Effect of the isosteric replacement of the phenyl motif with furyl (or thienyl) of 4-phenyl-N-arylsulfonylimidazolones as broad and potent anticancer agents†
Vinay K.
Sharma
a,
Dang The
Hung
a,
Ki-Cheul
Lee
a,
P.
Thanigaimalai
a,
Jong Seong
Kang
a,
Hwan-Mook
Kim
b and
Sang-Hun
Jung
*a
aCollege of Pharmacy and Institute of Drug Research and Development, Chungnam National University, Daejeon, 305-764, Korea. E-mail: jungshh@cnu.ac.kr; Fax: +82 42 823 6566; Tel: +82 42 821 5939
bBio-Evaluation Center, Korea Research Institute of Bioscience and Biotechnology, 685-1 Yangcheong-ri, Ochang-eup, Cheongwon-gun, Chungcheongbuk-do, 363-883, Korea
First published on 10th June 2011
Abstract
To investigate the effect of isosteric replacement of phenyl with heteroaromatics of 4-phenyl-N-arylsulfonylimidazolones (1–4) as broad and potent anticancer agents, a series of 4-furyl (5a–e, 7a–c) or thienyl-N-arylsulfonylimidazolones (6a–e) were designed and synthesized to evaluate their in vitro anticancer activity against HCT116, A549 and NCI-H460 cancer cell lines. Replacement of phenyl with furyl or thienyl slightly reduced the activity, however, these analogs showed comparable activity to doxorubicin. The thienyl analogs exhibited more potent activity than furyl analogs due to their more isosteric similarity with the phenyl moiety. In addition, the carbamate derivatives (5a–e and 6a–e) of both the analogs had better activities than the carbamoyl derivatives (7a–c). Therefore, the size and planarity of the aryl motif at 4-position of N-arylsulfonylimidazolones are crucial for their anticancer activity.
Introduction
Anticancer agents that interfere with microtubulin function are effective against both hematological malignancies and solid tumors. These agents function by affecting microtubulin dynamics to bring about microtubulin aggregation or dissociation at low or high concentrations, respectively.1 A number of tubulin inhibitors like colchicine, combretastatin A-4P, paclitaxel, epothilone A and vinblastine exhibit anticancer properties by interfering with the dynamics of tubulin polymerization and depolymerization resulting in mitotic arrest.2 However, the clinical use of all these antitubulin agents is associated with problems of drug resistance, toxicity, and bioavailability.3According to recent reports, some antitubulin agents binding at colchicine sites like combretastatin A-4P (Fig. 1) act as a vascular disrupting antitumor (VDA) agent,4 which causes the vascular structure inside a solid tumor to collapse and thus starve the tumor to death.5,6 In addition some small molecules such as N-pyridinyl sulfonamide (ABT-751),7–9chloroindolyl sulfonamide10 and styrylpyridine N-oxide sulfonamide11–13 (Fig. 1) have been reported as potent anticancer agents and are currently in clinical trials for different types of cancers.14–18 Though a large number of anticancer agents are under clinical trial still there is an urgent need to find novel tubulin inhibitors which are effective in treatment of multidrug-resistant (MDR) tumors.
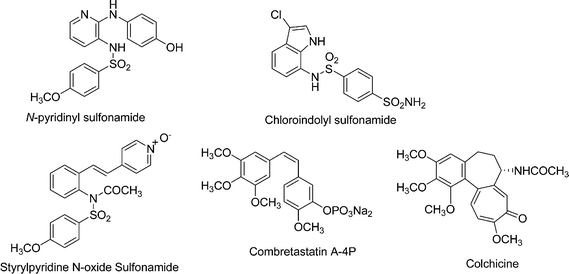 | ||
| Fig. 1 Known tubulin inhibitors. | ||
For a decade our group has been extensively involved in the designing, synthesis and SAR studies of 4-phenyl-1(N)-arylsulfonyl-4,5-dihydroimidazolones 1 for activity against various human cancer cell lines. As a result, we have discovered 4-phenyl-1-benzenesulfonyl-4,5-dihydroimidazolone as a basic pharmacophore for these derivatives.19–22 The activity of these analogs has been varied by substitution around the phenyl group of sulfonyl. STERIMOL L parameters of the substituent at 4-position of the benzenesulfonyl group are well correlated with the activity of these analogs.19 To study the role of the phenyl ring at 4-position of 4,5-dihydroimidazolones, the effects of various substituents for their in vitro anticancer activity were studied. It was found that replacement of a phenyl ring with cyclohexyl at this position abolished the activity.23 In addition, the presence of naphthyl and benzyl analogs also reduced the activity of arylsulfonyl-4,5-dihydroimidazolones remarkably.24 These results implied that the increment of size and volume at this position exerts an adverse effect on the activity of these analogs. In this regard, it was necessary to investigate the effect of smaller planar motifs on the activity. Thus, we synthesized a number of derivatives with a vinyl group replacing the phenyl one, but the activity decreased.25
In addition to the above substitutions, the role of 4,5-dihydroimidazolone (2) and 1H-imidazol-2(3H)-one (3) ring (Fig. 2) in the basic pharmacophore moiety was investigated.26 Interestingly, both the rings showed almost similar activity against various cell lines. Therefore, we concluded that only the adequate size and volume of the aromatic substituent at 4-position of 4,5-dihydroimidazolones or imidazolones are important as pharmacophore for their anticancer activity.
