Potent sirtuin inhibition bestowed by L-2-amino-7-carboxamidoheptanoic acid (L-ACAH), a Nε-acetyl-lysine analog†
Brett M.
Hirsch
a,
Zhanwen
Du‡
b,
Xiaopeng
Li‡
a,
Jorge A.
Sylvester
c,
Chrys
Wesdemiotis
ad,
Zhenghe
Wang
b and
Weiping
Zheng
*a
aDepartment of Chemistry, University of Akron, Akron, OH 44325, USA. E-mail: wzheng@uakron.edu; Tel: +1 330 972 2193
bDepartment of Genetics and Case Comprehensive Cancer Center, Case Western Reserve University, Cleveland, OH 44106, USA
cDepartment of Biomedical Engineering, University of Akron, Akron, OH 44325, USA
dDepartment of Polymer Science, University of Akron, Akron, OH 44325, USA
First published on 24th February 2011
Abstract
Inhibitors of sirtuin-catalyzed NAD+-dependent proteinlysine deacetylation reaction possess great value for a fuller exploration of sirtuin biology/pharmacology and as potential therapeutic agents for metabolic and age-related diseases and cancer. In the current study, we discovered that L-2-amino-7-carboxamidoheptanoic acid (L-ACAH), a Nε-acetyl-lysine analog, could confer potent SIRT1 inhibition in both the peptidic and the peptidomimetic inhibitors. This inhibition was demonstrated to be mechanism-based since the in vitro studies with the peptidic inhibitor revealed that (i) the inhibitor was competitive versus the acetyl-lysine substrate, (ii) the inhibitor was of the slow, tight-binding type, and (iii) the inhibitor was able to be converted to at least one longer-lived catalytic intermediate and perhaps all the way to the product (similar to 2′-O-acetyl-ADP-ribose) as well. The in vivo characterization of the peptidomimetic inhibitor revealed its capability of inhibiting the SIRT1 inside the human colon cancer cell line HCT116. This peptidomimetic compound was also shown to be an in vitroinhibitor against SIRT2 and SIRT3 with comparable potency to that against SIRT1. These results point to the potential of developing further potent cell permeable L-ACAH-based inhibitors for sirtuins.
Introduction
Silent information regulator 2 (Sir2) enzymes or sirtuins are a family of intracellular protein lysine deacetylases present in all kingdoms of life, with the yeast Sir2 being the founding family member.1 In mammals including humans, seven sirtuins, i.e. SIRT1–7, have been identified. Sirtuins can catalyze the acetyl group removal from the side chain of the specific Nε-acetyl-lysine (AcK) residues on histone and a variety of non-histone proteins such as transcription factors and metabolic enzymes (Fig. 1).1e,2 Moreover, this enzymatic deacetylation reaction has an absolute requirement of the coenzyme nicotinamide adenine dinucleotide (NAD+) as a cosubstrate. Consistent with this substrate/cosubstrate selection pattern, the sirtuin-catalyzed deacetylation reaction has been shown to play important roles in multiple biological processes such as gene transcription, apoptosis, DNA repair, metabolism, aging, neurodegeneration, and HIV-1 replication.1d,3 In this regard, the inhibitors of this enzymatic reaction could be useful molecular entities toward developing novel therapeutics for the metabolic and age-related diseases and cancer. These inhibitors could also be useful as research tools toward a fuller exploration of sirtuin biology and pharmacology.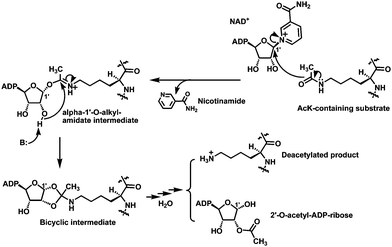 | ||
| Fig. 1 The proposed chemical mechanism for the sirtuin-catalyzed lysine deacetylation reaction. ADP, adenosine diphosphate. | ||
Our laboratory has been interested in identifying AcK analog-based inhibitors for the sirtuin-catalyzed deacetylation reaction.4 Considering that the coenzyme NAD+ is also involved in a myriad of biochemical reactions in a biological system besides the sirtuin-catalyzed reaction, such as a variety of oxidation–reduction reactions, our focus has been on developing AcK analogs as sirtuin inhibitory warheads. During the past a few years, we and others have been disclosing a family of potent sirtuin inhibitors containing Nε-thioacetyl-lysine (ThAcK).2e However, the potential liability of a thioamide compound to the metabolic activation and the ensuing cytotoxicity,5 especially the hepatotoxicity, could be a drawback for the ThAcK-based therapeutics, especially those intended to be administered orally due to the first-pass effect.6 This toxicity was thought to be derived from the metabolic S-oxidation of the thioamide functionality with the formation of the reactive S,S-dioxide electrophile that can react with nucleophilic species on biological macromolecules.5 Therefore, it has also been our interest to identify other AcK analog(s) that could also confer potent sirtuin inhibition. Here we would like to report our discovery of L-2-amino-7-carboxamidoheptanoic acid (L-ACAH) (Fig. 2) as one such novel sirtuin inhibitory warhead.
