modeling was performed on an Ubuntu 10.04 LTS system. For visualization and building the structures, Maestro from Schrödinger (Maestro version 9.1, Schrödinger, LLC, New York, NY, 2010) was used. The illustrations of modeling were generated by PyMol (version 0.99rc6). The docking studies were performed using the Induced Fit Docking protocol from Schrödinger (Schrödinger Suite 2010 Induced Fit Docking protocol; Glide version 5.6, Schrödinger, LLC, New York, NY, 2010; Prime version 2.2, Schrödinger, LLC, New York, NY, 2010.) The PDB codes and references for the used X-ray structures are mentioned in the caption of the figures. In all docking studies mentioned here, the XP mode was used.
For the conformational analysis the tool “Search Small Molecule Conformations” from Accelrys Discovery Studio 2.5 was used. All possible conformations were generated using the stochastic search method Boltzman Jump. The threshold was set to 20 kcal mol−1 and the maximum number of conformations was set to 250. Only the bond between the imidazole and the sulfur was selected as rotatable. The energy plot of Fig. 6 was plotted using Discovery Studio.
4.2 General procedure for the synthesis of compounds 1–3 (a–h)
The COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundcarbonic acid was dissolved in 5–10 ml COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundN-methylpyrrolidinone. After addition of 1.1 eq COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundcarbonyldiimidazole the mixture was stirred for 20 h at room temperature. 0.3 eq of the 2-amino pyridine derivative was added and the reaction mixture was heated to 120 °C for 4 h. The reaction mixture was quenched with a solution of concentrated sodium bicarbonate and extracted with COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundethyl acetate. The crude product was purified by flash chromatography.
N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}benzamide (1a).
(0.147 g, 0.35 mmol, 55%); HPLC purity (254 nm): 99.5%; 1H NMR (400 MHz, DMSO-D6) δ ppm 3.96 (t, J = 6.82 Hz, 2 H, 2-H) 4.26 (t, J = 6.82 Hz, 2 H, 3-H) 7.12 (m, 3 H, meta-4FPhH, para-PhH) 7.48 (m, 4 H, ortho-/meta-PhH) 7.58 (m, 1 H, Pyr.5-H) 8.00 (d, J = 7.83 Hz, 2 H, ortho-4FPhH) 8.21 (s, 1 H, Pyr.3-H) 8.38 (d, J = 4.55 Hz, 1 H, Pyr.6-H) 10.88 (s, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 34.7 (CH2), 46.0 (CH2), 113.6 (CH), 115.3 (d, JC-F = 22.0 Hz, CH), 118.9 (Cq), 124.9 (Cq), 128.4 (CH), 128.3 (CH), 128.9 (d, JC-F = 8.1 Hz), 130.6 (Cq), 131.9 (d, JC-F = 2.9 Hz), 133.9 (Cq), 139.5 (Cq), 142.3 (Cq), 148.5 (CH), 150.4 (Cq), 152.8 (Cq), 157.8 (Cq), 160.3 (d, JC-F = 243.7 Hz), 166.1 (Cq); IR (ATR) [cm−1] 2924, 1670, 1585, 1527, 1508, 1454, 1292, 1261, 1230, 1073, 1011, 992, 883, 852, 834, 755, 680; FAB-MS 407 [M + H]+
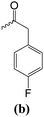
2-(4-fluorophenyl)-N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}acetamide (1b).
(0,155 g, 0.346 mmol, 54%); HPLC purity (254 nm): 99.1%;1H NMR (400 MHz, DMSO-D6) δ ppm 3.70 (s, 2 H, CH2) 3.92 (t, J = 6.82 Hz, 1 H, 2-H) 4.18 (t, J = 6.82 Hz, 1 H, 3-H) 7.03 (d, J = 4.55 Hz, 1 H, Pyr.6-H) 7.11 (m, 4 H, ortho-4FPhH, ortho4FBzH) 7.33 (m, 2 H, meta-4FBzH) 7.42 (m, 2 H, meta-4FPhH) 8.07 (s, 1 H, Pyr.3-H) 8.31 (d, J = 4.80 Hz, 1 H, Pyr.6-H) 10.80 (s, 1 H, NH); 13C NMR (110.67 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO-d6) δ ppm 34.7 (CH2), 41.9 (CH2), 46.0 (CH2), 112.2 (CH), 115.0 (d, JC-F = 23.3 Hz), 115.2 (d, JC-F = 23.3 Hz), 118.6 (CH), 124.8 (Cq), 128.9 (d, JC-F = 8.9 Hz), 130.6 (Cq), 131.0 (d, JC-F = 8.9 Hz),131.7 (Cq), 131.8 (Cq), 139.5 (Cq), 142.3 (Cq), 148.6 (CH), 150.3 (Cq), 152.6 (Cq), 159.9 (Cq), 170.1 (Cq); IR (ATR) [cm−1] 3681, 2923, 1605, 1507, 1455, 1402, 1220, 1156, 1033, 838, 782; FAB-MS 449 [M + H]+
N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}-2-phenylacetamide (1c).
