The p53-MDM2/MDMX axis – A chemotype perspective
Kareem
Khoury
a,
Grzegorz M.
Popowicz
b,
Tad A.
Holak
b and
Alexander
Dömling
*a
aUniversity of Pittsburgh, Department of Pharmaceutical Science, Drug Discovery Institute, Pittsburgh, PA, USA. E-mail: asd30@pitt.edu
bMax Planck Institut für Biochemie, München, Germany
First published on 9th March 2011
Abstract
The protein-protein interaction (PPI) of the tumor suppressor p53 and its negative regulator MDM2 consists of the most intense studied PPI with a group of small molecular weight antagonists described and many more disclosed in patent literature. Due to the Å-level structural insight into p53 interaction with MDM2 there is a reasonable understanding of the requirements of the molecules to bind. In contrast and despite the very close homology and 3-D similarity no potent MDMX antagonist has been disclosed up to date. The current review summarizes the different disclosed chemotypes for MDM2 including a discussion of the cocrystal structures. Structures and approaches to reconstitute functional p53 from mutated p53 are presented. Finally new screening methods and recent biotech deals based on p53 are discussed.
 Kareem Khoury | Kareem Khoury studied chemistry at the University of Pittsburgh. Currently he is pursuing his graduate degree in the field of medicinal chemistry at the University of Pittsburgh in the School of Pharmacy in the laboratory of Prof. Dömling. His interests include the design and synthesis of small molecular weight antagonists of protein-protein interactions and the discovery of novel agents for the treatment of neglected tropical diseases and other unmet medical needs. |
 Grzegorz M. Popowicz | Grzegorz Popowicz studied medical physics at the AGH University of Science and Technology, Krakow, Poland, and obtained his Master Degree in 2002. He then carried out his PhD research at the Max Planck Institute for Biochemistry, Martinsried, Germany, completing his PhD in 2006. He is currently a research associate scientist at this Institute. His research is focused on controlling protein-protein interactions for therapeutic purposes. Using NMR and X-ray crystallography techniques, he is developing small molecule and peptidic inhibitors for proteins involved in tumorigenesis. |
 Tad A. Holak | Tad A. Holak is the head of the Biological NMR Structure Group at the Max Planck Institute for Biochemistry, Munich-Martinsried, Germany. His research focuses on discovering three-dimensional structures and structure/function properties of proteins using the combination of multinuclear NMR, biochemistry and X-ray crystallography. The proteins studied are involved in cancer. From 1998–2009, he was a member of the editorial Board of the European Journal of Biochemistry and FEBS Journal. |
 Alexander Dömling | Alexander Dömling studied chemistry & biology at the Technical University Munich. After performing his PhD under the supervision of Ivar Ugi, he spent his postdoctoral year at the Scripps Research Institute on a Feodor Lynen stipend in the group of Nobel Laureate Barry Sharpless. He is founder of several biotech companies, including Morphochem and R&D Biopharmaceuticals. In 2004 he performed his habilitation in chemistry at the Technical University of Munich. Since 2006 he is associate professor at the University of Pittsburgh in the Department of Pharmacy and secondary appointments in Chemistry and Computational and Systems Biology. His research interests in chemistry are the design of new and stereoselective multicomponent reactions (MCR) and its applications to drug discovery, recently to discover antagonists of protein protein interactions. He is interested in the therapeutic areas of oncology, infectious and neglected tropical diseases. |
1 Introduction
p53 was discovered thirty years ago as the oncoprotein of a simian virus 40 large T-antigen.1 The first decade of p53 research focused on the cloning of p53 DNA; shortly after, however, it was realized that p53 is a tumor suppressor protein that is frequently mutated in human cancer.2,3 p53 was then uncovered as a transcription factor induced by stress, which can promote cell cycle arrest, apoptosis and senescence.4,5 In 1992 MDM2 was shown to bind tightly to p53 and inhibit its biological activity.6MDM2 has emerged as the key regulator of p53, and has been termed the “gatekeeper” of p53.7 Currently many pharmaceutical companies and academic institutions are focusing on ways to enhance p53 activity in tumor cells in hopes to develop a novel, more effective and better tolerated form of cancer treatment.8Currently, around 22 million people are living with a tumor that contain either an inactivating mutation of TP53 (the human gene that encodes p53) or have tumors in which the activity of p53 is partially negated through the inactivation of other signaling or effector components.9 Attention was recently drawn to a very aggressive brain tumor (Glioblastoma mutliformis) when Sen. Ted Kennedy died from it only 15 months after diagnosis.10 It is well established that many forms of Glioblastoma carry cells highly overexpressing the negative regulators of p53, MDM2 and/or MDMX, causing a severe decrease in p53 expression.11 Additionally, recent evidence suggests that combined use of MDM2 and MDMX antagonists could activate p53 more effectively then MDM2 antagonists alone.12,13
p53 is termed a tumor suppressor gene because its activity can stop the formation of tumors (Fig. 1). It belongs to a small family of related proteins that includes only two other members (p67 and p73), however of the three, p53 is the main one utilized by the cell to prevent tumor growth. p53 plays an essential role in guarding cells in response to various stresses, such as hypoxia or DNA damage, by inducing cell cycle arrest, repair, or apoptosis. It is specifically involved in the effects of survival of proteins in the mitochondria, regulating microRNA processing, DNA repair, and protein translation (to name a few). p53 prevents damaged cells from multiplying and passing mutated genes to the next generation, impairment allows for these processes to go unregulated.17 In fact a study showed that p53 deficient mice developed normally; however were prone to spontaneous tumor generation.18 Therefore cells that lack p53 have the potential to pass mutations on to the next generation which can facilitate tumor growth. p53 is a potent growth suppressive and proapoptotic protein that would harm normal proliferating cells if left uncontrolled.
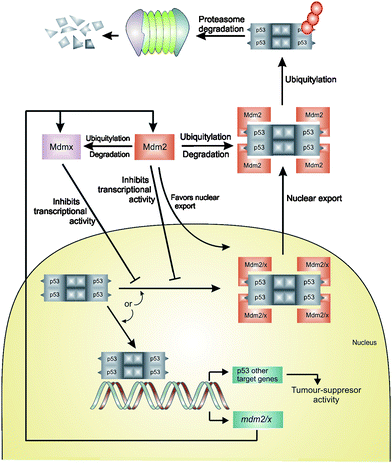 | ||
| Fig. 1 Regulation of p53 by MDM2 and MDMX through the autoregulatory feedback loop. The model above assumes that MDM2 and MDMX work independently to inhibit p53 activity.14,15 An alternative model assumes that MDM2 and MDMx form a heterodimer that is more effective at inhibiting p53 transactivation or enhancing p53 turnover. The second model is supported by the observation that in vitro the MDM2/MDMX heterodimers are more effective p53 ubiquitin ligases than MDM2 homodimers.16 | ||
The level of p53 protein in a cell is low and its concentration and activity are subjected to tight control both under normal physiological conditions and during stress. In an unstressed cell, this is accomplished by MDM2-mediated degradationvia the ubiquitin-proteosome pathway (regulation of p53 stability), and by inactivation of the p53 transcriptional activity primarily due to the MDMX-mediated occlusion of the p53 transactivation domain (inhibition of p53 activity). In addition MDM2 and MDMX modify the activity of p53 by transporting p53 into cytoplasm, away from nuclear DNA. Thus the activity of p53 as a transcription factor is out of reach. After stress, MDM2 degrades itself and MDMX, leading to the accumulation and activation of p53. Increased nuclear levels of p53 activate MDM2/X gene transcription, leading to elevated levels of MDM2 and MDMX. As activated p53 transactivates MDM2, the increasingly abundant MDM2 degrades MDMX more efficiently, enabling full p53 activation: the transcriptional stress response is at its peak. Following stress relief, the accumulated MDM2 preferentially targets p53 again; p53 levels decrease, and MDMX levels increase, p53 activity also decreases. Small amounts of p53 will reduce the amount of MDM2 protein and this will result in an increase of p53 activity, thus completing the loop. The switch that makes MDM2 preferentially target p53 for degradation in unstressed cells, then target itself and MDMX after stress relief, is not precisely understood.14,15
In cases where the cancer is not due to a mutation of p53 but due to an overexpression of a suppressor protein, such as MDM2/X, it is in principle possible to inhibit this interaction by a small molecule and release active p53 to the cell. In fact more than 50% of tumors show an overexpression and/or amplification of MDM2 and its gene. MDM2 is a special example of a protein that regulates p53 through an auto-regulatory feedback loop, in which p53 also regulates MDM2. p53 transcriptionally activates MDM2, and MDM2 in turn inhibits p53 in several ways.19MDM2 is an E3 ubiquitin ligase that either targets p53 for ubiquitin dependent degradation or inhibits p53 by modulating its activity and preventing interactions with other proteins.6 Other than MDM2, MDMX also plays an important role in regulation of p53 however is much less characterized (MDMX is also known as MDM4, and human versions as HDMX, and HDM4). MDMX is structurally homologous to MDM2 with a high degree of sequence homology and structural similarity (Fig. 2). MDMX can either act alone or form a heterocomplex with MDM2 and enhance ubiquitination of p53.7,20,21 There has been extensive validation of MDM2 as a target showing that even a small reduction in MDM2 is significant enough to increase p53 activity.7
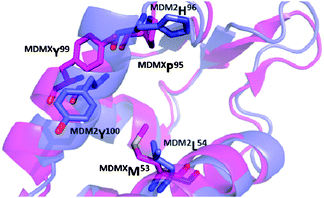 | ||