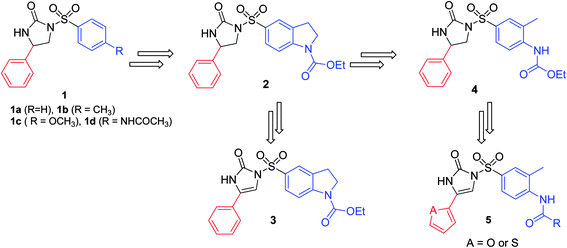 | ||
| Fig. 2 Structural modification at the sulfonyl position of N-arylsulfonylimidazolones. | ||
One of our previous studies on the mechanism of action of arylsulfonyl-4,5-dihydroimidazolones proved them to be tubulin inhibitors.27 These compounds were not only the potent inhibitors of tubulin polymerization but also maintained activity against multidrug resistant tumor cell lines, which implies that they are not a substrate for p-glycoprotein mediated transport like taxanes and vinca alkaloid derivatives. In the case of substitution at the sulfonyl group, the indoline (2) and 4-acylamino-3-methylbenzene (4) (Fig. 2) moieties with the carbamate or carbamoyl group at 1-position showed the most potent activity.27 However, in our previous studies26,27 the phenyl motif was the most appropriate group at 4-position of 4,5-dihydroimidazolones (2) or imidazolones (3) (Fig. 2) for their anticancer activity but still the optimization of this position is needed. Thus, in continuation of our search for the optimum size of the motif at this position, investigation of the effect of five membered heteroaromatics on the activity of these analogs due to their similar size and planarity in this π-system was conducted. Therefore, here in the current article we have designed and synthesized novel analogs 5 (Fig. 1) and evaluated in vitro inhibitory activities against three human cancer cell lines such as human colon carcinoma (HCT116) and human non-small cell lung cancer cell lines (A549 and NCI-H460).
Results and discussion
The general synthetic route to prepare arylsulfonylimidazolone derivatives 5a–e, 6a–e and 7a–c is illustrated in Scheme 1.23–25 In brief, the intermediate 11 was prepared from 2-acetylfuran (8). The reaction of 8 with bromine in ether produced 9 which was then converted to 10 by treatment with hexamethylenetetramine in chloroform, followed by hydrolysis under acidic conditions. Cyclization of compound 10 by treatment with potassium cyanate in water produced intermediate 11. Reaction of 11 with the corresponding arylsulfonyl chlorides 12 in the presence of sodium hydride in DMF produced the N-arylsulfonylimidazolones 5a–e and 6a–e as listed in Table 1. The compounds 7a–c were obtained from the compound 5a. The hydrolysis of 5a has been done using 10% sodium hydroxide solution in methanol to get the intermediate 13. Treatment of 13 with the appropriate alkyl (or aryl) isocyanate in anhydrous toluene gave compounds 7a–c (Table 1). All these synthesized compounds were characterized by physical and spectral analysis data that confirmed their assigned structures.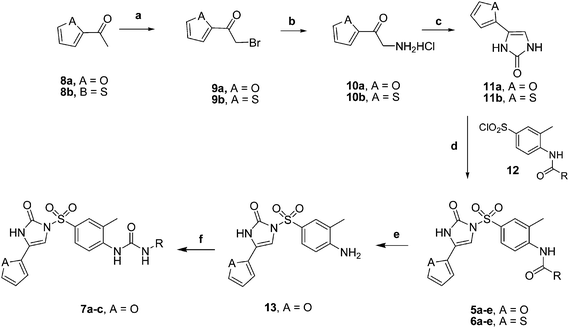 | ||
| Scheme 1 Synthesis of N-arylsulfonylimidazolones 5, 6 and 7.Reagents and conditions: (a) Br2/ether; (b) (CH2)6N4, HCl/EtOH; (c) KOCN/H2O; (d) sodium hydride in DMF; (e) NaOH/MeOH, H2O; (f) toluene, aryl or alkyl isocyanate. | ||
|
|
|||||
|---|---|---|---|---|---|
| No. | A | R | IC50/μMa | ||
| A549 | HCT116 | NCI-H460 | |||
| a IC50 values are taken as a mean from 3 experiments. | |||||