(0.530 g, 1.231 mmol, 96%); HPLC purity (254 nm): 94.9%; 1H NMR (200 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO-d6) δ ppm 3.70 (s, 2 H, CH2) 3.92 (t, J = 7.45 Hz, 2 H, 2-H) 4.18 (t, J = 6.82 Hz, 2 H, 3-H) 7.05 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.27 (m, 5 H, PhH) 7.42 (m, 2 H, ortho-4FPhH) 8.08 (s, 1 H, Pyr.3-H) 8.31 (d, J = 5.18 Hz, 1 H, Pyr.6-H) 10.82 (s, 1 H, NH); 13C NMR (50.33 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO-d6) δ ppm 35.0 (CH2), 43.3 (CH2), 46.5 (CH2), 112.6 (CH), 115.6 (d, JC-F = 21.4 Hz), 118.9 (CH), 125.2 (CH), 128.6 (CH), 129.3 (d, JC-F = 8.1 Hz), 129.5 (CH), 129.8 (Cq), 130.9 (d, JC-F = 2.9 Hz), 136.0 (Cq), 142.7 (Cq), 148.9 (CH), 150.7 (Cq), 153.0 (Cq), 161.6 (d, JC-F = 244.3 Hz), 170.6; IR (ATR) [cm−1] 2931, 1674, 1603, 1544, 1500, 1410, 1287, 1260, 1220, 1157, 1094, 838, 817, 759; FAB-MS: 431 [M + H]+
N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}-2-furamide (1d).
(0.210 g, 0.517 mmol, 80%); HPLC purity (254 nm): 99.5%; 1H NMR (200 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO-d6) δ ppm 3.95 (t, J = 7.07 Hz, 2 H, 2-H) 4.26 (t, J = 7.01 Hz, 1 H, 3-H) 6.68 (m, 1 H, Fur.4-H) 7.12 (m, 3 H, meta-4F-PhH, Fur.3-H) 7.46 (m, 2 H, ortho-4FPhH) 7.58 (d, J = 3.66 Hz, 1 H, Fur.5-H) 7.93 (m, 1 H, Pyr.5-H) 8.13 (s, 1 H, Pyr.3-H) 8.37 (d, J = 5.18 Hz, 1 H, Pyr.6-H) 10.67 (s, 1 H, NH); 13C NMR (50.33 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO-d6) δ ppm 35.1 (CH2), 46.4 (CH2), 112.5 (CH), 113.7 (CH), 115.7 (d, JC-F = 21.1 Hz), 116.1 (CH), 119.2 (CH), 129.3 (d, JC-F = 8.1 Hz), 130.9 (Cq), 139.9 (Cq), 142.8 (Cq), 146.9 (Cq), 149.0 (Cq), 150.9 (Cq), 152.6 (Cq), 156.9 (Cq), (meta-/ipso-4FPhC n.d.); IR (ATR) [cm−1] 3681, 3344, 2941, 2161, 1666, 1602, 1544, 1525, 1494, 1362, 1268, 1248, 1158, 1094, 1056, 836, 690; FAB-MS 407 [M + H]+
3-fluoro-N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}-4-methylbenzamide (1e).
(0.134 g, 0.298 mmol, 49%); HPLC purity (254 nm): 98.6%; 1H NMR (400 MHz, DMSO-D6) δ ppm 2.29 (s, 3 H, CH3) 3.96 (t, J = 6.82 Hz, 2 H, 2-H) 4.26 (t, 6.82 Hz, 2 H, 3-H) 7.12 (m, 3 H, ortho-4FPhH, Ar.2-H) 7.41 (t, J = 7.83 Hz, 1 H, Ar.6-H) 7.48 (m, 2 H, meta-4FPhH) 7.78 (m, 2 H, Ar.5H, Pyr.5-H) 8.18 (s, 1 H, Pyr.3-H) 8.39 (d, J = 5.05 Hz, 1 H, Pyr.6-H) 10.93 (s, 1 H, NH); 13C NMR (110.67 MHz, DMSO-D6) δ ppm 14.2 (CH3), 34.7 (CH2), 46.1 (CH2), 113.6 (CH), 114.4 (d, JC-F = 24.2 Hz), 115.2 (d, JC-F = 22.0 Hz), 119.1 (CH), 124.0 (d, JC-F = 3.7 Hz), 124.9 (CH), 128.7 (Cq), 128.9 (d, JC-F = 8.1 Hz), 130.6 (d, JC-F = 2.2 Hz), 131.6 (d, JC-F = 5.1 Hz), 133.5 (d, JC-F = 7.3 Hz), 139.5 (Cq), 142.4 (Cq), 148.5 (CH), 150.4 (Cq), 152.7 (Cq), 160.1 (d, JC-F = 243.7 Hz), 162.5 (d, JC-F = 244.4 Hz); FAB-MS 449 [M + H]+
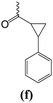
N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}-2-phenylcyclopropanecarboxamide (1f).
(0.171 g, 0.375 mmol, 59%); HPLC purity (254 nm): 98.6%; 1H NMR (400 MHz, DMSO-D6) δ ppm 3.94 (t, J = 7.07 Hz, 1 H, 2-H) 4.21 (t, J = 7.07 Hz, 2 H, 3-H) 7.14 (m, 9 H, meta-4FPhH, PhH) 7.44 (m, 2 H, ortho-4FPhH) 8.12 (s, 1 H, Pyr.3-H) 8.29 (d, J = 5.31 Hz, 1 H, Pyr.6-H) 10.91 (s, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 34.7 (CH2), 46.0 (CH2), 112.3 (CH), 115.2 (d, JC-F = 21.2 Hz), 118.5 (CH), 124.9 (Cq), 126.0 (CH), 126.1 (CH), 128.3 (CH), 128.8 (d, JC-F = 8.8 Hz), 130.6 (d, JC-F = 2.9 Hz), 139.5(Cq), 140.4 (Cq), 142.2 (Cq), 148.5 (CH), 150.3 (Cq), 152.5 (Cq), 161.2 (d, JC-F = 243.7 Hz), 171.0 (Cq); IR (ATR) [cm−1] 2982, 1977, 1684, 1606, 1534, 1506, 1406, 1327, 1260, 1221, 1190, 1157, 1033, 839, 761, 702; FAB-MS 457 [M + H]+
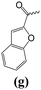
N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}-1-benzofuran-2-carboxamide (1g).