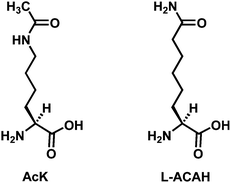 | ||
| Fig. 2 Structural comparison of Nε-acetyl-lysine (AcK) and L-2-amino-7-carboxamidoheptanoic acid (L-ACAH). | ||
Results and discussion
L-ACAH was designed as an AcK analog from the isosteric swapping of the NH and the CH3 groups of the acetamide portion in AcK (Fig. 2). This structural change was also made on the consideration that carboxamide is a common functional group in drug molecules.7 In order to examine the effect of this structural change on the sirtuin-catalyzed deacetylation reaction, we prepared an L-ACAH-containing penta-peptide (i.e.CH3CONH-HK-(L-ACAH)-LM-CONH2) and subjected it to an in vitro sirtuin deacetylase activity inhibition assay with SIRT1, the prototypical human sirtuin. Of note, the above 5-mer peptide sequence was derived from the known SIRT1 peptide substrate H2N-HK-AcK-LM-COOH.8Scheme 1 depicts the synthesis of this penta-peptide 1 starting from L-α-aminosuberic acid. The selective methylesterification of the side chain carboxyl of this α-amino acid by the method of Glenn et al.9 and the subsequent installation of the 9-fluorenylmethoxycarbonyl (Fmoc) group at the α-amino group afforded L-α-(9-fluorenylmethoxycarbonyl-amino)-suberic acid 8-methyl ester (the second intermediate in Scheme 1) that was used during the peptide sequence assembly on the Rink amide resin by the Fmoc chemistry-based solid phase peptide synthesis (SPPS).10 The L-ACAH residue was finally generated via the solution phase aminolysis11 of the side chain methylester at the central position of the unprotected penta-peptide 2 released from the resin with a cleavage cocktail. Penta-peptides 1 and 2 were both purified by preparative high pressure liquid chromatography (HPLC).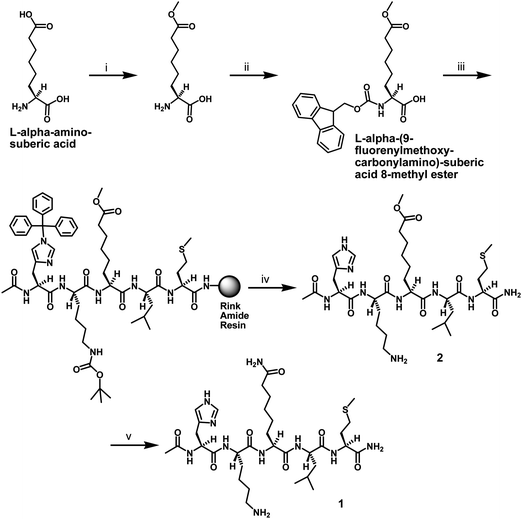 | ||
| Scheme 1 Synthesis of the L-ACAH-containing peptide 1. Reagents: (i) SOCl2, MeOH; (ii) Fmoc-OSu, 10% (w/v) aq. Na2CO3, 1,4-dioxane; (iii) Fmoc-based SPPS; (iv) TFA-containing peptide cleavage cocktail; (v) 28–30% (w/w) NH4OH. | ||
When peptide 1 was examined in our HPLC-based SIRT1 inhibition assay, it was found to be a fairly potent inhibitor against the lysine deacetylase activity of SIRT1, with an IC50 value (11.7 ± 10.0 μM) not dramatically higher than that of the 18-mer ThAcK-containing peptide inhibitor (1.7 ± 0.4 μM)4b (Table 1) and those of our previously reported ThAcK-containing penta-peptides.4c Of note, all of these inhibitors roughly have single-digit IC50 values under our SIRT1 inhibition assay condition. However, the methylester peptide 2 did not exhibit any SIRT1 inhibition at 500 μM (Table 1). These results indicated that the carboxamide side chain of L-ACAH somehow is able to confer a strong SIRT1 inhibition.
| Compound | ThAcK b | Peptide 1 | Peptide 2 | 3 | 4 | |
|---|---|---|---|---|---|---|
| a See ESI for the SIRT1/2/3 inhibition assay details.† b ThAcK, the 18-mer ThAcK-containing peptide: H2N-KKGQSTSRHK-ThAcK-LMFKTEG-COOH.4a,4b c Taken from ref. 4b. d N.I., No inhibition at 500 μM. e N.D., Not determined in the current study. | ||||||
| IC50/μM | SIRT1 | 1.7 ± 0.4c | 11.7 ± 10.0 | N.I.d | >1000 | 290.0 ± 88.0 |
| SIRT2 | N.D.e | N.D. | N.D. | N.D. | 190.2 ± 1.7 | |
| SIRT3 | N.D. | N.D. | N.D. | N.D. | 213.5 ± 31.2 | |
To explore the mechanism of SIRT1 inhibition by peptide 1, we first performed a series of inhibition experiments against SIRT1-catalyzed deacetylation of H2N-HK-AcK-LM-COOH at different concentrations of peptide 1. We found that the inhibition data could be best fit to the linear competitive mode of SIRT1 inhibition by peptide 1versus the acetylated substrate, as revealed by the pattern of intersecting double-reciprocal plots at the 1/v0 axis (Fig. 3a).12 This analysis also yielded an inhibition constant (Kis) of 2.6 μM for peptide 1.