| Fig. 2 Alignment of the p53 receptors MDM2 (blue, PDB-ID 1YCR) and MDMX (pink, PDB-ID 3DAB). The changes in amino acids L54M and H96P, as well as a rotation of Y101 create a distinct sub binding pocket of MDMX and can help to explain the differences in binding of small molecule to MDM2 and not MDMX. | ||
Activated p53 does not necessarily induce apoptosis in normal cells. It has been shown that tumor cells show a greater propensity to die in response to p53 as opposed to normal cells. Therefore there is great hope that specific p53/MDM2 inhibitors will act selectively on cancer cells. In certain animal experiments, however a side effect of p53/MDM2 inhibitors was the cause of death of thymocytes and gut epithelium cells.17 However the abnormal proliferation, loss of normal cell environment, and stress of a tumor cell can lead to enhance sensitivity to induce apoptosis in cancer cells.17
2 p53-MDM2/X interactions
The p53/MDM2 interface is defined as a protein-protein interaction (PPI). PPIs prove to be a difficult case for drug discovery in that the interface between two proteins is generally large (between 600 and 1300 Å2) and involve contacts from as many as 30 side chains from each protein.22 In typical drug discovery targets smaller molecules are preferred (to enhance absorption, distribution, metabolism, and excretion (ADME) characteristics) and prove to be a problem to inhibit such a large area (Fig. 3). However it has been shown that though two proteins make many contacts with each other there is generally a small pocket of a few amino acids that make up for the majority of the binding energy. This pocket has been termed a “hot spot” and can be used to target small molecule inhibition of PPIs.23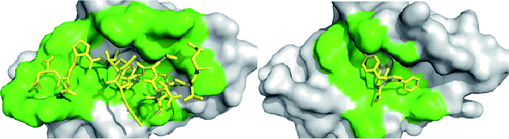 | ||
| Fig. 3 Receptor contact surface covered by the native p53 peptide and a corresponding small molecule antagonist. The target protein (MDM2) is represented as a grey solid surface, and peptide binder p53 is shown as yellow sticks (PDB-ID 1YCR). The contact surface on the target protein with the peptide and small molecule binder is shown in green. The small molecule (PDB-ID 3LBK) shows much less interaction to MDM2. Contact surface was defined as an amino acid within 4 Å of the peptide or small molecule. | ||
The 3D structure of MDM2 and MDMX with and without p53 derived peptides and small molecules is extensively elaborated in more than 20 high resolution X-ray and NMR structures. In a seminal work in 1996 Pavletich et al. described the first crystal structure of the interaction of p53/MDM2.24MDM2 has a deep and structured binding pocket for p53 (Fig. 4). The binding pocket measures only ∼18 Å2 along the long edge, the size of a typical small molecule.24 The p53/MDM2 complex has a “hot spot triad” made up of p53's Trp23, Leu26, and Phe19. The three hydrophobic amino acids fit into three shape and electrostatic complementary hydrophobic pockets, and the indole nitrogen of p53's Trp23 forms a hydrogen bond with Leu54 of MDM2 (Met53 in MDMX). In fact much of the binding energy resides in these three amino acids. Alanine scan studies show that mutation of any of the three hot-spot amino acids destroys the affinity between p53 and MDM2.25 A prerequisite for high affinity MDM2 antagonists is therefore that certain moieties of the molecule must mimic the three amino acids of p53's hot spot triad Trp23, Leu26, and Phe19. In fact an illustrative model termed “three finger pharmacophore” has been created.26
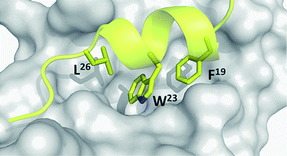 | ||
| Fig. 4 The p53-MDM2 hot spots. MDM2 (grey surface) and the key three amino acids (Leu26, Phe19, and Trp23, yellow sticks) mounted on a small amphipathic p53 derived α-helix (PDB-ID: 1YCR). | ||
3 Small molecule-MDM2 cocrystals
The first reported small molecule p53/MDM2 antagonist with in vivo activity is a representative of the class of cis-imidazoline compounds termed nutlins. The nutlins showed IC50s down to the low nanomolar range (18 nM) as determined by surface plasmon resonance (Table 1). The crystal structure shows the nutlin-2 binding to MDM2 in the p53 pocket in a way that mimics the three essential amino acids Try23, Leu26, and Phe19 (Fig. 5). The bromophenyl moieties in positions 4 and 5 bind deeply into the Leu26, and Trp23 pockets, respectively. The 2-ethoxy phenyl moiety of nutlin-2 mimics p53's Phe19, whereby the phenyl groups serves as a connector to place the ethoxy group to fill the Phe19 pocket but does not deeply penetrate itself. Additionally, the 4-methoxy function of nutlin-2, to mimics p53's Leu22. Nutlin-2 is synthesized via an 8 step sequential synthesis and requires separation of enantiomers via a supercritical chiral HPLC.27–29 An enantioselective shorter synthesis has recently been described.113| Name | MDM2 Ki (mM) | MDMX Ki (mM) | Reference |
|---|---|---|---|
| a Screened via Biacore's surface plasmon resonance technology. b Screened via ThermoFluor Screen. c Screened via Fluorescence Polarization. d Screened viaHTRF. | |||
| Nutlin-2 (1)a | 0.14 (IC50) | — | 28 |
| Benzodiazepindione (2)b | 0.080 | — | 30 |
| Spiroindolone (analog) (3)c | 0.003 | 55 | 33,34 |
| Chromenotriazolopyrimidine (4)d | 1.23 (IC50) | — | 36 |
| Imidazole (5)c | 0.916 | 36 | 35 |
| Imidazole (6)c | 0.109 | 11 | 35 |
 | ||
| Fig. 5 Co-crystal structure of Hoffmann La-Roche's nutlin-2 1 (green sticks), (PDB-ID: 1RV1) and MDM2 (grey surface) superimposed on the key amino acid side chain residues of the p53/MDM2 structure (yellow sticks, PDB-ID: 1YCR). | ||
A precursor of benzodiazepindione 2 (Fig. 6) by Johnson & Johnson was discovered through a high throughput ThermoFluor screen to inhibit p53/MDM2 (Table 1). Similar to nutlin-2, a high resolution X-ray structure exists, giving valuable structural insight into the binding interaction of this small molecule to MDM2. The substituents of the benzodiazepine show a strong mimic of p53's binding into the MDM2 pocket (Fig. 6). The two chlorophenyls bind to the Leu26 and Trp23 pocket, and the iodophenyl moiety binds in the Phe19 pocket. This molecule was synthesized by a two-step one pot Ugi reaction. Many derivatives are easily accessible and an in depth structure activity relationship has been published.30 For example, the introduction of a pentenylicacid to the N1 of the benzodiazepine ring improved potency, solubility, cell-based activity and enabled access to modify ADME properties. Compound 2 has been shown to be efficacious in animal models and acts in synergy with doxorubicin.31,32
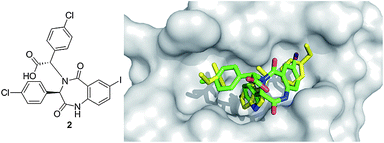 | ||
| Fig. 6 Co-crystal structure of Johnson & Johnson's benzodiazepine small molecule 2 (green sticks, PDB ID: 1T4E) and MDM2 (grey surface) superimposed on the key amino acid side chain residues of the p53/MDM2 structure (yellow sticks, PDB ID: 1YCR). | ||
A spiroindolone compound 3 was discovered by Ding et al. and is currently the most potent p53/MDM2 inhibitor scaffold published with a Ki of 3 nM derived from fluorescence polarization.33 In addition it has good cell based activity showing inhibition of cell growth in cancer cells with wild-type p53, excellent specificity in cancer cells with p53 knock-out and shows minimal toxicity in normal cells.34 Structural analysis of the crystal structure of an equipotent diastereoisomer is showing the oxindole group occupying the Trp26 pocket and also forming a hydrogen bond to MDM2's Leu54, much like p53's Trp26 (Fig. 7).35 This scaffold can be easily modified via an amidation step to allow for the synthesis of additional compounds with very potent inhibitor activity and good water solubility. The morpholino ethyl side chain does not show electron density but can be placed over the Leu22 of p53 based on the carbonyl His96 hydrogen bond interaction. Though this compound showed great affinity for MDM2, it had reduced cell permeability. A compound of the same class with a non-basic solubilizing side chain has recently been reported.34 This compound inhibits p53/MDM2, activates the p53 pathway in cells with wild type p53 and leads to cell cycle arrest in all cells and selective apoptosis in tumor cells. It has been shown to be active in cancerous animal tissue, resulting in inhibition of cell reproduction, induction of apoptosis and complete tumor growth inhibition.35
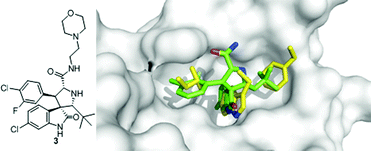 | ||
| Fig. 7 Co-crystal structure of Wang's spiroindolone small molecule 3 (green sticks, PBD-ID: 3LBL) and MDM2 (grey surface) superimposed on the key amino acid side chain residues of the p53/MDM2 structure (yellow sticks, PDB-ID: 1YCR). | ||
A chromenotriazolopyrimidine 4 was discovered to be a low μM inhibitor of p53/MDM2via homogeneous time-resolved fluorescence (HTRF) high-throughput screening by Amgen workers.36 Crystal structure analysis shows that the small molecule is able to mimic the key amino acids of p53 (Fig. 8). The two bromophenyl moieties bind to the Leu 26, and Trp23 pockets. The bromophenyl binding to the Leu26 pocket shows a weak π-stacking interaction with the nearby rim His96 imidazole of MDM2. The benzene ring on the backbone of the chromenotriazolopyrimidine binds into the Phe19 pocket. Efforts to optimize this compound included substitution of the two halogens, and additions to the benzene ring bound the Phe19 pocket. Such efforts showed an improvement in binding affinity with substitution of bromine to chlorine in the Trp23 binding site and addition of a methoxy group added to the 4 position of the benzene ring.36 Recently derivatives with improved pharmacokinetic properties were disclosed.37
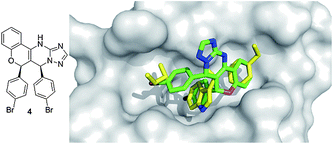 | ||
| Fig. 8 Co-crystal structure of Amgen's chromenotriazolopyrimidine small molecule 4 (green sticks, PDB ID: 3JZK) and MDM2 superimposed on the key amino acid side chain residues of the p53/MDM2 structure (yellow sticks, PDB ID: 1YCR). | ||
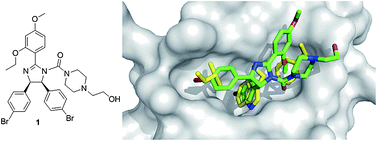