| 4 27 | –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– CH– |
OEt | 0.07 | 0.048 | 0.07 |
| 5a | O | OEt | 0.76 | 0.97 | 0.76 |
| 5b | O | OPr-n | 0.86 | 0.93 | 0.73 |
| 5c | O | OBu-n | 1.90 | 2.60 | 1.90 |
| 5d | O | OPr-i | 0.20 | 0.39 | 0.22 |
| 5e | O | OBu-i | 0.66 | 1.5 | 0.66 |
| 6a | S | OEt | 0.14 | 0.17 | 0.17 |
| 6b | S | OPr-n | 0.23 | 0.30 | 0.23 |
| 6c | S | OBu-n | 0.27 | 0.43 | 0.36 |
| 6d | S | OPr-i | 0.09 | 0.14 | 0.14 |
| 6e | S | OBu-i | 0.13 | 0.15 | 0.13 |
| 7a | O | NHPr-i | 0.74 | 0.93 | 0.54 |
| 7b | O | NHPh | >10 | >10 | >10 |
| 7c | O | NHcyHex | 3.35 | 3.35 | 2.68 |
| Doxorubicin | 0.477 | 0.400 | 0.456 | ||
The in vitro anticancer activity of compounds 5a–e, 6a–e and 7a–c was measured against human lung carcinoma (A549), human colon cancer (HCT 116) and small lung cancer (NCI-H460) cell lines using sulforhodamine B (SRB) colorimetric assay.28–30 The results from these tests are shown as IC50 values in Table 1.
The phenyl group of 1 was replaced with furyl (5a–e) and thienyl (6a–e) motifs. In addition, 4-acylamino-3-methylbenzene moieties were introduced at the sulfonyl function since these were the best motifs observed at this position.27 In the first set of experiments, increasing the carbon chain length in furyl analogs 5 at the carbamate moiety from ethyl (5a, IC50 = 0.76, 0.97 and 0.76 μM respectively), to propyl (5b, IC50 = 0.86, 0.93 and 0.73 μM respectively) and butyl (5c, IC50 = 1.90, 2.60 and 1.90 μM respectively) decreased the activity. The compounds 5a,b showed potent inhibitory activity comparable to doxorubicin but the n-butyl derivative 5c showed less potent inhibition. Interestingly, the bulkier alkyl carbamates such as isopropyl 5d (IC50 = 0.20, 0.39 and 0.22 μM respectively) and isobutyl 5e (IC50 = 0.66, 1.5 and 0.66 μM respectively) exhibited far better activity than the compounds 5a,b. These results indicate that the bulkier groups at the carbamate moiety are responsible for good inhibition.
In the second set of experiments, by replacing the phenyl group at the 4-position of 4 with the thienyl group, the carbamate analogs ethyl (6a, IC50 = 0.14, 0.17 and 0.17 μM respectively), propyl (6b, IC50 = 0.23, 0.30 and 0.23 μM respectively), butyl 6c (IC50 = 0.27, 0.43 and 0.36 μM respectively), isopropyl (6d, IC50 = 0.09, 0.14 and 0.14 μM respectively) and isobutyl (6e, IC50 = 0.13, 0.16 and 0.13 μM respectively) showed improved activity compared to the corresponding furyl derivatives. These results also follow the same trend as observed in the case of furyl derivatives. The bulkier isopropyl derivative 6d showed the best potency among the synthesized derivatives and is comparable to the phenyl derivative 4 especially against human lung carcinoma (A549). The above results indicate that these heteroaromatic moieties appear to be isosteres of phenyl. However, the furyl analogs 5a–e have shown less potent activity than thienyl ones 6a–e. This difference could be interpreted as closer isosteric similarity of thienyl to phenyl than furyl.