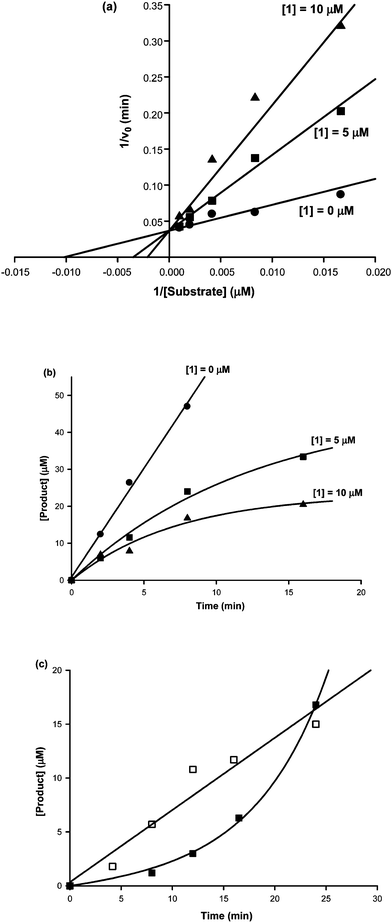 | ||
| Fig. 3 SIRT1 inhibition kinetics of peptide 1. (a) Competitive inhibition of peptide 1versus the SIRT1 substrate H2N-HK-AcK-LM-COOH. 1, peptide 1. (b) Time-dependent SIRT1 inhibition by peptide 1. Product, the deacetylated SIRT1 substrate, i.e.H2N-HK-K-LM-COOH. 1, peptide 1. (c) Activity recovery of the SIRT1 inhibited by peptide 1. Product, the deacetylated SIRT1 substrate, i.e.H2N-HK-K-LM-COOH. □: product formation versus time after the rapid dilution of the free GST-SIRT1 into the SIRT1 deacetylation assay solution; ■: product formation versus time after the rapid dilution of the pre-formed GST-SIRT1·peptide 1 complex into the SIRT1 deacetylation assay solution. | ||
We subsequently performed the following two more assays: (i) the time course assay of SIRT1-catalyzed deacetylation of H2N-HK-AcK-LM-COOH at different concentrations of peptide 1; (ii) the activity recovery assay of SIRT1 inhibited by peptide 1. When the time course assay data were plotted, a pattern indicative of the time-dependent SIRT1 inhibition by peptide 1 was obtained,13 as shown in Fig. 3b. This time-dependent inhibition is consistent with the notion that peptide 1 behaves as a slow binding inhibitor of SIRT1. However, the observed capability of the SIRT1 inhibited by peptide 1 to fully regain its deacetylase activity to the level exhibited by the un-inhibited SIRT1, as shown in Fig. 3c, argued against the possible formation of an irreversible covalent adduct of peptide 1 with SIRT1. The observed lag in the formation of the deacetylated product in the activity recovery curve shown in Fig. 3c indicates that peptide 1 also behaves as a tight binding inhibitor of SIRT1.13
Taken together, the above data indicated that the L-ACAH-containing peptide 1 is a potent SIRT1 inhibitor that is competitive versus the AcK-containing substrate and also additionally displays the slow, tight-binding kinetics. We believe that the potency and the mode of the SIRT1 inhibition by peptide 1 is conferred by the L-ACAH residue because the methylester peptide 2 (Scheme 1) was found to be a much weaker SIRT1 inhibitor despite having the same peptide sequence as that of peptide 1. Our kinetic analysis of the SIRT1 inhibition by peptide 1 also supported the idea that the side chain of L-ACAH is able to bind to the AcK binding tunnel in the SIRT1 active site and this initial binding is able to somehow promote the formation of a tight binding complex between peptide 1 and SIRT1.
Given the observed potent SIRT1 inhibition by peptide 1 and the previously demonstrated mechanism-based sirtuin inhibition by a ThAcK-containing compound—specifically, its ThAcK residue is able to be processed by a sirtuin with the formation of the stalled α-1′-S-alkylamidate intermediate (a close analog of the α-1′-O-alkylamidate intermediate formed during the normal sirtuin catalysis, Fig. 1)—4a,14 we were also interested in examining whether peptide 1 also behaved similarly as a mechanism-based SIRT1 inhibitor, since L-ACAH also has an amide functionality on its side chain. To explore this possibility, we used mass spectrometry in an attempt to detect any possibly stalled (or longer-lived) catalytic species from an incubation of peptide 1 with NAD+ in the presence of SIRT1. When the incubation mixture was subjected to a matrix-assisted laser desorption ionization (MALDI) mass spectral analysis in negative reflector mode, a species with m/z 1278.3 was observed, as shown in Fig. 4A, from this incubation mixture under a non-quenching condition which was also used by Smith and Denu to detect the stalled α-1′-S-alkylamidate intermediate from an incubation of a ThAcK-containing peptide and NAD+ in the presence of yeast sirtuin Hst2.14a We further found that the cluster of the peaks starting at m/z 1278.3 were not detectable under the same experimental conditions by omitting SIRT1, peptide 1, or NAD+ from the incubation mixture (Fig. S1A, S1B, and S1C†), and were not detectable from the MALDI matrix (i.e.α-cyano-4-hydroxycinnamic acid, CHCA) either (Fig. S1D†). Therefore, the species with m/z 1278.3 appeared to be exclusively formed from the SIRT1-catalyzed reaction of NAD+ with peptide 1. The species with m/z 1278.3 could represent the [M − H]− anion derived from intermediate I (an α-1′-O-alkylamidate species) or from intermediate II (a bicyclic species) (Fig. 5) since the neutral form of I has an identical chemical formulae (C48H79N15O20P2S) and an identical exact mass (1279.5) as compared to II. In an effort to ascertain which of these two could be more likely the stalled catalytic species and thus detectable with mass spectrometry, we subsequently performed a methanolysis assay according to the literature procedure,15 and 20% (v/v) of methanol was included in the above incubation mixture of peptide 1 with NAD+ and SIRT1. Our hypothesis is that, if intermediate I were stalled, then the nucleophilic methanol would have a higher chance to intercept this intermediate by attacking at the electrophilic C1′ position, leading to the diminished signal at m/z 1278.3 and the appearance of the mass spectral signal for β-1′-O-methyl-ADP-ribose (exact mass = 572.1). However, when the methanolysis reaction mixture was subjected to both MALDI (negative reflector mode) and electrospray ionization (ESI) (positive mode) mass spectral analysis, neither of these two signal changes was observed when compared with the corresponding mass spectral data from the reaction mixture in the absence of methanol (Fig. 4A vs. 4B and Fig. S2Avs. S2B†). Given the well established literature use of this kind of the methanolysis approach to detect the stalled α-1′-O-alkylamidate intermediate (Fig. 1) in the assays with the yeast Hst2[H135A] mutant and PfSir2 from Plasmodium falciparum,15 our result strongly suggested that the intermediate II was more likely the stalled catalytic species.