A recent crystal structure of 5 based on a threefold substituted imidazole scaffold (Fig. 9) discovered independently and at the same time by NOVARTIS and Dömling et al. from University of Pittsburgh adds to the p53/MDM2 structures.35 This one step, multicomponent reaction product shows high nM activity in fluorescence polarization (Table 1). The structure shows the indole of the small molecule mimicking the Trp26 pocket of p53, and a para-chlorobenzyl group mimicking the Leu22 of p53, and a para-fluorophenyl group mimicking the Phe19 of p53. The alignment of the indole group of the small molecule and the indole moiety of the Trp23 is perfect. The ligand indole-NH forms a hydrogen bond (2.73 Å) to the backbone carbonyl of Leu54. The 6-chloro group in the indole resides in a hydrophobic pocket, which is not filled in the p53-MDM2 complex, brings additional hydrophobic interactions. The carboxyl group in 2 position of the indol forms two bridged hydrogen bond to two crystallographic water of distances 2.57 Å and 3.35 Å.35
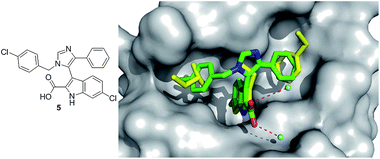 | ||
| Fig. 9 Co-crystal structure of Dömling's imidazole small molecule 5 (green sticks, PDB ID: 3LBK) and MDM2 superimposed on the key amino acid side chain residues of the p53/MDM2 structure (yellow sticks, PDB ID: 1YCR). | ||
4 MDMX – cocrystals
Much less structural biology information is available for MDMX. Only one small molecule has been co-crystallized up to date. Alignment of crystal structures of both MDM2 and MDMX can potentially draw pathways to selectively inhibit one or the other, or create a dual acting small molecule inhibitor for both proteins. Crystal structures reveal (Fig. 10) that although principal features of the p53/MDM2 interaction are still present in p53/MDMX, the MDMX has a significantly altered hydrophobic cleft on which p53 binds compared to MDM2. MDMX's Met53 and Tyr99 protrude into the pocket causing a smaller and differently shaped, shallower binding pocket. Though the Met54 side chain of MDMX is in the same position as MDM2's Leu54, the bulkiness of methionine compared to leucine creates a smaller binding cleft. A shift of MDMX's helix α2 causes a conformational change, which contribute significantly to the alterations in the p53 binding pocket of MDMX. The conformation of MDMX's Tyr99 is altered compared to MDM2's Tyr100. MDM2's Tyr100 is flipped away from the p53 binding pocket compared to MDMX's Tyr99, also contributing to a smaller binding pocket. (Fig. 10).38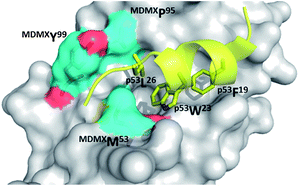 | ||
| Fig. 10 Co crystal structure MDMX with p53 (yellow sticks). Highlighted in blue (surface and sticks) are the three key amino acids that contribute to major differences in binding site between MDM2 and MDMX (PDB ID: 3DAB). | ||
Popowicz et al. recently published the first co-crystal structure of a small molecule inhibitor 6 and MDMX (Fig. 11).35 This imidazole compound 6 is similar to compound 5 binding to MDM2, and binds to MDMX in a way that mimics parts of the binding of p53. The central imidazole scaffold directs the three ligands into the three key sub binding pockets corresponding to p53Trp23, p53Phe19 and p53Leu26. The p53Trp23 pocket is filled with the 6-chloroindole substituent whose NH forms a hydrogen bond with the carbonyl atom of MDMXMet53 of 2.64 Å and thus mimics the same molecular interaction as seen with p53Trp23 and MDMX (2.77 Å). Additionally, the oxygen of the 2-carboxamide forms a solvent exposed hydrogen bond to the MDMX His54 of 3.28 Å. The 6-chloroindole substituent resides very similar to the corresponding MDM2 structure (Fig. 11) in the deep and hydrophobic Trp23 pocket of MDMX making extensive hydrophobic contacts to the receptor amino acids Leu98, Phe90, Leu56, Ile60, Gly57, and Val92. The 4-chlorobenzyl ring of the imidazole penetrates the Leu26 pocket and the imidazole's phenyl ring fills the Phe19 pocket; however, the plane of this ring is nearly perpendicular to the plane of p53's Phe19. The carboxamide moiety of the molecule bends over the phenyl group and comprises an additional filling of the large and hydrophobic Phe19 pocket, thus shielding the hydrophobic region of Met61 from solvent. Similar compounds without the dimethylpropylamine side chain show much weaker inhibition of p53/MDMX. The amino acid exchanges from MDM2 to MDMX (L45M and H95P) form a distinct sub binding pocket and can explain the difference in binding of small molecules.35
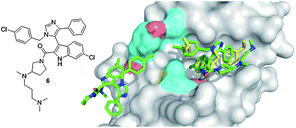 | ||
| Fig. 11 First co-crystal structure of MDMX (grey surface) with a small molecule inhibitor 6 (green sticks, PDB ID: 3LBJ) superimposed on the key amino acid side chain residues of the co-crystal of p53/MDMX (PDB ID: 3DAB). It is interesting to note that due to the slight difference in the amino acids of MDM2 and MDMX, MDMX forms an additional second pocket for a second small molecule inhibitor to bind to. This could allow for selectivity of MDMX over MDM2 when designing inhibitors. | ||
5 p53 Mutation
Downregulation of p53 can also be caused by mutations of p53. The most frequent is a mutation of Tyr220 to cysteine (Y220C). p53, in its wild type state, has a melting temperature of 44 °C and a half-life of approximately 9 min. The Y220C mutation lowers the melting temperature and stability of p53 in its DNA binding domain, causing it to denaturate very rapidly and is either too unstable to function in the body or depleted by denaturing agents. Recently, researchers have been attempting to create small molecules that selectively bind to the mutated p53 to restore its function. This principle of p53 reactivation was first shown using peptide CDB3, which binds to the DNA binding domain of p53, to stabilize both wild-type and mutant p53 in vitro, and elevated the activity of mutant p53 in cancer cell lines.39,40Recently, Boeckler et al. from Cambridge UK has solved the first co-crystal structures of small molecules binding to and stabilizing mutant p53. Their first molecule, PhiKan083 (8), was discovered by in silico screening methods. It can be seen that the central carbazole moiety of 8 is largely buried in the cleft, with the 9-ethyl group occupying the deepest part of the hydrophobic pocket (Fig. 12). The planar carbazole ring system is sandwiched between the hydrophobic side chains of Pro-222 and Pro-223 on one side, and Val-147 and Pro-151 on the other side of the binding cleft (Fig. 12). The ring nitrogen sits close to the position of the hydroxyl group of the tyrosine residue in the wild-type structure. The N-methylamine moiety forms a hydrogen bond with the main-chain carbonyl of Asp-228.39
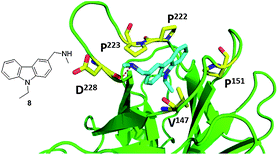 | ||
| Fig. 12 Co-crystal structure of T220C (green cartoon) p53 mutant and 8 (blue sticks) (PDB ID: 2VUK). Key interacting amino acids of p53 binding shown in yellow sticks. | ||
The same group discovered – via fragment based screening – a second type of compounds that bind to the mutated p53. The structure of fragment 9 bound to mutated p53 shows that the molecule sits deeply within the mutation-induced cavity (Fig. 13). The sulfur atom on the thiazole ring is positioned near the mutated Cys220 residue at the deepest part of the cleft. In addition to hydrophobic packing interactions, fragment 9 forms specific hydrogen bonds with the protein and water molecules. Most significantly, the substituent amine in the 2-position on the thiazol ring makes a hydrogen bond with the carbonyl oxygen of Leu145.40
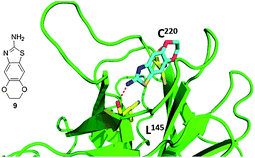 | ||
| Fig. 13 Co-crystal structure of T220C p53 mutant (green cartoon) and fragment 9 (blue sticks). This compound was discovered via fragment screening (PDB-ID: 2XOU). Key interacting amino acids of p53 binding shown in yellow sticks. | ||
Fragment 10, which also contains an aromatic benzothiazole ring system, binds in the same plane as fragment 8 within the cavity but sits in a more upright position (Fig. 14). Positioned as such, the thiazole nitrogen is within hydrogen-bonding distance of the hydroxyl group of Thr150. The methoxy moieties of the molecule sit at the bottom of the cavity in close proximity to Cys220.40
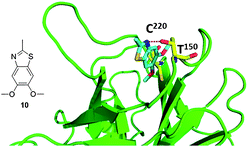 | ||
| Fig. 14 Co-crystal structure of T220C p53 mutant and fragment 10, this compound was discovered via fragment screening (PDB ID: 2XOW). Key interacting amino acids of p53 binding shown in yellow sticks. | ||
The binding mode of fragment 11 is unique in that two molecules are bound within the cavity (Fig. 15). The molecules are positioned at opposite sides of the cavity with the trifluoromethyl groups of each pointing toward the mutated Cys220 residue. Binding of one of the molecules is stabilized by the interaction of the amine at position 2 on the aromatic ring with the carbonyl oxygen of, similar to the binding of fragment 10. This molecule also makes hydrophobic interactions with Val147 and Pro222. The second fragment packs between the hydrophobic side chains of Pro153 and Pro222 at a site that is not occupied by the other fragments. The fluorine atoms likely play a significant role in binding by coordinating strongly with the protein and can indirectly strengthen the binding interaction of the fragment by polarizing the amine groups attached to the benzene ring.40
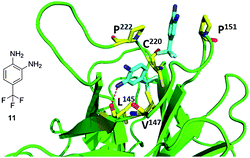 | ||
| Fig. 15 Co-crystal structure of T220C p53 mutant (green cartoon) and fragment 11 (blue sticks). This compound was discovered via fragment screening (PDB ID: 2XOV). Key interacting amino acids of p53 binding shown in yellow sticks. | ||
When we look at the overlap of mutated p53 (Y220C) in comparison to wild type p53, we see that the Trp220 fills the pocket that all of the small molecules bind to in the mutant p53 (Fig. 16). Therefore these molecules will selectively bind to mutated p53 and should have no effect on wild type p53. Restoring mutated p53 with small molecules is therefore a promising approach to also address mutated p53 cancers in the future. Clearly the current fragments are not potent enough and have to be grown and optimized for affinity and other drug-like properties in the future.