In addition, we also synthesized some carbamoyl derivatives (with furyl moiety) and observed that the activity level of isopropyl carbamoyl analogs (7a, IC50 = 0.74, 0.93 and 0.54 μM respectively) is retained. But phenyl (7b, IC50 > 10, >10 and >10 μM respectively) and cyclohexyl (7c, IC50 = 3.35, 3.35 and 2.68 μM respectively) analogs showed a drastic decrease in the activity. Thus, increasing the volume on the carbamoyl group exerts an adverse effect on their cytotoxicity.
Conclusion
To investigate the effect of isosteric replacement of phenyl with heteroaromatics of 4-phenyl-N-arylsulfonylimidazolones (1–4) as broad and potent anticancer agents, a series of 4-furyl (5a–e, 7a–c) or thienyl-N-arylsulfonylimidazolones (6a–e) were designed and synthesized to evaluate their in vitro anticancer activity against HCT116, A549 and NCI-H460 cancer cell lines. Replacement of phenyl with furyl or thienyl slightly reduced the activity, however, these analogs showed comparable activity to doxorubicin. Further, the thienyl analogs exhibited more potent activity than furyl analogs and the difference could be interpreted as closer isosteric similarity of thienyl to phenyl than furyl. Moreover, the bulkier isopropyl derivative 6d showed the best potency among the synthesized derivatives and comparable to phenyl derivative 4 especially against human lung carcinoma (A549). In addition, the carbamate derivatives (5a–e and 6a–e) of both the analogs had better activities than the carbamoyl derivatives (7a–c). Therefore, the size and planarity of the aryl motif at 4-position of N-arylsulfonylimidazolones are crucial for their anticancer activity.Acknowledgements
This work was supported by Priority Research Centers Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2009-0093815).References and notes
- F. Pellegrini and D. R. Budman, Cancer Invest., 2005, 23, 264 CrossRef.
- N. Mahindroo, J.-P. Liou, J.-Y. Chang and H.-P. Hsieh, Expert Opin. Ther. Pat., 2006, 16, 647 Search PubMed.
- M. Sridhare, M. J. Macapinlac, S. Goel, D. Verdier-Pinard, T. Fojo, M. Rothenberg and D. Colevas, Anti-Cancer Drugs, 2004, 15, 553 CrossRef CAS.
- M. A. Jordan and L. Wilson, Nat. Rev. Cancer, 2004, 4, 253 CrossRef CAS.
- N. E. Mealy and L. M. Balcells, Drugs Future, 2006, 31, 541 Search PubMed.
- P. D. Davis, G. J. Dougherty, D. C. Blakey, S. M. Galbraith, G. M. Tozer, A. L. Holder, M. A. Naylor, J. Nolan, M. R. L. Stratford, D. J. Chaplin and S. A. Hill, Cancer Res., 2002, 62, 7247 CAS.
- H. Yoshino, N. Ueda, J. Nijima, H. Sugumi, Y. Kotake, N. Koyanagi, K. Yoshimatsu, M. Asada, T. Watanabe, T. Nagaau, K. Tsukahara, A. Iijima and K. Kitoh, J. Med. Chem., 1992, 35, 2496 CrossRef.
- N. Koyanagi, T. Nagasu, F. Fujita, T. Watanabe, K. Tsukahara, Y. Funahashi, M. Fujita, T. Taguchi, H. Yoshino and K. Kitoh, Cancer Res., 1994, 54, 1702 CAS.
- K. Yoshimatsu, A. Yamaguchi, H. Yoshino, N. Koyanagi and K. Kitoh, Cancer Res., 1997, 57, 3208 CAS.
- T. Owa, H. Yoshino, T. Okauchi, K. Yoshimatsu, Y. Ozawa, N. H. Sugi, T. Nagasu, N. Koyanagi and K. Kitoh, J. Med. Chem., 1999, 42, 3789 CrossRef CAS.
- H. Hideki Tanaka, N. Ohshima, M. Ikenoya, K. Komori, F. Katoh and H. Hidaka, Cancer Res., 2003, 63, 6942.