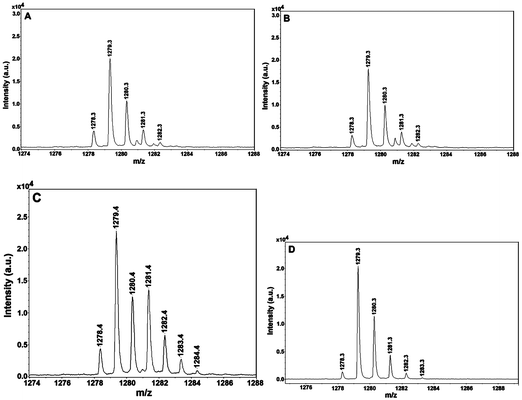 | ||
| Fig. 4 Mass spectrometry detection of the catalytic species from the assay mixtures of peptide 1 with SIRT1 or Sir2Tm. (A)Peptide 1 with SIRT1; (B)peptide 1 with SIRT1 in the presence of 20% (v/v) methanol; (C)peptide 1 with SIRT1 in the presence of 63% (v/v) H218O; (D)peptide 1 with Sir2Tm. All the assay mixtures were subjected to the MALDI-MS analysis in a negative reflector mode with CHCA as the matrix. See ESI for further experimental conditions.† | ||
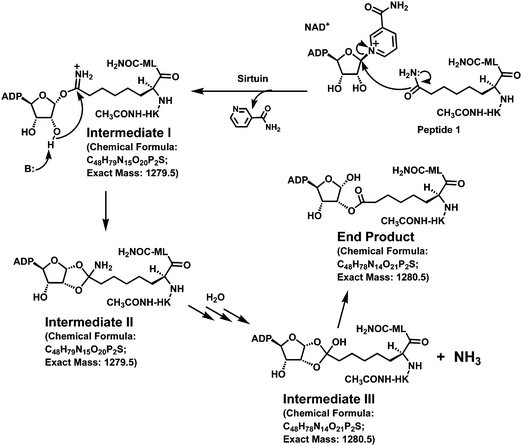 | ||
| Fig. 5 Proposed sirtuin processing of peptide 1. The chemical formula and the exact mass for Intermediate I are those for its neutral form. | ||
By inspecting closely the cluster of peaks starting at m/z 1278.3 in Fig. 4A, we also noticed with interest that the peak at m/z 1279.3 was significantly more intense than that at m/z 1278.3. This could be interpreted as that the species with m/z 1279.3 represents the M−˙ free radical anion derived from intermediate II. Alternatively, it could represent the [M − H]− anion derived from intermediate III or the end product (Fig. 5) since these two catalytic species also have identical chemical formulae (C48H78N14O21P2S) and an identical exact mass (1280.5). To explore these possibilities, we performed another incubation of peptide 1 with NAD+ and SIRT1 in the presence of 63% (v/v) of H218O. Our hypothesis is that, if the peak at m/z 1279.3 was derived from intermediate III and/or the end product, then a mass increase by 2 atomic mass units (amu) for this peak and its associated isotopic peaks should be observed following the transfer of the 18O label of H218O onto these two catalytic species. Since it has been established in the literature that the 18O label of H218O was transferred onto the 2′-acetoxy carbonyl oxygen of 2′-O-acetyl-ADP-ribose (Fig. 1),15a,16 it is reasonable to predict that the 18O label of H218O could also be transferred onto the 2′-acyloxy carbonyl oxygen of the end product (Fig. 5). Even though it is still currently unknown how the bicyclic intermediate collapses to form the deacetylated product and 2′-O-acetyl-ADP-ribose (Fig. 1),2e the obligatory involvement of H2O at this stage of the reaction coordinate supports the view that the 18O label of H218O will be transferred onto the free OH group of the hemi-orthoester portion of intermediate III if it is indeed formed. When the incubation mixture containing H218O was subjected to the MALDI (negative reflector mode) mass spectral analysis, we clearly observed significant changes of the pattern of the cluster of the peaks starting at m/z 1278.3 (Fig. 4A vs. 4C). The most notable changes included the significantly enhanced signal level at m/z 1281.3 and 1282.3, as well as the appearance of two significant peaks at m/z 1283.4 and m/z 1284.4. This data thus strongly suggested that the species with m/z 1279.3 (Fig. 4A) represents the [M − H]− anion derived from the intermediate III and/or the end product (Fig. 5).
Even though we are currently still unable to determine exactly which of these two species or both are the longer-lived catalytic species detectable by mass spectrometry, our current data nevertheless have provided strong support for the idea that the L-ACAH-containing peptide 1 behaves as a mechanism-based SIRT1 inhibitor, in that it can be converted by SIRT1 to at least one longer-lived catalytic intermediate (i.e. II and perhaps III as well, Fig. 5) and perhaps all the way to the end product shown in Fig. 5 as well. Our observed potent SIRT1 inhibition by peptide 1 could therefore be accounted for by the formation of these relatively stabilized catalytic species that also likely behave as tight-binding bisubstrate analog inhibitors since they are basically the covalent conjugates between NAD+ and peptide 1 (Fig. 5). Of note, the observed relatively weaker signal at m/z 1278.3 than that at m/z 1279.3 could be due to the differential abundance of the corresponding catalytic species and/or their differential ionization efficiency under our MALDI-MS experimental conditions.
Following the above experiments with SIRT1, we also performed a mass spectral analysis of the incubation mixture of peptide 1 and NAD+ with the bacterial sirtuin Sir2Tm under the same experimental conditions. As shown in Fig. 4, essentially the same pattern of the peak cluster starting at m/z 1278.3 was also observed (Fig. 4A vs. 4D). Again, it was found that the formation of this cluster of peaks depended on the presence of Sir2Tm, peptide 1, and NAD+ since the omission of any of these three components from the incubation mixture led to the absence of this peak cluster (Fig. S1E, S1F, and S1G†). Our results with SIRT1 and Sir2Tm further suggested that different sirtuin enzymes are able to process peptide 1 in a similar manner.
Given the close structural similarity between the side chains of AcK and L-ACAH, our current study has also provided another piece of evidence supporting the existence of the proposed bicyclic intermediate along the sirtuin-catalyzed lysine deacetylation reaction coordinate (Fig. 1). In this regard, a L-ACAH-containing peptide (e.g.peptide 1 in the current study) could be an invaluable biochemical/biophysical probe for further dissecting the intermediate events for the sirtuin-catalyzed lysine deacetylation reaction, via trapping a longer-lived catalytic intermediate (e.g. II in Fig. 5) within the active site of a sirtuin crystal, and solving the corresponding co-crystal structure by X-ray crystallography. This type of study will be able to not only provide direct evidence for the existence of a sirtuin deacetylation reaction intermediate (e.g. the proposed bicyclic intermediate or an intermediate in the path of the collapse of the bicyclic intermediate toward the deacetylated product and 2′-O-acetyl-ADP-ribose (Fig. 1)), but also shed light on how the bicyclic intermediate collapses in the presence of water toward the deacetylated product and 2′-O-acetyl-ADP-ribose, which is currently unknown. The previous success14b in using a ThAcK-containing peptide to structurally elucidate the sirtuin deacetylation reaction intermediate steps involving α-1′-O-alkylamidate via solving the co-crystal structure of Sir2Tm (a bacterial sirtuin) with a molecule of the stalled α-1′-S-alkylamidate intermediate trapped within its active site should encourage similar endeavors using a L-ACAH-containing peptide.