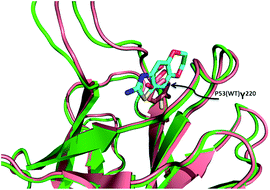 | ||
| Fig. 16 Alignment of T220C p53 mutant (green cartoon) and wildtype p53 (pink cartoon). In the wildtype the tyrosine (pink sticks) fills in the binding pocket of the fragments from the T220C p53 mutant (PDB ID: 2PCX and 2XOU). | ||
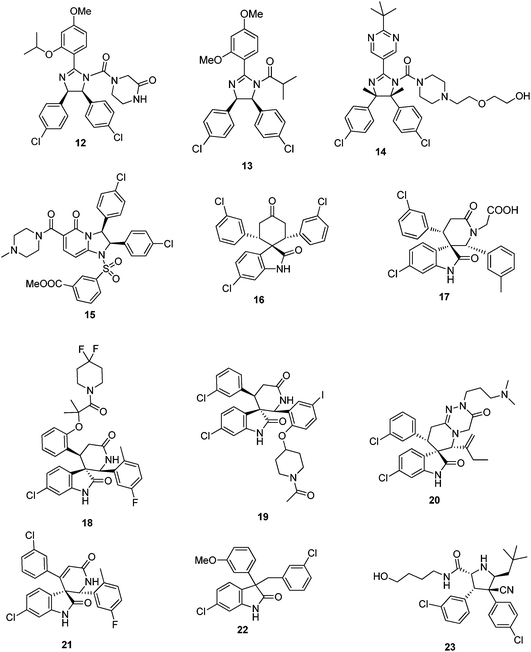
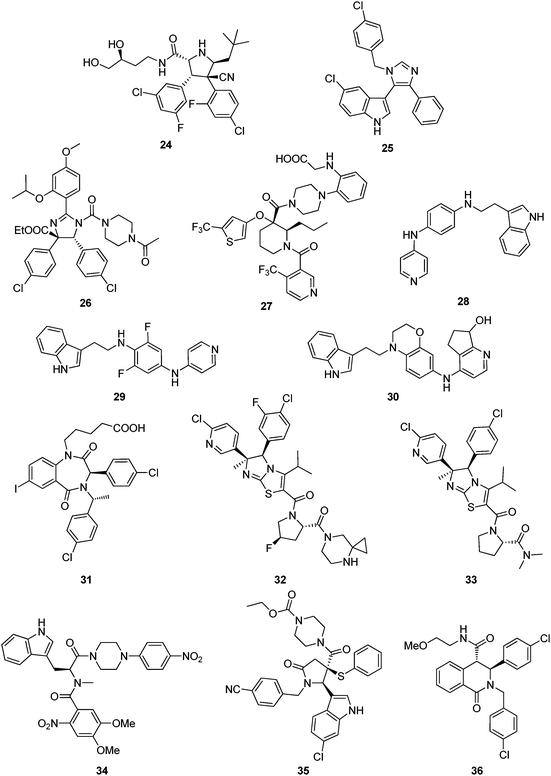
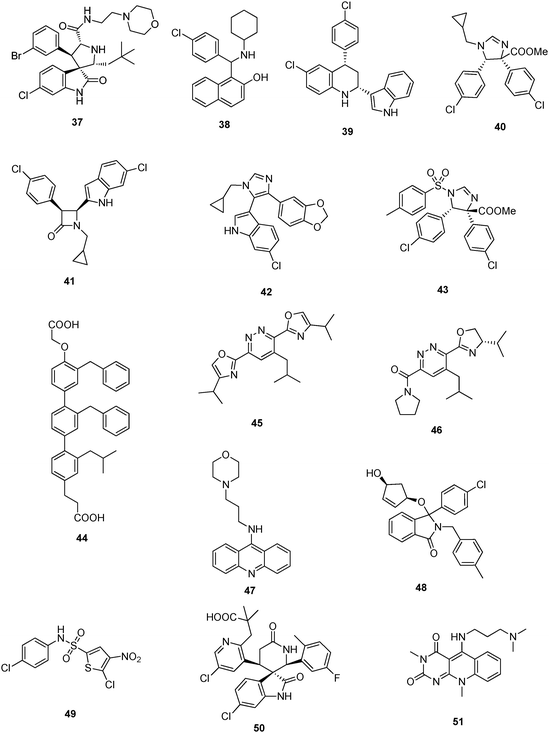
6 The patent landscape of p53-MDM2 and -MDMX inhibitors
The commercial attractiveness of the target area of p53-MDM2 and p5-MDMX is reflected in the vivid protection of intellectual property by industry and academic institutions. To date (October 2010) more than 150 patents and applications containing the key words p53 and MDM2, MDMX, HDM2, HDM4 or MDM4 where found. The key information including general scaffold examples, use and biological assays are summarized and discussed below. The information is clustered according to the inventor/institutions in order to provide an overview on the IP landscape. The plethora of discovered small molecular weight compounds antagonizing the p53 binding site in MDM2 strongly suggests that this PPI is druggable. Therefore higher molecular weight peptide derivatives such as miniature proteins, cyclic peptides, stapled peptides, pavaian pancreatic polypeptides, D-peptides, peptoids, and β-peptides are not subject of the review. Other matter such as siRNA, aptamers, drug fusions and conjugates, antibodies, affybodies, gene technological approaches, immunogenic oligopeptides, antisense, adeno- or herpes-viral methods will also not be discussed here. For other recent summaries of the patents in the area of p53 MDM2 the reader is also directed to the reviews of Weber and Deng et al.41,42Hoffmann-La Roche
Doubtless Hoffmann-La Roche has the largest patent portfolio in the area of p53-MDM2 antagonists. The scaffold of 1,2,4,5-tetrasubstituted 4,5-cis-imidazolidines received the trivial name nutlin, according to the site of discovery Nutley. A derivative nutlin-3 (12) served as a proof-of-concept compound (a probe) to elucidate the p53 pathway and its involvement in cancer.28 With the successful entering of the nutlin derivative RG-7112 (RO5045337) into clinical trial, Roche is also the most advanced company in this field. RG-7112 (Fig. 17) is currently undergoing early clinical trials for haematological neoplasms and advanced solid tumor.43,44,108 The candidate compound differs from the initial generation of nutlins by an extended N-3-propyl-methylsulfone piperazine side chain possibly adding additional hydrogen bonding opportunities between the sulfone fragment and Tyr67 and Asn72. Noteworthy also is the introduction of two additional methyl groups on the imidazolidine ring, replacing the hydrogens of the first generation nutlins.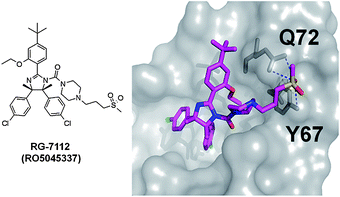 | ||
| Fig. 17 Proposed binding mode of RG-7112 docked into PDB-ID 1YCR. | ||
The nutlins of the first generation (13) are described as N-acylated or unsubstituted cis-2,4,5-triphenylimidazolines.45–47 The synthesis of nutlins comprise a lengthy multistep synthesis.48 Derivatives of the scaffold were structurally characterized in MDM2 and many publications suggested the usefulness of these compounds in cancer therapy.48 In more than 300 scientific publications the commercially available nutlin-3 is used as a probe compound to elucidate the p53 pathway and its implications in cancer. Nutlin derivatives with heteroaromatic substituents were described in WO2009047161. The compounds were produced as racemates and the enantiomers were separated subsequently by supercritical fluid chromatography. Compound 14 inhibited the interaction of p53 and MDM2 which showed IC50 of 0.031 μM.49
From the diphenyldihydroimidazopyridinones derivatives described by Roche compound 15 inhibited the interaction of MDM2 with p53 with an IC50 value of 0.46 μM. The synthesis involves a multistep synthesis starting from 3-(tert-butyldimethylsilyloxy)glutaric anhydride and 1,2-bis(4-chlorophenyl)ethylene-diamine (a key building block also in the classical nutlin and derivative synthesis).50
The ability of the Hoffmann La Roche's spirocyclic indolinone derivatives to inhibit the interaction between p53 and MDM2 proteins was measured by homogeneous time-resolved fluorescence (HTRF) assay, and compound 16 showed MDM2 antagonist activities of 94.4% inhibition at 1.4 μM. Another series of spirooxindoles disclosed by Roche retained a free keto group in a central cyclohexanone. 17 as a racemic compound in vitro inhibited the interaction between p53 and MDM2 proteins with IC50 of 0.37 μM.51,52
Spiroindolinone derivatives as interaction inhibitors between p53 and MDM2 proteins and their preparation via cycloadditon methodology, pharmaceutical compositions and use in the treatment of cancer was disclosed by Chen et al.53 It was determined that 18 exhibited the IC50 value of 0.054 μM. Compound 19, from a related series, inhibited the binding of recombinant GST-tagged MDM2 protein to a p53 peptide (biotinylated on its N-terminal end) with IC50 of 0.034 μM.54 The lead compound of Roche's spiroindolepyridotriazinediones derivative 20, inhibited the interaction between p53 and MDM2 proteins with IC50 = 0.1 μM. Roche's potent spiroindolinone pyridine derivatives 21 can be accessed in great diversity by condensing oxindoles with benzaldehydes, followed by a [4 + 2] cycloaddition with a suitably substituted 3-trimethylsilyoxy-2-aza-1,3-butadiene. 21, for example displayed an IC50 of 66 nM in a HTRF assay in the presence of 0.02% BSA.55
A compound series lacking the central cyclic scaffold representing a 3,3-disubstituted oxindole fragment gave compound 22 as racemate and its (S)-isomer had IC50 of 0.46 μM and 0.28 μM, respectively.56
Recently pyrrolidine-2-carboxamides were claimed by Roche as anticancer agents and synthesized by a cycloaddition reaction followed by amidation.5723 and 24 for example are potent MDM2 antagonist exhibiting IC50s (measured in the presence of 0.02% BSA) of 309 and 165 nM, respectively.56
Novartis
Novartis was amongst the first companies to recognize the potential of the target p53-Mdm2 for cancer. In fact the first optimized peptides antagonizing p53-MDM2 incorporating the non-classical 6-chlorotryptophane where developed by Garcia-Echeverria, Chène, Furet et al.58–60 However, only recently Novartis disclosed their first class of small molecular weight scaffolds, 3-imidazolylindoles for treatment of proliferative diseases and their preparation.61 A representative of the scaffold is compound 25, prepared by a van Leusen multicomponent reaction. Very similar compounds have been previously described as anticancer and antibacterial agents, therefore questioning the novelty of the disclosure.62,63Preparation of substituted dihydroimidazole derivatives 26 for use as antitumor agents was disclosed recently.64 Compounds based on this scaffold demonstrated IC50 values from about 70 nM to about 2 μM. This scaffold shows many similarities to Roche's nutlin scaffold.