- M. Takagi, T. Honmura, S. Watanabe, R. Yamaguchi, M. Nogawa, I. Nishimura, F. Katoh, M. Matsuda and H. Hidaka, Invest. New Drugs, 2003, 21, 387 CrossRef CAS.
- M. A. DiMaio, A. Mikhailov, C. L. Rieder, D. D. Von Hoff and R. E. Palazzo, Mol. Cancer Ther., 2009, 8, 592 CrossRef CAS.
- A. M. Mauer, E. E. Cohen, P. C. Ma, M. F. Kozloff, L. Schwartzberg, A. I. Coates, J. Qian, A. E. Hagey and G. B. Gordon, J. Thorac. Oncol., 2008, 3, 631 Search PubMed.
- E. Fox, J. M. Maris, B. C. Widemann, W. Goodspeed, A. Goodwin, M. Kromplewski, M. E. Fouts, D. Medina, S. L. Cohn, A. Krivoshik, A. E. Hagey, P. C. Adamson and F. M. Balis, Clin. Cancer Res., 2008, 14, 1111 CrossRef CAS.
- A. S. Zandvliet, J. H. M. Schellens, C. Dittrich, J. Wanders, J. H. Beijnen and A. D. R. Huitema, Br. J. Clin. Pharmacol., 2008, 66, 485 CrossRef CAS.
- W. S. Siegel-Lakhai, A. S. Zandvliet, A. D. R. Huitema, M. M. Tibben, G. Milano, V. Girre, V. Diéras, A. King, E. Richmond, J. Wanders, J.h. Beijnen, J.h. Schellens and M. Brit, Br. J. Cancer, 2008, 98, 1320 Search PubMed.
- L. L. Garland, C. Taylor, D. L. Pilkington, J. L. Cohen and D. D. Von Hoff, Clin. Cancer Res., 2006, 12, 5182 CrossRef CAS.
- H.-S. Lee, K.-L. Park, S.-U. Choi, C.-O. Lee and S.-H. Jung, Arch. Pharmacal Res., 2000, 23, 579 Search PubMed.
- S.-H. Jung, K.-L. Park, H.-S. Lee and J.-S. Whang, Arch. Pharmacal Res., 2001, 24, 499 Search PubMed.
- I.-W. Kim and S.-H. Jung, Arch. Pharmacal Res., 2002, 25, 421 Search PubMed.
- H.-Y. Choo Park, S. Choi, S.-H. Jung, H.-Y. Koh and A.-N. Pae, Bioorg. Med. Chem., 2003, 11, 4585 Search PubMed.
- S.-H. Jung and S.-J. Kwak, Arch. Pharmacal Res., 1997, 20, 283 Search PubMed.
- S.-H. Jung, S.-J. Kwak, N. D. Kim, S.-U. Lee and C.-O. Lee, Arch. Pharmacal Res., 2000, 23, 35 Search PubMed.
- S.-H. Kwak, S.-C. Bang, H.-H. Seo, H.-R. Shin, K.-C. Lee, L. T. A. Hoang and S.-H. Jung, Arch. Pharmacal Res., 2006, 29, 721 Search PubMed.
- S. J. Yoon, Y. H. Chung, M. S. Lee, D. R. Choi, J. A. Lee, H. S. Lee, H. R. Yun, D. K. Lee, E. Y. Moon, H. S. Hwang, C. H. Choi and S.-H. Jung, US pat., 592910327, 1999.
- S. Kim, J.-H. Park, S.-Y. Koo, J.-I. Kim, M.-H. Kim, E.-J. Kim, K. Jo, H.-G. Choi, S.-B. Lee and S.-H. Jung, Bioorg. Med. Chem. Lett., 2004, 14, 6075 Search PubMed.
- L. V. Rubinstein, R. H. Shoemaker, K. D. Paull, R. M. Simon, S. Toshini, P. Skehan, D. A. Scudiero, A. Monks and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1113 CrossRef CAS.
- P. Skehan, R. Storeng, D. A. Scudiero, A. Monks, J. McMahon, D. T. Vistica, J. T. Warren, H. Bokesch, F. Kenny and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1107 CrossRef CAS.
- C. W. Lee, D. H. Hong, S. B. Han, S.-H. Jung, H. C. Kim, R. L. Fine, S.-H. lee and H. M. Kim, Biochem. Pharmacol., 2002, 64, 473 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0md00219d |
| This journal is © The Royal Society of Chemistry 2011 |