Since the lysine Nε-deacetylation reaction can also be catalyzed by the Zn2+-containing classical family of protein lysine deacetylases including the 11 human HDACs, i.e.HDAC1–11 (HDAC stands for histone deacetylase),1b,1c in order to examine if the L-ACAH residue can be processed by a classical deacetylase enzyme, we used human HDAC8 as the typical classical member to perform the time course and the inhibition experiments with the following pair of peptides: CH3CONH-RH-AcK-(AcK)-CONH2 and CH3CONH-RH-AcK-(L-ACAH)-CONH2. Of note, the former peptide was used in our previous studies as a substrate for the HeLa nuclear extract enriched in HDAC1 and HDAC2.17 However, we further found that this AcK-containing peptide was also a HDAC8 substrate, therefore it was used in the two experiments we performed in the current study as the positive control or the substrate. Moreover, we believe that the AcK residue in the parentheses of this peptide substrate was the one to be deacetylated since (i) the side chain of this AcK was found to bind to the AcK binding tunnel within the active site of HDAC8 in a co-crystal structure of HDAC8 and H2N-RH-AcK-(AcK)-CONH-AMC (AMC stands for 7-amino-4-methylcoumarin)18 and (ii) the above second peptide containing L-ACAH at the corresponding AcK position was found in the current study not to be processed by HDAC8 even though it still contains an AcK residue (vide infra).
When the above two peptides were subjected to a HPLC-based HDAC8 time course assay, we observed a robust HDAC8-catalyzed deacetylation from CH3CONH-RH-AcK-(AcK)-CONH2, in that around 8.3% of substrate turnover was already observed following the incubation of this substrate peptide with HDAC8 at room temperature for 1 h; however, no enzymatic transformation was detected from CH3CONH-RH-AcK-(L-ACAH)-CONH2 even after a 4 h incubation with HDAC8 at room temperature (see ESI†). Furthermore, only ∼12% of inhibition of the HDAC8-catalyzed deacetylation of CH3CONH-RH-AcK-(AcK)-CONH2 by 500 μM of CH3CONH-RH-AcK-(L-ACAH)-CONH2 was observed. Our observed lack of the enzymatic transformation of CH3CONH-RH-AcK-(L-ACAH)-CONH2 suggested that the side chain of L-ACAH is either unable to bind to the AcK binding tunnel within the HDAC8 active site or is able to bind but with its side chain amide functionality incorrectly positioned for the amide hydrolysis to occur. Moreover, this lack of the enzymatic transformation of CH3CONH-RH-AcK-(L-ACAH)-CONH2 contrasts with the capability of HDAC8 to efficiently catalyze the dethioacetylation and detrifluoroacetylation reactions previously demonstrated respectively for the ThAcK- and trifluoroacetyl-lysine-containing sirtuin inhibitors.4a,4b,17,19 The observed very weak HDAC8 inhibition by CH3CONH-RH-AcK-(L-ACAH)-CONH2 further suggested that the L-ACAH residue can selectively confer potent inhibition against the sirtuin family members versus the classical family members of the proteinlysine deacetylase enzymes (at least SIRT1versusHDAC8).
Having identified peptide 1 as a fairly potent peptidic SIRT1 inhibitor, we were also interested in exploring the possibility of identifying the L-ACAH-containing non-peptidic or peptidomimetic SIRT1 inhibitors, as has been done by us and others for the identification of non-peptidic or peptidomimetic ThAcK-containing SIRT1 inhibitors.2e For this purpose, compounds 3 and 4 were designed. Specifically, in compound 3, L-ACAH was incorporated into the non-peptidic scaffold that Suzuki et al. used previously to develop their non-peptidic ThAcK-containing human sirtuin inhibitor;20 in compound 4, L-ACAH was incorporated into the peptidomimetic scaffold that we used previously to develop our peptidomimetic ThAcK-containing human sirtuin inhibitors.4c
The synthesis of compound 3 as depicted in Scheme 2 also started with L-α-aminosuberic acid. The selective methylesterification of the side chain carboxyl of this α-amino acid by the method of Glenn et al.9 and the subsequent aminolysis11 of the side chain methylester afforded L-ACAH itself. The installation of benzyloxycarbonyl (Cbz) at the α-amino group of L-ACAH and the subsequent anilide formation with aniline afforded compound 3. The second intermediate in Scheme 1 (i.e.L-α-(9-fluorenylmethoxycarbonyl-amino)-suberic acid 8-methyl ester) was also used to prepare compound 4 as depicted in Scheme 3. Specifically, compound 4 was obtained by the initial SPPS on the Rink amide resin and the subsequent solution phase aminolysis11 of the methylester-containing intermediate released from the resin.