Schering Corp.
Schering Corp. disclosed the preparation of substituted piperidines 27 that increase p53 activity and their use.65 Compound 27 with a molecular weight of 619 Da, for example, exhibit FP IC50, FP Ki, and cell viability CO50 values of less than 50.0 μM.Johnson & Johnson
Researchers from Johnson & Johnson followed a different strategy by aiming to inhibit the interface of MDM2 with a proteasome.66 For example the tryptamine-derived (4-pyridino)-1,4-diaminophenyl 28 binds to MDM2 and interferes with MDM2 proteolysisvia trypsin. 28 prevented binding of MDM2 to the proteasome in 293 T cells. In glioblastoma cells, 28 induced p53 and activated downstream signaling. This compound was recently reported to be advanced into phase I human clinical trials in patients with advanced or refractory solid tumors under the designation JNJ-26854165.67Several groups of indole derivatives were synthesized by J&J.68 Compound 29 were assayed for human ovarian carcinoma cell (type A2780) proliferation inhibition and found to possess pIC50 values from <5–7.13. Other indole compounds 30 attached to a stiff bicyclic 1,4-diaminophenol scaffold were also disclosed.69 In anti-proliferative tests using human A2780 ovarian cancer cells, the pIC50 of 30 was 6.37.
One of the first scaffolds in the p53-MDM2 arena with potent activity was the benzodiazepinedione series discovered at 3D Pharmaceuticals (a J&J company).70 A co-crystal structure of a derivative with MDM2 discussed in the previous chapter provides insight into the binding mode of the scaffold and hints optimization of the series by structure based approaches. Despite extensive trials to optimize the series and initial success, it seems to be abandoned for further progress to clinical trials.31,71,72 Benzodiazepinediones such as 31 were made by a short sequence involving a Ugi multicomponent reaction as a key step.30 Compounds based on the general scaffold possess IC50 between 10 nM and 1 μM.70,73 Attempts to optimize the affinity and PKPD properties have led to related series.
Daiichi Sankyo company
Imidazothiazole compounds, containing proline moiety, tested in a non-specified MDM2/p53 binding inhibition assays, showed an IC50 was 2.8 nM (compound 32).74 A similar compound 33, inhibited the His-p53/GST-MDM2 interaction with IC50 of 0.014 μM and the proliferation of human lung cancer cell NCI-H460 and human colon cancer cell DLD-1 with IC50 of 1.38 and 40.7 μM, respectively.75Zeneca Ltd
Amino acid and peptidyl piperazine-4-phenyl derivatives as inhibitors of the interaction between MDM2 and p53 were disclosed by Zeneca in 2000.76 Compounds exemplified by 34 possess an IC50 in the range from 0.03 to 200 μM. However no other patents or publications have been published since then based on those compounds.Nexuspharma Inc
Libraries of pyrrolidine-2-ones, synthesized via multicomponent reactions, have been described by Nexuspharma.77,78 Compound 35, for example, with a molecular weight of 642 Da showed an IC50 = 1 μM. The multicomponent reaction chemistry employed allows for the rapid assembly of the scaffold in only two steps. The sulfur atom, being essential part of the scaffold, is however, potentially metabolically instable. Compounds where the sulfur is replaced by oxygen and methylene have also been disclosed.NexusPharma Inc also synthesized a series of tetrahydroisoquinolin-1-one derivatives by an efficient multicomponent reaction between homophthalicacid anhydride, primary amines and aldehydes, followed by a subsequent amidation.79 Compounds were claimed as therapeutics for treating stroke, myocardial infarction, ischemia, multi-organ failure, spinal cord injury, Alzheimer's disease, injury from ischemic events and heart valvular degenerative disease. Derivative 36, for example, inhibits p53/MDM2 as shown by FP and HSQC-NMR.80 Interestingly similar compounds based on the same scaffold have been described as inhibitors of another transcription factor HOXA13, questioning the selectivity of this scaffold for MDM2.81
University of Michigan
Wang et al. from the University of Michigan described a series of highly potent spirooxindoles as small molecular weight inhibitors of MDM2.33,34,82 Compounds out of the spirooxindole series (37) showed rhabdomyosarcoma, chronic lymphocytic leukemia, adult acute myelogenous leukemia and xenograft activity.83–85 Compounds were sub-licensed to Sanofi-Aventis from biotech company Ascenta Therapeutics for clinical development.University of Pittsburgh
Dömling et al. from University of Pittsburgh recently described selective and dual-action p53/MDM2/MDMX antagonists (38–43).86 These compounds were found from a new rational structure based approach and are all based on efficient multicomponent reaction chemistry.87 Interestingly the class of imidazoindoles were independently and at the same time disclosed by Novartis, however apparently discovered by a HTS approach.61 Other scaffolds discovered by this approach include Ugi thiohydantoines, Orru imidazolidines and others. Selected compounds showed IC50 ranging from 60 μM to 30 nM. For example 42 has a Ki of 1 μM and 40 has a Ki 3 μM.The Scripps Research Institute
Rebek et al. from The Scripps Research Institute (TSRI) disclosed a general pyridazine scaffold mimicking an α-helix mediated PPI.88 However, no biological activities are given in the patents. The underlying idea can be regarded as a derivative of Hamilton's terphenyl backbone, however incorporating heterocycles (oxazole, pyridazine), thus potentially improving water solubility and synthetic access. Hamilton's compound 44, for example, has a Ki of 0.97μM by FP.89 Similar ideas of α-helix mimetic heterocyclic scaffolds have been brought forward recently using van Leusen's imidazole chemistry.90Similarly, Rebek et al. disclosed further pyridazine scaffolds including the heterocycles pyrrolidine and oxazolidine bound to the central pyridazine in a 3,6-fashion as general α-helix mimics91,92 Compound 45 and 46, for example, was prepared in 6 steps from 1,4-di-methyl-1,2,4,5-tetrazine-3,6-dicarboxylate, 4-methylpentyne, pyrrolidine, and L-valinol. No biological data are given: neither target examples nor affinities.
University of Pennsylvania
Acridine type compounds for the activation of p53 have been patented by the University of Pennsylvania, 47.93 The compound clearly does not fit into the current p53 MDM2 small molecule binding model. Presumably the mode-of-action is different from MDM2 interaction.Cancer Research Technology Limited
2,3-Dihydroisoindol-1-one derivatives as inhibitors of the interaction of the MDM2 protein with p53 and their preparation, pharmaceutical compositions and use in the treatment of cancer are claimed by the Cancer Research Technology Limited.94 Compound 48, for example, has an IC50 of 1.4 μM.Cyclacel Limited
Researchers from the biotech company Cyclacel focus on bisarylsulfonamide compounds 49.95 The chemotype of these compounds does not fit the current pharmacophore model of p53/MDM2 antagonists. Thus the compounds likely possess another mode-of-action.Chen Li (China)
Spiroindolinone pyridine derivatives have been claimed by Chen Liet al. for antagonizing p53 MDM2.96 The ability of the compounds to inhibit the interaction between p53 and MDM2 proteins was evaluated in vitro (HTRF assay). From these assays compound 50 was found to display IC50 = 0.145 μM, 0.02% BSA. Interestingly, in 50 the ubiquitous phenyl group is exchanged against a pyridine ring system, suggesting that the three binding pockets can be optimized using heterocycles and thus useful to improve PKPD properties.National Institute of Health
Highly soluble pyrimidoquinolinedione compounds for treating cancer have been claimed by Weismann et al. from the National Institute of Health (NIH).97 Examples given include 51 which accumulate MDM2 and p53 in non-transformed cells; p53 accumulated by 51 is transcriptionally active, and selectively induced apoptosis in transformed cells. The compounds do not fit the currently accepted pharmacophore model of p53-MDM2 antagonists binding into the MDM2 pocket and therefore likely have a different mode-of-action.7 Therapeutically useful methods involving p53 MDM2/X interacting compounds
One method of treating cancer in a subject with a plurality of cancer cells, is administering to the subject, a therapeutically effective amount of a compound including an MDM2 binding component and a membrane resident component bound to it. Another method includes providing a method of selectively necrosing cancer cells, a method of causing membranolysis in cancer cells, and a cancer treatment compound.98A combination therapy approach involving a DNA alkylating agent, a DNA repair inhibitor, and a p53 agent has been claimed beneficial for the treatment of cancer.99 In some instances the combination drug therapy allows the drugs to be administered to a patient at therapeutically effective submyeloablative levels.
A collaboration between the Forschungszentrum Karlsruhe and Janssen Pharmaceutical allowed for a screening methodology for the MDM2-proteasome interaction.100 The invention is based on characterization of the interaction of MDM2 or related protein with the proteasome subunits S6A, S6B, S5A, S2, and S4. This invention allows the discovery of compounds involved in the ubiquitin-proteasome proteolysis (UPS) pathway. Clinical candidate JNJ-26854165 resulted from this project.
The biotech company Cellumen disclosed a biosensor assay in living cells for the detection of compounds able to disrupt a p53-MDM2 construct in the nucleus of cancer cells.101 Adenoviral expression of p53-GRP and MDM2-GFP constructs can be used to discover cell membrane and nucleus viable compounds by observing the subcellular localization of the constructs. HTS applications of the screening system have been described (Fig. 18). This screening system has merits since compounds can only be observed if they can are “drug-like”, e.g. they effectively intervene with the target under realistic cellular conditions, and they show membrane penetration, nuclear enrichment, low or no cellular metabolism and are no or poor efflux pump substrates.102,103
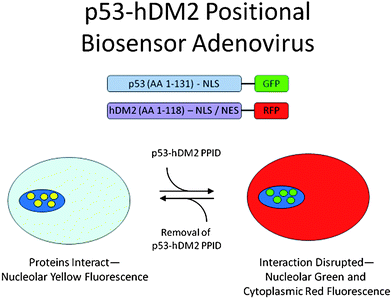 | ||
| Fig. 18 Biosensor assay for detecting p53/MDM2 small molecule inhibitory effects in cells (reprint with permission of Assay and Drug Development Technologies). | ||
A related live cell based screening based on Y2H technology has been disclosed by workers from Aventis.104
The crystal structure of the human double minute 2 protein (HDM2) in complexes with the inhibitor (4-chloro-phenyl)-[3-(4-chloro-phenyl)-7-iodo-2,5-dioxo-1,2,3,5-tetrahydro-[e][1,4]diazepin-4-yl]-acetic acid is claimed by Schering as a tool that permit the design of ligands which can function as active agents which function as inhibitors of MDM2 and MDMX oncoproteins.105
St. Jude Children's Research Hospital's Kriwacki et al. characterized the interaction domains of Arf and MDM2. The invention further exploits this discovery by providing unique methods for identifying and/or designing compounds that mimic, inhibit and/or enhance the effect of Arf on MDM2.106
Wahl from the Salk Institute discovered that the nuclear export signal of tumor suppressor proteins can be impeded from mediating export of the tumor suppressor protein from the cell nucleus.107 Accordingly, peptides are provided which elevate tumor suppressor function within the cell nucleus, yet are impeded from exiting the cell nucleus to the cytoplasm. Invention peptides are impeded from being exported from the cell nucleus by alteration of a nuclear export signal of a tumor suppressor protein or by compounds which inhibit the nuclear export signal from complexing with the nuclear export machinery. In accordance with another embodiment of the present invention, methods are provided for treating a neoplastic condition, such as tumors, by administering compounds which elevate tumor suppressor function within the cell nucleus.
8 Clinical trials
Currently there are two p53/MDM2 inhibitor small compounds that have undergone or are undergoing phase 1 clinical trials, one from Johnson & Johnson and one from Hoffmann-La Roche. The Johnson & Johnson compound (JNJ-26854165) started in November of 2006 and was completed in February of 2010 (ClinicalTrials.gov Identifier: NCT00676910). Patients with advanced or refractory solid tumors were examined. There are no results published about this phase clinical trial and there is no evidence of this compound going into phase 2 trials.67Hoffmann-La Roche's compound RG7112 is undergoing two different trials, one for patients with advanced solid tumors and one for patients with hematologic neoplasms.43,44 The study start date for the solid tumors trial was December 2007, and it is expected to be completed in November of 2011 (final data collection date for primary outcomes measure). The start date for the patients with hematologic neoplasms was May 2008, and will be completed in March 2012. The compound is dosed orally. The respective trials are performed in parallel to the development of p53 chip based diagnosis in order to identify potential responders to the therapy. First preliminary trial results with RG7112 look promising.108 Patients with relapsed/refractory leukemia were treated for 10 days orally in a dose escalation study from 20 mg m−2 d−1 to 1920 mg m−2 d−1 with continuous escalation. The p53 transcriptional target and secreted protein, MIC-1 served as a pharmacodynamic marker and increased with increasing drug concentration. One patient with relapsed AML achieved complete remission that is ongoing for more than 9 month. The study therefore comprise a proof-of-principle showing p53 stabilization, activation of p53 targets and the p53 pathway. Future improved (better solubility, stability, and absorption) MDM2 antagonists hold the promise of even more selective and effective therapies.43,44
9 Conclusion
Large and medium sized pharma companies with a cancer franchise seem to be actively engaged in the discovery and development of low molecular weight p53-MDM2 and/or -MDMX antagonists. Several small biotech companies are also engaged in p53-MDM2 antagonist discovery which recently led to three big pharma biotech deals (Table 2). Interestingly, in all cases the deals are based on preclinical compounds. Some academic institutions have built an impressive IP portfolio in the p53-MDM2 field which might lead to more biotech/university pharma deals in the near future.There is ample evidence that dual action MDM2 and MDMX antagonists could be superior to single target MDM2 or MDMX antagonists.12,14 This is also in line with the biological complexity of cancer and therefore polypharmacology approaches might turn out to be superior to single target compounds. Similar findings are now well established in the field of kinase inhibitors. The design and discovery of dual action MDM2/X antagonists however turns out to be more complex than anticipated based on the high sequence and structural homology of the two targets especially in the p53 binding site.109 All known p53-MDM2 antagonists show a factor 103 or higher selectivity for MDM2. Selective MDMX antagonists might be of high interest by their own right. For example, only MDMX is highly overexpressed in retinoblastoma.109 On the other hand it has been found that MDMX is overexpressed in chronic lymphocytic leukaemia (CLL) and marks a subset of p53 wild-type CLL with a poor cytotoxic response to Nutlin-3.110
Although a first high resolution structure of a small molecule in MDMX has been disclosed recently,111 no potent low nM compound is known so far. However, once high resolution structures are available, it is often a matter of time until the first potent MDMX antagonists will emerge and dual action inhibitors will follow.112
Disfunctional p53 pathway is a hallmark of all cancer cells. The transcription factor p53 is leading irreparably damaged cells into apoptosis, e.g. cancer cells during radiation and chemotherapy. However, most cancers have severe defects in the p53 pathway thus unable to undergo apoptosis, even if with strong signals for undergoing apoptosis are present. Half of the disfunctional p53 results from a strong overexpression of the negative p53 regulator proteins MDM2 and MDMX. Inhibiting the negative p53 regulator proteins MDM2 and MDMX has therefore recently evolved as a promising new approach to fight cancer. The discovery and design of compounds able to restore mutated p53, a decade ago deemed to be impossible has now become a attractive research field. This is due to the recent fragment screening showing several cocrystal structures of small molecules in specific binding sites of mutated p53. MDM2/X antagonists and p53 restoring agents in a broader sense are a rapidly emerging drug discovery area comparable the kinase field 15 years ago.
| Biotech Company | Pharma Company | Compounds | Deal Size |
|---|---|---|---|
| a http://www.boehringer-ingelheim.com/news/news_releases/press_releases/2010/10_january_2010.html. b http://en.sanofi-aventis.com/binaries/20100604_ASCENTA_en_tcm28-28695.pdf. c http://www.aileronrx.com/pdf/RocheGoesShoppingForStaples_083010.pdf. | |||
| Priaxon/NexusPharma | Boehringer Ingelheim | Pyrrolidin-2-ones, tetramic aicds, tetra-hydroisoquinoline-1-ones; preclinical compound | EUR 86 million in milestone payments; royalties on potential future net sales of products; undisclosed upfront paymenta |
| Ascenta Therapeutics | Sanofi-Aventis | Spirooxindoles; compounds licensed from University of Michigan; preclinical compounds | Undisclosed upfront payment; milestone payments could add up to US$ 398 million; royalties on worldwide salesb |
| Aileron Therapeutics | Hoffmann La-Roche | Stapled peptides against five targets; preclinical compounds | $25 million upfront and R&D funding; technology access fee up to US$ 1.1 billion; US$ 40 million investment from Roche corporate venture fundc |
References
- D. P. Lane and L. V. Crawford, Nature, 1979, 278, 261–263 CrossRef CAS.
- G. Matlashewski, P. Lamb, D. Pim, J. Peacock, L. Crawford and S. Benchimol, EMBO J, 1984, 3, 3257–3262 CAS.
- M. Oren and A. J. Levine, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 56–59 CrossRef CAS.
- E. Yonish-Rouach, D. Resnitzky, J. Lotem, L. Sachs, A. Kimchi and M. Oren, Nature, 1991, 352, 345–347 CrossRef CAS.
- L. Diller, J. Kassel, C. E. Nelson, M. A. Gryka, G. Litwak, M. Gebhardt, B. Bressac, M. Ozturk, S. J. Baker and B. Vogelstein, et al. , Mol Cell Biol, 1990, 10, 5772–5781 CAS.
- J. Momand, G. Zambetti, D. Olson, D. George and A. Levine, Cell, 1992, 69, 1237–1245 CrossRef CAS.
- C. Brown, S. Lain, C. Verma, A. Fersht and D. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS.
- A. J. Levine and M. Oren, Nat. Rev. Cancer, 2009, 9, 749–758 CrossRef CAS.
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS.
- L. M. Liau and C. H. Weaver, Ted Kennedy's Recent Diagnosis of Glioblastoma Raises Awareness of Brain Cancer Search PubMed.
- M. E. Halatsch, U. Schmidt, A. Unterberg and V. I. Vougioukas, Anticancer Res, 2006, 26, 4191–4194 CAS.
- M. Wade and G. M. Wahl, Mol. Cancer Res., 2009, 7, 1–11 CrossRef CAS.
- F. Bernal, M. Wade, M. Godes, T. N. Davis, D. G. Whitehead, A. L. Kung, G. M. Wahl and L. D. Walensky, Cancer Cell, 2010, 18, 411–422 CrossRef CAS.
- J.-C. W. Marine, M. A. Dyer and A. G. Jochemsen, J. Cell Sci., 2007, 120, 371–378 CrossRef CAS.
- F. Toledo and G. M. Wahl, Nat. Rev. Cancer, 2006, 6, 909–923 CrossRef CAS.
- K. Linke, P. D. Mace, C. A. Smith, D. L. Vaux, J. Silke and C. L. Day, Cell Death Differ., 2008, 15, 841–848 CrossRef CAS.
- K. H. Vousden and X. Lu, Nat. Rev. Cancer, 2002, 2, 594–604 CrossRef CAS.
- L. A. Donehower, M. Harvey, B. L. Slagle, M. J. McArthur, C. A. Montgomery, Jr, J. S. Butel and A. Bradley, Nature, 1992, 356, 215–221 CrossRef CAS.
- X. Wu, J. H. Bayle, D. Olson and A. J. Levine, Genes Dev., 1993, 7, 1126–1132 CrossRef CAS.
- D. Danovi, E. Meulmeester, D. Pasini, D. Migliorini, M. Capra, R. Frenk, P. de Graaf, S. Francoz, P. Gasparini, A. Gobbi, K. Helin, P. G. Pelicci, A. G. Jochemsen and J. C. Marine, Mol. Cell. Biol., 2004, 24, 5835–5843 CrossRef CAS.
- M. J. Riemenschneider, R. Buschges, M. Wolter, J. Reifenberger, J. Bostrom, J. A. Kraus, U. Schlegel and G. Reifenberger, Cancer Res, 1999, 59, 6091–6096 CAS.
- O. Sperandio, C. H. Reynes, A. C. Camproux and B. O. Villoutreix, Drug Discovery Today, 2010, 15, 220–229 CrossRef CAS.
- T. Clackson and J. A. Wells, Science, 1995, 267, 383–386 CrossRef CAS.
- P. H. Kussie, S. Gorina, V. Marechal, B. Elenbaas, J. Moreau, A. J. Levine and N. P. Pavletich, Science, 1996, 274, 948–953 CrossRef CAS.
- S. M. Picksley, B. Vojtesek, A. Sparks and D. P. Lane, Oncogene, 1994, 9, 2523–2529 CAS.
- A. Domling, Curr. Opin. Chem. Biol., 2008, 12, 281–291 CrossRef CAS.
- L. T. Vassilev, Trends Mol. Med., 2007, 13, 23–31 CrossRef CAS.
- L. T. Vassilev, B. T. Vu, B. Graves, D. Carvajal, F. Podlaski, Z. Filipovic, N. Kong, U. Kammlott, C. Lukacs, C. Klein, N. Fotouhi and E. A. Liu, Science, 2004, 303, 844–848 CrossRef CAS.
- Z. Wang, M. Jonca, T. Lambros, S. Ferguson and R. Goodnow, J. Pharm. Biomed. Anal., 2007, 45, 720–729 CrossRef CAS.
- D. J. Parks, L. V. Lafrance, R. R. Calvo, K. L. Milkiewicz, V. Gupta, J. Lattanze, K. Ramachandren, T. E. Carver, E. C. Petrella, M. D. Cummings, D. Maguire, B. L. Grasberger and T. Lu, Bioorg. Med. Chem. Lett., 2005, 15, 765–770 CrossRef CAS.
- B. L. Grasberger, T. Lu, C. Schubert, D. J. Parks, T. E. Carver, H. K. Koblish, M. D. Cummings, L. V. LaFrance, K. L. Milkiewicz, R. R. Calvo, D. Maguire, J. Lattanze, C. F. Franks, S. Zhao, K. Ramachandren, G. R. Bylebyl, M. Zhang, C. L. Manthey, E. C. Petrella, M. W. Pantoliano, I. C. Deckman, J. C. Spurlino, A. C. Maroney, B. E. Tomczuk, C. J. Molloy and R. F. Bone, J. Med. Chem., 2005, 48, 909–912 CrossRef CAS.
- H. K. Koblish, S. Zhao, C. F. Franks, R. R. Donatelli, R. M. Tominovich, L. V. LaFrance, K. A. Leonard, J. M. Gushue, D. J. Parks, R. R. Calvo, K. L. Milkiewicz, J. J. Marugan, P. Raboisson, M. D. Cummings, B. L. Grasberger, D. L. Johnson, T. Lu, C. J. Molloy and A. C. Maroney, Mol. Cancer Ther., 2006, 5, 160–169 CrossRef CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, G. Wang, S. Qiu, S. Shangary, W. Gao, D. Qin, J. Stuckey, K. Krajewski, P. P. Roller and S. Wang, J. Med. Chem., 2006, 49, 3432–3435 CrossRef CAS.
- S. Shangary, D. Qin, D. McEachern, M. Liu, R. S. Miller, S. Qiu, Z. Nikolovska-Coleska, K. Ding, G. Wang, J. Chen, D. Bernard, J. Zhang, Y. Lu, Q. Gu, R. B. Shah, K. J. Pienta, X. Ling, S. Kang, M. Guo, Y. Sun, D. Yang and S. Wang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3933–3938 CrossRef CAS.
- G. M. Popowicz, A. Czarna, S. Wolf, K. Wang, W. Wang, A. Domling and T. A. Holak, Cell Cycle, 2010, 9 Search PubMed.
- J. G. Allen, M. P. Bourbeau, G. E. Wohlhieter, M. D. Bartberger, K. Michelsen, R. Hungate, R. C. Gadwood, R. D. Gaston, B. Evans, L. W. Mann, M. E. Matison, S. Schneider, X. Huang, D. Yu, P. S. Andrews, A. Reichelt, A. M. Long, P. Yakowec, E. Y. Yang, T. A. Lee and J. D. Oliner, J. Med. Chem., 2009, 52, 7044–7053 CrossRef CAS.
- H. P. Beck, M. Degraffenreid, B. Fox, J. G. Allen, Y. Rew, S. Schneider, A. Y. Saiki, D. Yu, J. D. Oliner, K. Salyers, Q. Ye and S. Olson, Bioorg Med Chem Lett, 2010 Search PubMed.
- G. M. Popowicz, A. Czarna and T. A. Holak, Cell Cycle, 2008, 7, 2441–2443 CAS.
- F. M. Boeckler, A. C. Joerger, G. Jaggi, T. J. Rutherford, D. B. Veprintsev and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10360–10365 CrossRef CAS.
- N. Basse, J. L. Kaar, G. Settanni, A. C. Joerger, T. J. Rutherford and A. R. Fersht, Chem. Biol., 2010, 17, 46–56 CrossRef CAS.
- J. Deng, R. Dayam and N. Neamati, Expert Opin. Ther. Pat., 2006, 16, 165–188 Search PubMed.
- L. Weber, Expert Opin. Ther. Pat., 2010, 20, 179–191 Search PubMed.
- ClinicalTrials.gov, A Study of RO5045337 [RG7112] in Patients With Advanced Solid Tumors.
- ClinicalTrials.gov, 2010.
- WO2003051359, 2003.
- WO/2003/051360, 2003.
- WO/2007/063013, 2007.
- D. C. Fry, S. D. Emerson, S. Palme, B. T. Vu, C. M. Liu and F. Podlaski, J. Biomol. NMR, 2004, 30, 163–173 CrossRef CAS.
- WO/2009/047161, 2009.
- WO/2008/125487, 2008.
- WO/2008/055812, 2006.
- US - 11/846597 2008.
- US 2009163512 2009.
- WO/2009/077357, 2008.
- WO/2008/141975, 2008.
- WO/2006/136606, 2006.
- US2010075948, US 2010-702402] 2010.
- WO/2005/097820, 2004.
- NZ333609, WO9801467, 2000.
- P. Chene, Nat. Rev. Cancer, 2003, 3, 102–109 CrossRef CAS.
- PIXXD2 WO 2008119741, 2008.
- B. Beck, A. Mueller and A. Domling, QSAR Comb. Sci., 2006, 25, 527–535 CrossRef CAS.
- WO 2001025213, 2001.
- WO/2008/065068, 2008.
- US 2008004287, 2008.
- R. Kulikov, J. Letienne, M. Kaur, S. R. Grossman, J. Arts and C. Blattner, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 10038–10043 CrossRef CAS.
- ClinicalTrials.gov, A Research Study of JNJ-26854165 to Determine the Safety and Dose in Patients With Advanced Stage or Refractory Solid Tumors.
- WO/2009/037308, 2009.
- WO/2009/037343, 2009.
- WO/2004/096134, 2004.
- K. Leonard, J. J. Marugan, P. Raboisson, R. Calvo, J. M. Gushue, H. K. Koblish, J. Lattanze, S. Zhao, M. D. Cummings, M. R. Player, A. C. Maroney and T. Lu, Bioorg. Med. Chem. Lett., 2006, 16, 3463–3468 CrossRef CAS.
- D. J. Parks, L. V. LaFrance, R. R. Calvo, K. L. Milkiewicz, J. J. Marugan, P. Raboisson, C. Schubert, H. K. Koblish, S. Zhao, C. F. Franks, J. Lattanze, T. E. Carver, M. D. Cummings, D. Maguire, B. L. Grasberger, A. C. Maroney and T. Lu, Bioorg. Med. Chem. Lett., 2006, 16, 3310–3314 CrossRef CAS.
- WO/2003/041715, WO/2003/041715, 2003.
- WO/2010/082612, WO/2010/082612, 2009.
- JP 2009298713, JP 2009298713, 2009.
- WO/2000/015657, 2000.
- WO/2010/028862, WO/2010/028862, 2010.
- J. Wei and J. T. Shaw, Org. Lett., 2007, 9, 4077–4080 CrossRef CAS.
- WO/2008/034039, WO/2008/034039, 2008.
- U. Rothweiler, A. Czarna, M. Krajewski, J. Ciombor, C. Kalinski, V. Khazak, G. Ross, N. Skobeleva, L. Weber and T. A. Holak, ChemMedChem, 2008, 3, 1118–1128 CrossRef CAS.
- P. Y. Ng, Y. Tang, W. M. Knosp, H. S. Stadler and J. T. Shaw, Angew. Chem., Int. Ed., 2007, 46, 5352–5355 CrossRef CAS.
- K. Ding, Y. Lu, Z. Nikolovska-Coleska, S. Qiu, Y. Ding, W. Gao, J. Stuckey, K. Krajewski, P. P. Roller, Y. Tomita, D. A. Parrish, J. R. Deschamps and S. Wang, J. Am. Chem. Soc., 2005, 127, 10130–10131 CrossRef CAS.
- J. A. Canner, M. Sobo, S. Ball, B. Hutzen, S. DeAngelis, W. Willis, A. W. Studebaker, K. Ding, S. Wang, D. Yang and J. Lin, Br. J. Cancer, 2009, 101, 774–781 CrossRef CAS.
- C. Saddler, P. Ouillette, L. Kujawski, S. Shangary, M. Talpaz, M. Kaminski, H. Erba, K. Shedden, S. Wang and S. N. Malek, Blood, 2008, 111, 1584–1593 CAS.
- J. Long, B. Parkin, P. Ouillette, D. Bixby, K. Shedden, H. Erba, S. Wang and S. N. Malek, Blood, 2010, 116, 71–80 CrossRef CAS.
- WO/2008/130614, WO/2008/130614, 2008.
- A. Czarna, B. Beck, S. Srivastava, G. M. Popowicz, S. Wolf, Y. Huang, M. Bista, T. A. Holak and A. Domling, Angew. Chem., Int. Ed., 2010, 49, 5352–5356 CrossRef CAS.
- US2006205728, US2006205728, 2009.
- H. Yin, G. I. Lee, H. S. Park, G. A. Payne, J. M. Rodriguez, S. M. Sebti and A. D. Hamilton, Angew. Chem., Int. Ed., 2005, 44, 2704–2707 CrossRef CAS.
- W. Antuch, S. Menon, Q. Z. Chen, Y. Lu, S. Sakamuri, B. Beck, V. Schauer-Vukasinovic, S. Agarwal, S. Hess and A. Domling, Bioorg. Med. Chem. Lett., 2006, 16, 1740–1743 CrossRef CAS.
- WO/2010/042758, WO/2010/042758, 2010.
- WO/2010/011864, WO/2010/011864, 2010.
- WO/2008/010984, WO 2007-US16122 2008.
- WO/2009/156735, WO/2009/156735, 2009.
- WO/2004/005278, WO/2004/005278, 2004.
- US 2010204257, US 2010204257, 2010.
- WO/2007/146375, WO/2007/146375, 2007.
- WO/2009/070650, WO/2009/070650, 2009.
- WO/2009/062128, WO/2009/062128, 2009.
- WO/2008/132175, WO/2008/132175, 2008.
- WO/2008/060483, WO/2008/060483, 2008.
- D. D. Dudgeon, S. N. Shinde, T. Y. Shun, J. S. Lazo, C. J. Strock, K. A. Giuliano, D. L. Taylor and P. A. Johnston, Assay Drug Dev. Technol., 2010, 8, 437–458 CrossRef CAS.
- D. D. Dudgeon, S. Shinde, Y. Hua, T. Y. Shun, J. S. Lazo, C. J. Strock, K. A. Giuliano, D. L. Taylor and P. A. Johnston, J. Biomol. Screening, 2010, 15, 766–782 CrossRef CAS.
- WO/2006/045941, WO/2006/045941, 2006.
- US 2004197893, US 2004197893, 2004.
- US2004248198, US2002045192, 2004.
- AU3896899, WO9958135, 1999.
- M. Andreeff, K. Kojima, S. Padmanabhan, R. Strair, M. Kirschbaum, P. Maslak, P. Hillmen, S. O'Brien, F. Samaniego, G. Borthakur, M. Konopleva, L. Vassilev and G. Nichols, Abstract #657 presented in part at the American Society of Hematology Annual Meeting, San Diego, CA, 2010 Search PubMed.
- N. A. Laurie, S. L. Donovan, C. S. Shih, J. Zhang, N. Mills, C. Fuller, A. Teunisse, S. Lam, Y. Ramos, A. Mohan, D. Johnson, M. Wilson, C. Rodriguez-Galindo, M. Quarto, S. Francoz, S. M. Mendrysa, R. K. Guy, J. C. Marine, A. G. Jochemsen and M. A. Dyer, Nature, 2006, 444, 61–66 CrossRef CAS.
- M. D. Bo, P. Secchiero, M. Degan, D. Marconi, R. Bomben, G. Pozzato, G. Gaidano, G. Del Poeta, F. Forconi, G. Zauli and V. Gattei, Br. J. Haematol., 2010, 150, 237–239.
- G. M. Popowicz, A. Czarna, U. Rothweiler, A. Szwagierczak, M. Krajewski, L. Weber and T. A. Holak, Cell Cycle, 2007, 6, 2386–2392 CrossRef CAS.
- G. M. Popowicz, A. Dömling and T. Holak, Angew. Chem., Int. Ed., 2011, 50 DOI:10.1002/anie.201003863.
- T. Davis and J. Johnston, Chem. Sci., 2011, 2 10.1039/C1SC00061F.
| This journal is © The Royal Society of Chemistry 2011 |