(0.202 g, 0.442 mmol, 72%); HPLC purity (254 nm): 99.2%;1H NMR (400 MHz, DMSO-D6) δ ppm 3.96 (t, J = 6.82 Hz, 2 H, 2-H) 4.28 (t, J = 7.07 Hz, 1 H, 3-H) 7.13 (m, 3 H, ortho-4FPhH, Pyr.5-H) 7.35 (t, J = 7.33 Hz, 1 H, BzFr5-H) 7.49 (m, 3 H, Bzfr6-H, meta-4FPhH) 7.69 (d, J = 8.08 Hz, 1 H, Bzfr7-H) 7.80 (d, J = 7.83 Hz, 1 H, BzFr4-H) 7.99 (s, 1H, Bzfr3-H) 8.19 (s, 1 H, Pyr.3-H) 8.41 (d, J = 4.80 Hz, 1 H, Pyr.6-H) 10.94 (m, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 34.7 (CH2), 46.1 (CH2), 111.3 (CH), 111.9 (CH), 113.5 (CH), 115.2 (d, JC-F = 21.2 Hz), 119.1 (CH), 123.0 (CH), 123.8 (CH), 124.7 (Cq), 126.9 (Cq), 127.3 (CH), 128.9 (d, JC-F = 8.05 Hz), 130.5 (d, JC-F = 3.66 Hz), 139.6 (Cq), 142.5 (Cq), 147.7 (Cq), 148.6 (CH), 150.5 (Cq), 152.0 (Cq), 154.5 (Cq), 157.1 (Cq), 161.3 (d, JC-F = 244.4 Hz); IR (ATR) [cm−1] 3681, 3416, 2973, 1673, 1576, 1520, 1504, 1412, 1285, 1258, 1216, 1206, 1166, 1114, 1097, 1055, 1014, 837, 818, 745; FAB-MS 457 [M + H]+
4-fluoro-N-{4-[6-(4-fluorophenyl)-2,3-dihydroimidazo[2,1-b][1,3]thiazol-5-yl]pyridin-2-yl}benzamide (1h).
(0.031 mg, 0.071 mmol, 12%); HPLC purity (254 nm): 99.0%; 1H NMR (200 MHz, DMSO-D6) δ ppm 3.96 (t, J = 7.08, 6.95 Hz, 1 H, 2-H) 4.27 (t, J = 7.2 Hz, 1 H) 7.13 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.33 (m, 2 H, meta-4FPhAcH) 7.48 (m, 2 H, ortho-4FPhAcH) 8.08 (m, 2 H, ortho-4FPhH) 8.19 (s, 1 H, Pyr.3-H) 8.39 (d, J = 4.80 Hz, 1 H, Pyr.6-H) 10.96 (s, 1 H, NH); 13C NMR (50.33 MHz, DMSO-D6) δ ppm 35.1 (CH), 114.0 (CH), 115.7 (21.9Hz), 119.4 (CH), 125.2 (Cq), 129.2 (8.1 Hz), 130.7 (3.1 Hz), 131.0 (d, JC-F = 3.5 Hz), 131.2 (d, JC-F = 9.2 Hz), 139.9 (Cq), 142.7 (Cq), 148.9 (CH), 150.8 (Cq), 161.7 (d, JC-F = 244.3 Hz), 162.2 (Cq), 165.4 (Cq); IR (ATR) [cm−1] 2924, 2162, 1659, 1603, 1547, 1499, 1413, 1288, 1266, 1225, 1158, 1094, 1014, 839, 812, 758, 678; FAB-MS: 435 [M + H]+
N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}benzamide (2a).
(0.132 g, 0.315 mmol, 49%); HPLC purity (254 nm): 98.8%;1H NMR (400 MHz, DMSO-D6) δ ppm 2.65 (s, 3 H, S-CH3) 3.44 (s, 3 H, N-CH3) 7.10 (m, 2 H, meta-4FPhH) 7.15 (d, J = 5.05 Hz, 1 H, Pyr.5-H) 7.44 (m, 2 H, meta-PhH) 7.50 (m, 2 H, ortho-4FPhH) 7.59 (t, J = 7.96 Hz, 1 H, para-PhH) 8.00 (d, J = 7.33 Hz, 2 H, ortho-PhH) 8.18 (s, 1 H, Pyr.3-H) 8.48 (d, J = 5.05 Hz, 1 H, Pyr.6-H) 10.96 (s, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 15.3 (CH), 31.6 (CH), 115.0 (d, JC-F = 21.2 Hz), 115.5 (CH), 120.9 (CH), 127.8 (CH), 128.0 (CH), 128.3 (CH), 128.4 (CH), 130.4 (d, JC-F = 2.9 Hz), 132.0 (CH), 133.9 (Cq), 137.2 (Cq), 139.8 (Cq), 144.0 (Cq), 148.8 (CH), 152.9 (Cq), 161.0 (d, JC-F = 243.7 Hz), 161.2, 166.1; IR (ATR) [cm−1] 2924, 1676, 1603, 1544, 1521, 1504, 1408, 1375, 1219, 1156, 1060, 838, 708; FAB-MS: 419 [M + H]+
2-(4-fluorophenyl)-N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}acetamide (2b).