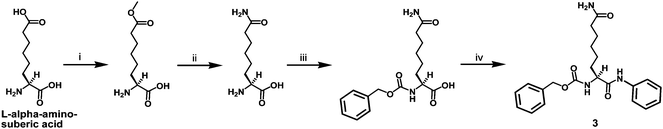 | ||
| Scheme 2 Synthesis of the L-ACAH-containing compound 3. Reagents: (i) SOCl2, MeOH; (ii) 28–30% (w/w) NH4OH; (iii) Cbz–OSu, 10% (w/v) aq. Na2CO3, 1,4-dioxane; (iv) aniline, EDC·HCl, HOBt. | ||
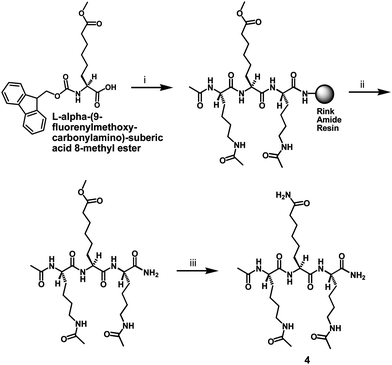 | ||
| Scheme 3 Synthesis of the L-ACAH-containing compound 4. Reagents: (i) Fmoc-based SPPS; (ii) TFA-containing peptide cleavage cocktail; (iii) 28–30% (w/w) NH4OH. | ||
Compounds 3 and 4 were then both subjected to the SIRT1 inhibition assay. While 3 was found to be a fairly weak inhibitor against SIRT1-catalyzed deacetylation of H2N-HK-AcK-LM-COOH with an IC50 > 1 mM (Table 1), 4 was found to be a reasonably potent SIRT1 inhibitor with IC50 ∼290 μM (Table 1). This observed difference in the SIRT1 inhibitory potency of compounds 3 and 4 further reinforced our previous observation that our peptidomimetic scaffold was able to confer stronger sirtuin inhibition than the non-peptidic scaffold used by Suzuki et al.4c Moreover, even though peptide 1 and the ThAcK-containing peptidic inhibitors (ThAcK in Table 1 and those penta-peptides in ref. 4c) are similarly potent SIRT1 inhibitors with roughly single-digit IC50 values under our SIRT1 inhibition assay conditions, compounds 3 and 4 were both found to be weaker SIRT1 inhibitors than their ThAcK-containing counterparts whose IC50 values were determined previously to be ∼140 μM and ∼2.1 μM, respectively, under our SIRT1 inhibition assay conditions.4c This observation suggested that ThAcK is more powerful in conferring sirtuin inhibition than L-ACAH so that the sirtuin inhibitory potency could be less contingent on the binding contributions from the neighboring structural motifs for ThAcK-containing inhibitors than those containing L-ACAH.
Compound 4 was further evaluated in the in vitroSIRT2 and SIRT3 inhibition assays. As shown in Table 1, 4 was also found to be an inhibitor against SIRT2 and SIRT3 with comparable potency to that against SIRT1. This observed roughly similar inhibitory potency of compound 4 against SIRT1, SIRT2, and SIRT3 is consistent with our previously observed similar inhibitory potency against these three sirtuins by the ThAcK-containing counterpart of 4, and suggested that the proteolytically stable and cell permeable peptidomimetic scaffold found in these two compounds is able to confer potent sirtuin inhibition, yet unable to confer decent selective inhibition among sirtuins (at least among SIRT1, SIRT2, and SIRT3), warranting the further structural optimization of this peptidomimetic scaffold. Moreover, the roughly similar inhibitory potency observed in the current study for compound 4 against SIRT1, SIRT2, and SIRT3 also suggested that L-ACAH is a general inhibitory warhead against multiple sirtuin deacetylase enzymes, a notion that has already been established for ThAcK.2e
Even though compound 4 is a quite less potent SIRT1 inhibitor than its ThAcK counterpart (IC50 ∼290 μM vs. ∼2.1 μM4c), its potency is nevertheless only ∼2-fold less than that for the ThAcK counterpart of 3 (IC50 ∼290 μM vs. ∼140 μM4c), which was shown to be able to inhibit the SIRT1 inside the human colon cancer cell line HCT116.20 Moreover, the ThAcK counterpart of 4 was also shown by us previously to be able to inhibit the SIRT1 inside the human colon cancer cell line HCT116 since the peptidomimetic scaffold of this ThAcK-containing compound was found to be proteolytically stable and cell permeable.4c Therefore, compound 4 was also further evaluated in a cell-based assay to examine its capability of achieving SIRT1 inhibition inside the cell. For this purpose, we also used the HCT116 human colon cancer cells that express the wild-type p53 protein and assessed the extent of enhancement of the level of the acetylation of this SIRT1 endogenous substrate protein at the K382 position21 following the treatment with compound 4. As shown in Fig. 6, compound 4 was also able to enhance the p53 K382acetylation level in a dose-dependent manner. Consistent with our in vitro finding that 4 was less potent than its ThAcK counterpart as a SIRT1 inhibitor, the former compound also exhibited a lower capacity of increasing the p53 K382acetylation level than the latter. Specifically, while the latter compound was shown by us previously to be capable of increasing the p53 acetylation level by ∼2-fold at 3 μM,4c essentially no p53 acetylation level increase was observed at a concentration of 5 μM of 4; however, 4 was shown to be able to dramatically (several fold) enhance the p53 acetylation level at a concentration of 50 μM (Fig. 6). Therefore, compound 4 could well serve as a lead compound for the further development of L-ACAH-based peptidomimetic or non-peptidic human SIRT1 inhibitors that will show potent inhibition in both in vitro and in vivo settings.
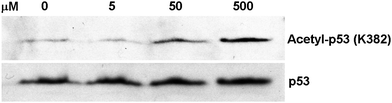 | ||
| Fig. 6 Western blot analysis of the p53 protein acetylation level change in HCT116 human colon cancer cells following the treatment with compound 4. | ||
Conclusions
In the current study, we discovered that replacing L-ACAH for AcK in a SIRT1 peptide substrate could confer potent SIRT1 inhibition. The in vitro studies with the peptidic inhibitor CH3CONH-HK-(L-ACAH)-LM-CONH2 revealed that (i) the inhibitor was competitive versus the AcK-containing substrate H2N-HK-AcK-LM-COOH, (ii) the inhibitor was of the slow, tight-binding type as suggested by the observed time-dependent inhibition and a lag in the formation of the deacetylated product in the activity recovery curve of the inhibited SIRT1 toward a full regain of its deacetylase activity, and (iii) the inhibitor was able to be converted to at least one longer-lived catalytic intermediate and perhaps all the way to the product (similar to 2′-O-acetyl-ADP-ribose) as well. The in vivo characterization of the L-ACAH-containing peptidomimetic inhibitor 4 revealed its capability of inhibiting the SIRT1 inside the human colon cancer cell line HCT116. Compound 4 was also shown to be an in vitroinhibitor against SIRT2 and SIRT3 with comparable potency to that against SIRT1. These results suggested that the sirtuin deacetylase inhibition by a L-ACAH-containing compound is mechanism-based and a general inhibitory strategy against sirtuin-catalyzed lysine deacetylation reaction, and point to the potential of developing further potent and cell permeable L-ACAH-based peptidomimetic or non-peptidic inhibitors for sirtuins. In our opinion, the L-ACAH-based sirtuin inhibitors shall also constitute an attractive class of compounds useful as chemical biological tools for the fuller exploration of sirtuin biology and pharmacology, as well as useful as potential therapeutics for the metabolic and age-related diseases and cancer.Note added in proof
Very recently we also investigated whether nicotinamide was able to inhibit the formation of the catalytic species from peptide 1 in the presence of NAD+ and a sirtuin (e.g.SIRT1). It was demonstrated that nicotinamide was able to do so in a concentration-dependent manner, as indicated by an increase in the intensity ratio of the MALDI-MS peaks derived from NAD+ versus those derived from the longer-lived catalytic species from peptide 1, when the exogenous nicotinamide added to the assay mixture was increased (see Fig. 7). This inhibitory effect of nicotinamide is reminiscent of the sirtuin-catalyzed nicotinamide base exchange reaction and the consequent sirtuin inhibition by nicotinamide, i.e.nicotinamide is able to intercept the α-1′-O-alkylamidate intermediate (Fig. 1) to regenerate NAD+ and the acetylated substrate, thus impeding the forward deacetylation chemistry from this intermediate.2e This data is therefore also fully consistent with the idea that peptide 1 was able to be processed by a sirtuin (e.g. SIRT1) according to the proposed catalytic pathway in Fig. 5.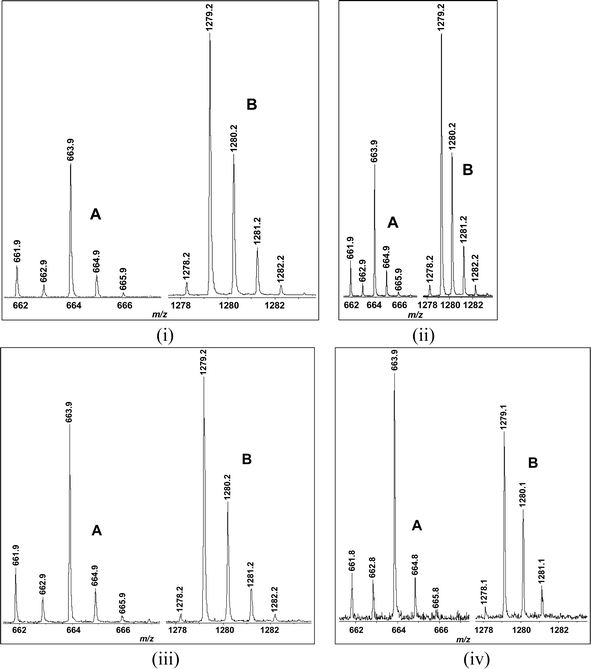 | ||
| Fig. 7 MALDI-MS analysis of the incubation mixtures of peptide 1 with SIRT1 in the presence of the exogenous nicotinamide added to the incubation mixture at the following final concentrations: (i) 0 μM; (ii) 50 μM; (iii) 250 μM; (iv) 1,250 μM. In each panel, the A and B clusters of peaks were respectively derived from NAD+ and the longer-lived catalytic species from peptide 1. This assay was performed in the same manner as that for the mass spectrometry detection of the catalytic species with peptide 1 and SIRT1 or Sir2Tm (see the ESI†). | ||
Acknowledgements
This study was supported by the James L. and Martha J. Foght Endowment, the University of Akron Research Foundation, and the U.S. National Institutes of Health (R15CA152972 and R01CA127590). We thank Prof. Tony Kouzarides (University of Cambridge, UK) for the GST-SIRT1 plasmid, and Prof. Cynthia Wolberger (Johns Hopkins University School of Medicine, USA) for the Sir2Tm protein. We would like to thank the high resolution mass spectrometry facility of the University of California, Riverside for the HRMS data. We also thank Prof. Chrys Wesdemiotis and his research group at the University of Akron for the assistance with mass spectrometric analysis (ESI-MS) of the peptides synthesized in the current study. David G. Fatkins is thanked for the synthesis of CH3CONH-RH-AcK-(AcK)-CONH2 used in the current study. We wish to thank The Goodyear Corporation for donation of the NMR instrument (Varian Mercury 300) used in this work. We also wish to thank The National Science Foundation (CHE-8808587) for funds used to purchase the NMR instrument (Varian GEMINI 300) used in this work.Notes and references
- (a) R. A. Frye, Biochem. Biophys. Res. Commun., 2000, 273, 793 CrossRef CAS; (b) S. Thiagalingam, K. H. Cheng, H. J. Lee, N. Mineva, A. Thiagalingam and J. F. Ponte, Ann. N. Y. Acad. Sci., 2003, 983, 84 CrossRef CAS; (c) I. V. Gregoretti, Y. M. Lee and H. V. Goodson, J. Mol. Biol., 2004, 338, 17 CrossRef CAS; (d) L. R. Saunders and E. Verdin, Oncogene, 2007, 26, 5489 CrossRef CAS; (e) B. C. Smith, W. C. Hallows and J. M. Denu, Chem. Biol., 2008, 15, 1002 CrossRef CAS; (f) S. Greiss and A. Gartner, Mol. Cells, 2009, 28, 407 CrossRef CAS.
- (a) A. A. Sauve and V. L. Schramm, Curr. Med. Chem., 2004, 11, 807 CrossRef CAS; (b) A. A. Sauve, C. Wolberger, V. L. Schramm and J. D. Boeke, Annu. Rev. Biochem., 2006, 75, 435 CrossRef CAS; (c) B. D. Sanders, B. Jackson and R. Marmorstein, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1604 CrossRef CAS; (d) A. A. Sauve, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804, 1591 CrossRef CAS; (e) B. M. Hirsch and W. Zheng, Mol. Biosyst., 2011, 7, 16 RSC.