(0.384 g, 0.852 mmol, 43%); HPLC purity (254 nm): 98.7%; 1H NMR (400 MHz, DMSO-D6) δ ppm 2.62 (s, 3 H) 3.36 (s, 3 H) 3.73 (s, 2 H, CH2) 7.08 (m, 5 H, meta-4FPhH, meta-4FBzH, Pyr.5-H) 7.35 (m, 4 H, ortho-4FPhH, ortho-4FBzH) 8.02 (s, 1 H, Pyr.3-H) 8.39 (d, J = 5.05 Hz, 1 H, Pyr.6-H) 10.94 (s, 1 H, NH); 13C NMR (100.67 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundDMSO) δ ppm 15.3 (CH3), 31.6 (CH3), 41.9 (CH2), 114.3 (CH), 114.9 (d, JC-F = 21.2 Hz), 115.1 (d, JC-F = 21.2 Hz), 120.5 (CH), 127.8 (Cq), 128.3 (d, JC-F = 8.1 Hz), 130.3 (d, JC-F = 2.9 Hz), 131.0 (d, JC-F = 8.1 Hz), 131.7 (d, JC-F = 3.0 Hz), 137.2 (Cq), 139.8 (Cq), 143.9 (Cq), 148.8 (CH), 152.7 (Cq), 161.0 (d, JC-F = 243.0 Hz), 161.1 (d, JC-F = 242.3 Hz), 170.2 (Cq); IR (ATR) [cm−1] 3031, 1657, 1608, 1571, 1540, 1507, 1412, 1270, 1255, 1216, 1159, 971, 883, 840, 787, 740, 702; LC-MS: ret.t.: 7.57, 451 [M + H]+
N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-phenylacetamide (2c).
(0.052 g, 0.118 mmol, 19%); HPLC purity (254 nm): 99.4%;1H NMR (400 MHz, DMSO-D6) δ ppm 2.62 (s, 3 H, S-CH3) 3.37 (s, 3 H, N-CH3) 3.71 (s, 2 H, CH2) 7.06 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.22 (m, 1 H, para-PhH) 7.30 (m, 4 H, ortho-/meta-PhH) 7.38 (m, 2 H, ortho-4FPhH) 8.03 (s, 1 H, Pyr.3-H) 8.40 (m, 1 H, Pyr.6-H) 10.85 (s, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 15.3 (CH3), 31.6 (CH3), 42.9 (CH2), 114.3 (CH), 115.1 (d, JC-F = 21.2 Hz), 120.5 (CH), 126.6 (CH), 127.8 (Cq), 128.2 (CH), 128.3 (d, JC-F = 8.1 Hz), 129.2 (CH), 130.3 (d, JC-F = 2.9 Hz), 135.6 (Cq), 137.2 (Cq), 139.9 (Cq), 143.9 (Cq), 148.8 (CH), 152.7 (Cq), 161.0 (d, JC-F = 243.7 Hz), 170.2 (Cq); IR (ATR) [cm−1] 3631, 2982,1683, 1607, 1538, 1506, 1408, 1380, 1315, 1268, 1216, 1159, 1033, 839, 817, 776,723,697, 656; FAB-MS 433 [M + H]+
N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-furamide (2d).
(0.202 g, 0.495 mmol, 78%); HPLC purity (254 nm): 97.8%; 1H NMR (200 MHz, DMSO-D6) δ ppm 2.64 (s, 3 H, S-CH3) 3.43 (s, 3 H, N-CH3) 6.68 (m, 1 H, Fur.5-H) 7.11 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.42 (m, 2 H, ortho-4FPhH) 7.60 (d, J = 3.54 Hz, 1 H, Fur.3-H) 7.93 (m, 1 H, Fur.4-H) 8.11 (s, 1 H, Pyr.3-H) 8.46 (d, J = 5.18 Hz, 1 H, Pyr.6-H) 10.75 (s, 1 H, NH); 13C NMR (50.33 MHz, DMSO-D6) δ ppm 15.3 (CH3), 31.6 (CH3), 112.1 (CH), 115.3 (CH), 115.2 (d, JC-F = 21.5 Hz), 115.7 (CH), 120.8 (CH), 127.8 (Cq), 127.8 (Cq), 128.4 (d, JC-F = 8.1 Hz), 130.3 (d, JC-F = 3.1 Hz), 137.3 (Cq), 139.8 (Cq), 144.1 (Cq), 146.5 (Cq), 146.6 (CH), 148.8 (CH), 152.3 (Cq), 161.1 (d, JC-F = 243.8 Hz); IR (ATR) [cm−1] 3122, 1677, 1584, 1544, 1519, 1504, 1413, 1377, 1287, 1217, 1161, 1010, 885, 845, 841, 815, 753, 666; FAB-MS: 409 [M + H]+
3-fluoro-N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-4-methylbenzamide (2e).
(0.173 g, 0.384 mmol, 60%); HPLC purity (254 nm): 100%; 1H NMR (400 MHz, DMSO-D6) δ ppm 2.31 (s, 3 H, CH3) 2.66 (s, 3 H, CH3) 3.45 (s, 3 H, CH3) 7.11 (m, 2 H, meta-4FPhH) 7.17 (d, J = 4.80 Hz, 1 H, Pyr.5-H) 7.44 (m, 3 H, ArH) 7.80 (m, 2 H, ortho-4FPhH) 8.17 (m, 1 H, Pyr.3-H) 8.50 (d, J = 4.80 Hz, 1 H, Pyr.6-H) 11.00 (s, 1 H, NH); 13C NMR (400 MHz, DMSO-D6) δ ppm 14.6 (CH3), 14.6 (CH3), 32.0 (CH) 114.8 (d, JC-F = 24.5 Hz), 115.5 (d, JC-F = 21.4 Hz), 116.0 (CH), 121.4 (CH), 124.4 (d, JC-F = 3.4 Hz), 128.2 (Cq), 128.7 (d, JC-F = 8.4 Hz), 129.0 (Cq), 129.4 (Cq), 130.7 (Cq), 130.8 (Cq), 132.0 (d, JC-F = 5.0 Hz), 133.8 (d, JC-F = 6.9 Hz), 137.6 (Cq), 140.2 (Cq), 144.4 (Cq), 149.2 (CH), 153.1 (Cq), 159.0 (Cq), 160.6 (d, JC-F = 243.5 Hz), 165.1 (Cq); IR (ATR) [cm−1] 3263, 1665, 1544, 1506, 1458, 1413, 1368, 1297, 1270, 1259, 1156, 879, 848, 836, 819, 727, 733, 698; FAB-MS: 451 [M + H]+
N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-phenylcyclopropanecarboxamide (2f).
(0.160 g, 0.348 mmol, 55%);HPLC purity (254 nm): 100%; 1H NMR (400 MHz, DMSO-D6) δ ppm 1.35 (m, 1 H, Cp1-H) 1.45 (m, 1 H, Cp2-H) 2.36 (m, 2 H, Cp3-H) 2.64 (s, 3 H, S-CH3) 3.39 (s, 3 H, N-CH3) 7.09 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.17 (m, 3 H, ortho-PhH, para-PhH) 7.28 (m, 2 H, meta-PhH) 7.40 (m, 2 H, ortho-4FPhH) 8.08 (s, 1 H, Pyr.3-H) 8.40 (d, J = 5.05 Hz, 1 H, Pyr.6-H) 10.97 (s, 1 H, NH); 13C NMR (100.67 MHz, DMSO-D6) δ ppm 15.3 (CH), 16.1 (CH2), 25.4 (CH), 25.8 (CH), 31.6 (CH), 114.3 (CH), 115.2 (d, JC-F = 22.0 Hz), 120.4 (CH),126.0 (CH), 126.1 (CH), 127.9 (Cq), 128.2 (CH), 128.3 (CH), 130.4 (Cq), 137.0 (Cq), 139.9 (Cq), 140.4 (Cq), 143.9 (Cq), 148.8 (CH), 152.6 (Cq), 158.7 (d, JC-F = 231 Hz), 162.2 (Cq), 171.1(Cq); IR (ATR) [cm−1] 3029, 1689, 1606, 1545, 1504, 1405, 1219, 1181, 1156, 957, 838, 817, 757, 697; FAB-MS: 459 [H + H]+
N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-1-benzofuran-2-carboxamide (2g).
(0.175 g, 0.379 mmol, 60%); HPLC purity (254 nm): 97.1%; 1H NMR (400 MHz, DMSO-D6) δ ppm 2.66 (s, 3 H, SCH3) 3.46 (s, 3 H, NCH3) 7.11 (m, 2 H, meta-4FPhH) 7.20 (d, J = 5.05 Hz, 1 H, Pyr.5-H) 7.36 (t, J = 7.58, 1 H, Bzfr.6-H) 7.45 (m, 2 H, ortho-4FPhH) 7.51 (dd, J = 8.34, 7.83 Hz, 1 H, Bzfr.5-H) 7.71 (d, J = 8.34 Hz, 1 H, Bzfr.7-H) 7.82 (d, J = 7.83 Hz, 1 H, BzFr.4-H) 8.02 (s, 1 H, Pyr.3-H) 8.17 (s, 1 H, Bzfr.3-H) 8.52 (d, J = 4.80 Hz, 1 H, Pyr.6-H) 11.05 (m, 1 H); 13C NMR (110.67 MHz, DMSO-D6) δ ppm 15.3 (CH3), 31.6 (CH3), 111.4 (CH), 111.9 (CH), 115.0 (d, JC-F = 22.0 Hz), 115.5 (CH), 121.1 (CH), 123.1 (CH), 123.8 (CH), 126.9 (CH), 127.4 (CH), 127.7 (Cq), 128.4 (d, JC-F = 8.05 Hz), 130.3 (d, JC-F = 2.9 Hz), 137.4 (Cq), 139.9 (Cq), 144.1 (Cq), 147.7 (Cq), 148.9 (CH), 152.1 (Cq), 154.6 (Cq), 157.2 (Cq), 160.1 (d, JC-F = 243.7 Hz); IR (ATR) [cm−1] 3030, 1676, 1580, 1542, 1520, 1505, 1405, 1295, 1221, 1174, 838, 751, 741, 656; FAB-MS: 459 [M + H]+
4-fluoro-N-{4-[4-(4-fluorophenyl)-1-methyl-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}benzamide (2h).
(0.216 g, 0.49 mmol, 78%); HPLC purity (254 nm): 100%; 1H NMR (200 MHz, DMSO-D6) δ ppm 2.65 (s, 3 H, SCH3) 3.43 (s, 3 H, NCH3) 7.11 (m, 3 H, meta-4FPhacH, Pyr.5-H) 7.32 (m, 2 H, meta-4FPhH) 7.44 (m, 2 H, ortho-4FPhacH) 8.08 (m, 2 H, ortho-4FPhH) 8.17 (s, 1 H, Pyr.3-H) 8.48 (d, J = 5.18 Hz, 1 H, Pyr.6-H) 11.03 (s, 1 H, NH); 13C NMR (50.33 MHz, DMSO-D6) δ ppm 15.3 (CH3), 31.6 (CH3), 114.3 (d, JC-F = 21.5 Hz), 114.9 (CH), 115.3 (d, JC-F = 21.9 Hz), 115.4 (CH), 120.9 (CH), 127.8 (Cq), 128.3 (d, JC-F = 8.1 Hz), 130.3 (d, JC-F = 3.1 Hz), 130.4 (d, JC-F = 3.1 Hz), 130.8 (d, JC-F = 9.2 Hz), 137.2 (Cq), 139.8 (Cq), 144.0 (Cq), 148.8 (CH), 152.8 (Cq), 161.1 (d, JC-F = 243.8 Hz), 161.8 (Cq), 164.3 (d, JC-F = 250.0 Hz), 165.1 (Cq); IR (ATR) [cm−1] 3245, 1659, 1603, 1545, 1501, 1456, 1413, 1368, 1291, 1272, 1224, 1159, 1094, 978, 840, 811, 759, 734, 703, 683; FAB-MS: 437 [M + H]+
N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}benzamide (3a).
(0.392 g, 0.969 mmol, 58%); HPLC purity (254 nm): 99.8%; 1H NMR (400 MHz, MeOD) δ ppm 2.64 (s, 3 H, S-CH3) 7.14 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.48 (m, 4 H, meta-PhH, ortho-4FPhH) 7.57 (m, 1 H, para-4FPhH) 7.92 (d, J = 7.58 Hz, 2 H, ortho-PhH) 8.19 (m, 1 H, Pyr.6-H) 8.31 (s, 1 H, Pyr.3-H); 13C NMR (100.67 MHz, MeOD) δ ppm 16.6 (CH), 113.8 (Cq), 116.8 (21.2 Hz), 119.2 (CH), 128.7 (CH), 129.7 (CH), 131.7 (8.8 Hz), 133.3 (CH), 135.6 (Cq), 149.1 (CH), 153.6 (Cq), 165.5 (Cq), 168.4 (Cq), IR (ATR) [cm−1] 3051, 2890, 1673, 1602, 1547, 1523, 1502, 1419, 1286, 1236, 1222, 1174, 1156, 1094, 1077, 837, 813, 693
2-(4-fluorophenyl)-N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}acetamide (3b).
(0.541 g, 1.238 mmol, 62%); HPLC purity (254 nm): 98.6%; 1H-NMR (200 MHz, DMSO-D6): δ ppm 2.59 (s, 3H, SCH3), 3.67 (s, 2H, CH2), 6.99 (d, J = 5.05 Hz, 1H, Pyr.5–H), 7.06–7.53 (m, 8H, ortho-/meta- 4FPhH, ortho-/meta- 4FPhAcH) 8.11 (d, J = 5.31 Hz, 1H, Pyr.6-H), 8.29 (s, 1H, Pyr.3-H), 10.58 (s, 1H, NH), 12.69 (s, 1H, ImNH); 13C-NMR (50.33 MHz, DMSO-D6): δ ppm 15.4, 42.4, 110.9, 115.3 (d, JC-F = 21.0 Hz), 116.2 (d, 2J(C,F) = 21.25 Hz), 116.9, 126.9, 130.5, 131.1 (d, JC-F = 8.5 Hz), 131.3 (d, JC-F = 8.0 Hz), 132.4 (d, JC-F = 3.0 Hz), 134.7, 142.6, 144.2, 148.0, 152.8, 161.5 (d, JC-F = 240.65 Hz), 162.3 (d, JC-F = 246.0 Hz), 170.0; IR (ATR) [cm−1] 3280, 3074, 1695, 1609, 1549, 1500, 1420, 1225, 1156, 1095, 998, 834, 797; LC-MS: ret.t.: 6.37, 437 [M + H]+
N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-phenylacetamide (3c).
(0.035 g, 0.083 mmol, 13%); HPLC purity (254 nm): 94.1%;1H NMR (400 MHz, MeOD) δ ppm 2.61 (s, 3 H, SCH3) 3.69 (s, 2 H, BzCH2) 7.11 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.25 (m, 1 H, para-BzH) 7.31 (m, 4 H, ortho-/meta-PhH) 7.42 (m, 2 H, ortho-4FPhH) 8.14 (m, 1 H, Pyr.6-H); 13C NMR (100.67 MHz, MeOD) δ ppm 16.0 (CH3), 44.7 (CH), 113.3 (CH), 116.8 (d, JC-F = 22.0 Hz), 119.1 (CH), 128.1 (CH), 129.7 (CH), 130.2 (CH), 131.7 (d, JC-F = 8.0 Hz), 136.4 (Cq), 145.1 (Cq), 149.1 (CH), 153.3 (Cq), 164.2 (d, JC-F = 246.7 Hz), 172.5 (Cq); IR (ATR) [cm−1] 2926, 2161, 1764, 1607, 1548, 1503, 1413, 1221, 1157, 969, 836, 814, 719, 694; FAB-MS: 419 [M + H]+
N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-furamide (3d).
(0.610 g, 1.547 mmol, 92.4%); HPLC purity (254 nm): 99.0%; 1H NMR (400 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmethanol-D4) δ ppm 2.64 (s, 3 H, S-CH3) 6.65 (m, 1 H, Fur.4-H) 7.15 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.29 (m, 1 H, Fur.5-H) 7.47 (m, 2 H, ortho-4FPhH) 7.75 (m, 1 H, Fur.3-H) 8.21 (d, J = 5.31 Hz, 1 H, Pyr.6-H) 8.28 (s, 1 H, Pyr.5-H); 13C NMR (50.33 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundCDCl3) δ ppm 15.8 (CH3), 110.9 (CH), 112.6 (CH), 116.1 (d, JC-F = 21.4 Hz), 117.0 (CH), 117.0 (CH), 130.3 (d, JC-F = 8.0 Hz), 143.4 (Cq), 144.5 (Cq), 145.7 (Cq), 146.4 (Cq), 150.2 (Cq), 156.2 (Cq), 160.5 (Cq); IR (ATR) [cm−1] 2930, 1660, 1607, 1579, 1554, 1529, 1503, 1421, 1309, 1298, 1243, 1226, 1157, 1014, 995, 842, 756, 738; LC-MS: ret.t.: 6.22, M= 395 [M + H]+
3-fluoro-N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-4-methylbenzamide (3e).
(0.437 g, 1.001 mmol, 51%); HPLC purity (254 nm): 99.8%; 1H NMR (400 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundmethanol-D4) δ ppm 2.32 (s, 3 H, ArCH3) 2.63 (s, 3 H, S-CH3) 7.13 (m, 3 H, meta-4FPhH, Pyr.5-H) 7.36 (m, 1 H, Ar.2-H) 7.46 (m, 2 H, ortho-4FPhH) 7.62 (m, 2 H, Ar.5-H, Ar.6-H) 8.18 (m, 1 H, Pyr.6-H) 8.27 (s, 1 H, Pyr.3-H); 13C NMR (100.67 MHz, MeOD) δ ppm 14.6 (CH), 16.6 (CH), 113.9 (d, JC-F = 4.4 Hz), 115.3 (d, JC-F = 24.2 Hz), 116.8 (d, JC-F = 21.2 Hz), 119.3 (CH), 124.4 (CH), 130.8 (d, JC-F = 17.5 Hz), 131.7 (d, JC-F = 8.7 Hz), 132.9 (CH), 135.2 (d, JC-F = 6.6 Hz), 149.2 (CH), 153.5 (Cq), 162.4 (d, JC-F = 244.5 Hz), 164.2 (d, JC-F = 247.4 Hz), 166.9 (Cq); IR (ATR) [cm−1] 3275, 2926, 2161, 1680,1608, 1546, 1497, 1415, 1294, 1265, 1223, 996, 839, 825, 766, 748, 679; LC-MS: ret.t.: 6.95, M= 437 [M + H]+
N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-2-phenylcyclopropanecarboxamide (3f).
(0.328 g, 0.738 mmol, 38%); HPLC purity (254 nm): 100%; 1H NMR (400 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundchloroform-D) δ ppm 1.37 (m, 1 H, Cp.3-H) 1.69 (m, 1 H, Cp.3-H) 1.89 (m, 1 H, Cp.1-H) 2.56 (m, 1 H, Cp.2-H) 2.65 (s, 3 H, S-CH3) 6.97 (d, J = 5.05 Hz, 1 H, Pyr.5-H) 7.06 (m, 4 H, meta-4FPhH, ortho-PhH) 7.19 (m, 1 H, para-PhH) 7.26 (m, 2 H, meta-ArH) 7.43 (m, 2 H, meta-4FPhH) 7.90 (d, J = 5.56 Hz, 1 H, Pyr.6-H) 8.32 (s, 1 H, Pyr.3-H) 9.55 (s, 1 H, NH); 13C NMR (100.67 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundchloroform-D) δ ppm 16.1 (CH), 16.8 (CH2), 26.7 (CH), 27.4 (CH), 111.1 (CH), 115.8 (d, JC-F = 22.0 Hz), 117.3 (CH), 126.1 (CH), 126.6 (CH), 128.6 (CH), 130.3 (d, JC-F = 8.0 Hz), 139.9 (Cq), 144.0 (Cq), 146.3 (Cq), 151.6 (Cq), 162.7 (d, JC-F = 248.1 Hz), 171.4 (Cq); IR (ATR) [cm−1] 3072, 2731, 1688, 1609, 1548, 1499, 1422, 1221, 1178, 997, 960, 892, 836, 815, 755, 697
N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}-1-benzofuran-2-carboxamide (3g).
(0.663 g, 1.492 mmol, 80%); HPLC purity (254 nm): 94.2%; 1H NMR (200 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundchloroform-D/DMSO-D6) δ ppm 2.63 (s, 3 H, S-CH3) 7.01 (m, 2 H, meta-4FPhH) 7.17 (m, 1 H, Bzfr) 7.25 (m, 1 H, Bzfr) 7.42 (m, 4 H, Bzfr, ortho-4FPhH) 7.58 (m, 1 H, Bzfr) 7.62 (m, 1 H, Bzfr.) 8.07 (d, J = 5.56 Hz, 1 H, Pyr.6-H) 8.50 (m, 1 H) 9.39 (s, 1 H, NH); 13C NMR (50.33 MHz, COMPOUND LINKS
Read more about this on ChemSpider
Download mol file of compoundchloroform-D/DMSO-D6) δ ppm 16.3 (CH3), 111.8 (CH), 112.4 (CH), 116.2 (d, JC-F = 21.5 Hz), 118.3 (CH), 123.2 (CH), 124.3 (CH), 127.8 (CH), 127.9 (CH), 130.8 (d, JC-F = 8.1 Hz), 144.2 (Cq), 146.6 (Cq), 146.7 (Cq), 148.2 (Cq), 151.1 (Cq), 155.4 (Cq), 156.9 (Cq), 163.0 (d, JC-F = 248.5 Hz); IR (ATR) [cm−1] 3407, 3326, 2161, 1669, 1607, 1575, 1547, 1520, 1502, 1427, 1291, 1221, 1173, 993, 975, 884, 836, 812, 750, 708; FAB-MS: 445 [M + H]+
4-fluoro-N-{4-[4-(4-fluorophenyl)-2-(methylthio)-1H-imidazol-5-yl]pyridin-2-yl}benzamide (3h).
(0.129 g, 0.305 mmol, 46%); HPLC purity (254 nm): 98.8%; 1H NMR (400 MHz, MeOD) δ ppm 2.62 (s, 3 H, SCH3) 7.12 (m, 3 H, meta-4FBzH, Pyr.5H) 7.20 (m, 2 H, meta-4FPhH) 7.45 (m, 2 H, ortho-4FBzH) 7.96 (m, 2 H, ortho-4FPhH) 8.16 (d, J = 5.05 Hz, 1 H, Pyr.6-H) 8.27 (s, 1 H, Pyr.3-H); 13C NMR (100.67 MHz, MeOD) δ ppm 16.6 (CH), 113.8 (CH), 116.6 (d, JC-F = 22.7 Hz), 116.9 (CH), 119.2 (CH), 131.5 (d, JC-F = 8.8 Hz), 131.7 (d, JC-F = 8.8 Hz), 132.0 (Cq), 149.1 (CH), 153.5 (Cq), 164.2 (d, JC-F = 248.1 Hz), 165.6 (d, JC-F = 251 Hz), 167.2 (Cq); IR (ATR) [cm−1] 2925, 2162, 1661, 1603, 1547, 1501, 1413, 1289, 1223, 1158, 1094, 995, 837, 812, 758, 686; FAB-MS: 423 [M + H]+
Acknowledgements
The authors would like to thank the Federal Ministry of Education and Research, Germany, Merckle GmbH, Ulm, Germany, and the Fonds der Chemischen Industrie, Germany, for their generous support of this work.
Notes and references
- J. C. Lee, D. E. Griswold and B. Votta, Int. J. Immunopharmacol., 1988, 10, 835 CrossRef CAS.
- I. Lantos, P. E. Bender, K. A. Razgaitis, B. M. Sutton, M. J. DiMartino, D. E. Griswold and D. T. Walz, J. Med. Chem., 1984, 27, 72 CrossRef CAS.
- D. M. Goldstein and T. Gabriel, Curr. Top. Med. Chem., 2005, 5, 1017 CrossRef CAS.
- Z. Wang, B. J. Canagarajah, J. C. Boehm, S. Kassisa, M. H. Cobb, P. R. Young, S. Abdel-Meguid, J. L. Adams and E. J. Goldsmith, Structure, 1998, 6, 1117–1128 CrossRef CAS.
- S. Laufer, D. Hauser, D. Domeyer, K. Kinkel and A. Liedtke, J. Med. Chem., 2008, 51, 4122 CrossRef CAS.
- K. Ziegler, D. Hauser, A. Unger, W. Albrecht and S. Laufer, ChemMedChem, 2009, 4, 1939 CrossRef CAS.
- S. Laufer, G. Wagner, D. Kotschenreuter and W. Albrecht, J. Med. Chem., 2003, 46, 3230 CrossRef CAS.
-
W. Albrecht; C. Greim; H. G. Striegel; K. Tollmann; P. Merckle; S. Laufer.; PCT Int. Appl. WO 2006089798 A1 20060831, 2006.
- M. Goettert, R. Graeser and S. Laufer, Anal. Biochem., 2010, 406, 233–234 CrossRef CAS.
- R. Selig, D. Schollmeyer, W. Albrecht and S. Laufer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o1132 CrossRef.
- Mercury 2.3 (Build RC4).
- R. Selig, D. Schollmeyer, T. Stegmiller, W. Albrecht and S. Laufer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o3284 CrossRef.
- K. Ziegler, D. Schollmeyer and S. Laufer, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, o3128 CrossRef.
-
Accelrys Discovery Studio 2.5.0.9164, Accelrys Software Inc., 2005–2009 Search PubMed.
-
Glide version 5.0, Schroedinger, LLC, New York, NY, 2005 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.