- (a) T. Liu, P. Y. Liu and G. M. Marshall, Cancer Res., 2009, 69, 1702 CrossRef CAS; (b) T. Finkel, C. X. Deng and R. Mostoslavsky, Nature, 2009, 460, 587 CrossRef CAS; (c) J. Yu and J. Auwerx, Ann. N. Y. Acad. Sci., 2009, 1173, E10 CrossRef CAS; (d) S. Imai and L. Guarente, Trends Pharmacol. Sci., 2010, 31, 212 CrossRef CAS; (e) M. C. Haigis and D. A. Sinclair, Annu. Rev. Pathol.: Mech. Dis., 2010, 5, 253 Search PubMed; (f) M. Lakshminarasimhan and C. Steegborn, Exp. Gerontol., 2011, 46, 174 CrossRef CAS; (g) E. Verdin, M. D. Hirschey, L. W. Finley and M. C. Haigis, Trends Biochem. Sci., 2010, 35, 669 CrossRef CAS; (h) K. L. Guan and Y. Xiong, Trends Biochem. Sci., 2010 Search PubMed [Epub ahead of print].
- (a) D. G. Fatkins, A. D. Monnot and W. Zheng, Bioorg. Med. Chem. Lett., 2006, 16, 3651 CrossRef CAS; (b) D. G. Fatkins and W. Zheng, Int. J. Mol. Sci., 2008, 9, 1 Search PubMed; (c) B. M. Hirsch, C. A. Gallo, Z. Du, Z. Wang and W. Zheng, Med. Chem. Commun., 2010, 1, 233 RSC.
- (a) R. A. Neal and J. Halpert, Annu. Rev. Pharmacol., 1982, 22, 321 Search PubMed; (b) M. J. Ruse and R. H. Waring, Toxicol. Lett., 1991, 58, 37 CrossRef CAS; (c) D. N. Cox, V. P. Davidson, C. E. Judd, C. Stodgell and G. J. Traiger, Toxicol. Appl. Pharmacol., 1992, 113, 246 CrossRef CAS; (d) G. M. Coppola, H. Anjaria and R. E. Damon, Bioorg. Med. Chem. Lett., 1996, 6, 139 CrossRef CAS; (e) J. S. Kang, H. Wanibuchi, K. Morimura, R. Wongpoomchai, Y. Chusiri, F. J. Gonzalez and S. Fukushima, Toxicol. Appl. Pharmacol., 2008, 228, 295 CrossRef CAS.
- N. H. G. Holford, in Basic and Clinical Pharmacology; B. G. Katzung, S. B. Masters and A. J. Trevor, ed.; McGraw-Hill Medical: USA 2009, 37–51, 11th edn Search PubMed.
- T. L. Lemke, Review of Organic Functional Groups: Introduction to Medicinal Organic Chemistry; Lippincott Williams & Wilkins: USA 2003, 4th edn Search PubMed.
- (a) A. L. Garske and J. M. Denu, Biochemistry, 2006, 45, 94 CrossRef CAS; (b) N. Jamonnak, B. M. Hirsch, Y. Pang and W. Zheng, Bioorg. Chem., 2010, 38, 17 CrossRef CAS.
- M. P. Glenn, M. J. Kelso, J. D. Tyndall and D. P. Fairlie, J. Am. Chem. Soc., 2003, 125, 640 CrossRef CAS.
- D. A. Wellings and E. Atherton, Methods Enzymol., 1997, 289, 44 CAS.
- P. Franchetti, L. Cappellacci, M. Grifantini, A. Barzi, G. Nocentini, H. Yang, A. O'Connor, H. N. Jayaram, C. Carrell and B. M. Goldstein, J. Med. Chem., 1995, 38, 3829 CrossRef CAS.
- I. H. Segel, Enzyme Kinetics: Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems; Wiley-Interscience: USA 1993 Search PubMed.
- J. F. Morrison and C. T. Walsh, Adv. Enzymol. Relat. Areas Mol. Biol., 1988, 61, 201 Search PubMed.
- (a) B. C. Smith and J. M. Denu, Biochemistry, 2007, 46, 14478 CrossRef CAS; (b) W. F. Hawse, K. G. Hoff, D. G. Fatkins, A. Daines, O. V. Zubkova, V. L. Schramm, W. Zheng and C. Wolberger, Structure, 2008, 16, 1368 CrossRef CAS; (c) L. Jin, W. Wei, Y. Jiang, H. Peng, J. Cai, C. Mao, H. Dai, W. Choy, J. E. Bemis, M. R. Jirousek, J. C. Milne, C. H. Westphal and R. B. Perni, J. Biol. Chem., 2009, 284, 24394 CrossRef CAS.
- (a) B. C. Smith and J. M. Denu, Biochemistry, 2006, 45, 272 CrossRef CAS; (b) J. B. French, Y. Cen and A. A. Sauve, Biochemistry, 2008, 47, 10227 CrossRef CAS.
- A. A. Sauve, I. Celic, J. Avalos, H. Deng, J. D. Boeke and V. L. Schramm, Biochemistry, 2001, 40, 15456 CrossRef CAS.
- D. G. Fatkins and W. Zheng, Anal. Biochem., 2008, 372, 82 CrossRef CAS.
- A. Vannini, C. Volpari, P. Gallinari, P. Jones, M. Mattu, A. Carfí, R. De Francesco, C. Steinkühler and S. Di Marco, EMBO Rep., 2007, 8, 879 CrossRef CAS.
- B. C. Smith and J. M. Denu, J. Biol. Chem., 2007, 282, 37256 CrossRef CAS.
- T. Suzuki, T. Asaba, E. Imai, H. Tsumoto, H. Nakagawa and N. Miyata, Bioorg. Med. Chem. Lett., 2009, 19, 5670 CrossRef CAS.
- H. Vaziri, S. K. Dessain, E. Ng Eaton, S. I. Imai, R. A. Frye, T. K. Pandita, L. Guarente and R. A. Weinberg, Cell, 2001, 107, 149 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, Figures S1 and S2, copies of HPLC traces. See DOI: 10.1039/c0md00212g |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |