Repairing faulty genes by aminoglycosides: Identification of new pharmacophore with enhanced suppression of disease-causing nonsense mutations†
Jeyakumar
Kandasamy‡
,
Dana
Atia-Glikin‡
,
Valery
Belakhov
and
Timor
Baasov
*
The Edith and Joseph Fischer Enzyme Inhibitors Laboratory, Schulich Faculty of Chemistry, Technion - Israel Institute of Technology, Haifa, 32000, Israel. E-mail: chtimor@tx.technion.ac.il; Fax: +972-4-829-5703; Tel: +972-4-829-2590
First published on 14th January 2011
Abstract
New pseudo-trisaccharide derivatives of aminoglycosides containing a chiral methyl group at the side-chain of the ribosamine ring (ring III) were designed, synthesized and their ability to readthrough nonsense mutations was examined in both in vitro and ex vivo systems, along with toxicity tests. Novel lead structures, exhibiting significantly reduced cell toxicity, enhanced specificity to eukaryotic cytoplasmic ribosome, and superior readthrough efficiency compared with those of gentamicin, were discovered. The comparative benefit of new leads was demonstrated in six different nonsense DNA-constructs underling the genetic diseases cystic fibrosis, Duchenne muscular dystrophy, Usher syndrome and Hurler syndrome.
Nonsense mutations are caused by single point alterations in DNA that give rise to in-frame UAA, UAG, or UGA stop codons in mRNA coding regions and lead to the formation of truncated, nonfunctional proteins.1,2 This type of mutation represents the underlying cause of more than 1800 inherited human diseases that affect children and adults,3 and for many of those diseases there is presently no effective treatment. Although gene therapy can in principle directly overcome the disease caused by nonsense mutation, this approach is still far from achieving clinical success. One alternative approach is the induction of selective readthrough of pathogenic nonsense mutations, but not of normal termination codons, so that the expression of the full-length, functional protein is restored to some extent.4Aminoglycoside antibiotics were the first small-molecule drugs that gave promising results and the validation of this approach: numerous in vitro and in vivo experiments including clinical trials have demonstrated the ability of selected aminoglycoside structures to induce mammalian ribosome to readthrough disease-causing nonsense mutations and partially restore full-size functional proteins.1,2,5
It is highly noteworthy that the full-size protein produced through this approach, even if only in low amounts, may be functionally significant. This is particularly relevant for recessive disorders like cystic fibrosis (CF), Duchenne muscular dystrophy (DMD), Hurler syndrome (HS), and many more, in which a single crucial protein expression is essentially absent and even 1% of normal protein function may restore a near-normal or clinically less severe phenotype.2,6,7 Accordingly, the greatest promise of aminoglycosides as potential pharmacogenetic agents is primarily for the treatment of recessive disorders caused by nonsense mutations.
Despite the promise of this approach, the high human toxicity of aminoglycosides and reduced readthrough efficiency at safe doses have limited their clinical benefit for suppression therapy.1,8 For example, in spite of its high toxicity, the clinical aminoglycoside antibiotic gentamicin (Fig. 1) was/is frequently used for proof-of-concept experiments in various disease models and in clinical trials, and almost no systematic study has been performed to tune aminoglycoside structures for better readthrough activity and low toxicity.6 In addition, while the recent X-ray crystal structures of the bacterial ribosome in complex with aminoglycosides shed light on the mechanism of action of aminoglycosides as antibiotics,9,10 no crystal structure of a human ribosome is presently available, and the mechanism of aminoglycoside-induced readthrough is largely obscure.6,11
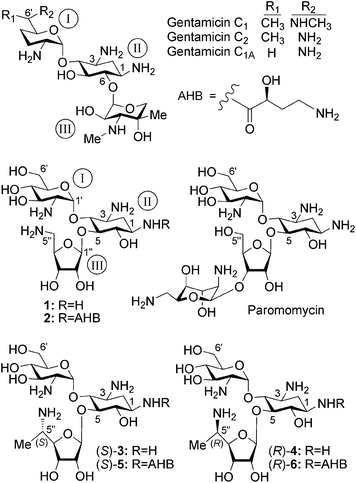 | ||
| Fig. 1 Chemical structures of a series of natural and synthetic 2-deoxystreptamine-derived (ring II) aminoglycosides, including compounds 1, 2, (S)-3, (R)-4, (S)-5 and (R)-6 that were investigated in this study. | ||
To address these issues, recently we hypothesized that by separating the structural elements of aminoglycosides that induce readthrough from those that affect toxicity we might reach potent derivatives with improved readthrough activity and reduced toxicity.6,12 By keeping this line of research, we initially developed the lead compounds 112 and 213 as novel pseudo-trisaccharide derivatives of the clinical aminoglycoside paromomycin (Fig. 1). Compound 1 exhibited significantly reduced cytotoxicity in comparison to gentamicin and paromomycin, and promoted dose-dependent suppression of nonsense mutations of the PCDH15 gene, the underlying cause of type 1 Usher syndrome (USH1), but its suppression potency was notably lower relative to that of gentamicin and paromomycin.14 Compound 2, which was developed as the second-generation lead structure, exhibited significantly reduced cell, cochlear and acute toxicities, and has substantially higher readthrough efficiency than those of gentamicin and paromomycin.13 Continuing systematic fine-tuning of the developed leads, in this communication we report on the development of new lead structures (S)-3 and (S)-5 (Fig. 1) that differ from the previous leads (1 and 2) by the presence of the side-chain (S)-5′′-methyl group on the ribosamine ring (ring III) and show significantly reduced cell toxicity while in parallel they exhibit substantially higher readthrough activity of disease-causing nonsense mutations than those of gentamicin. The comparative data observed also indicate that the installation of the (S)-5′′-methyl group either on compound 1 (to give compound (S)-3) or on compound 2 (to give compound (S)-5) does not significantly affect the cell toxicity of the resulting structure, while it greatly enhances the stop-codon readthrough activity and specificity to the eukaryotic ribosome of the resulting structures in comparison to those of the parent structures.
The rationale for selecting the ribosamine ring (ring III) as the modification target was to preserve the already well established impacts of the rings I and II in the previous leads (1 and 2), and in parallel to explore a new structural motif with significant suppression activity and perhaps with reduced toxicity. The choice for the installation of a chiral methyl group at the C5′′-position of the ribosamine ring of 1 and 2 was largely based on our very recent observations with derivatives of the natural aminoglycoside drug G418.15 In this study we demonstrated that the introduction of an (R)-6′-methyl group (originally present in G418) on the aminoglycoside structure (including the compounds 1 and 2) to yield the (R)-6′-secondary alcohol on the side-chain of ring I, significantly increases suppression activity while it has no significant influence on the cell toxicity of the resulting derivative. Identifying and securing this valuable pharmacophore, the (R)-6′-methyl group on ring I, we then anticipated that the exploration of the impact of the chiral methyl group at the side-chain C5′′-position of the ribosamine ring (ring III) could lead to a completely new pharmacophore with improved readthrough activity and probably with reduced toxicity. Since the installation of a methyl group at the C5′′-position of the ribosamine ring generates a new stereogenic center, we decided to prepare both C5′′-diastereomers with defined absolute configuration and compare their biological properties. For this purpose we synthesized two separate pairs of C5′′-diastereomers: (S)-3, (R)-4 and (S)-5, (R)-6 (Fig. 1), while each pair possesses an (S)-5′′-methyl and (R)-5′′-methyl on ring III and is derived from the parent 1 and 2, respectively.
Structures 3–6 were synthesized according to the general strategy that involves construction of the ring III as two individual compounds possessing an (S)-5-methyl and (R)-5-methyl with already established stereochemistry [(S)-17 and (R)-18], and using them as donors for the glycosylation reactions. These donors were readily accessible from the known thioglycoside 712 as illustrated in Scheme 1. Selective protection of C2- and C3-hydroxyls by isopropylidene (2,2-dimethoxypropane/acetone, CSA) was followed by oxidation of the remained primary alcohol using Dess–Martin periodinane (DMP, dichloromethane) to afford the aldehyde 8 in 70% isolated yield for two steps. Treatment of 8 with MeMgBr gave the corresponding secondary alcohol as a mixture of C5-diasteromers (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio) in 88% isolated yield. This mixture was separated by flash column chromatography and the major diastereomer was separately studied for the assignment of absolute stereochemistry at the C5-position (vide infra). This study established that the major and minor diastereomers exhibit (R)- and (S)-configuration, respectively [(R)-9 and (S)-10]. The following steps in Scheme 1 were separately performed on each diastereomer. Tosylation (TsCl, pyridine, 4-DMAP) of the secondary alcohol was followed by SN2 displacement of the corresponding tosylates [(R)-11 and (S)-12)] with NaN3 (DMF, HMPA) to furnish the azides (S)-13 and (R)-14 with inverted configurations. Hydrolysis of the isopropylidene ketal with aqueous acetic acid, followed by benzoylation of the resulting secondary alcohols, provided the benzoates (S)-15 and (R)-16. Our earlier studies on the assembly of the pseudo-trisaccharides 112 and 213 have demonstrated that the desired C5 acceptors are highly unreactive in glycosylation reactions and only trichloroacetimidate donors gave satisfactory results. Therefore, the thioglycosides (S)-15 and (R)-16 were converted to the corresponding trichloroacetimidates (S)-17 and (R)-18 in two successive steps; hydrolysis with NBS in aqueous acetone and treatment of the resulting hemiacetals with CCl3CN in the presence of DBU. The donors (S)-17 and (R)-18 were used in glycosylation reactions without further purification.
:1 ratio) in 88% isolated yield. This mixture was separated by flash column chromatography and the major diastereomer was separately studied for the assignment of absolute stereochemistry at the C5-position (vide infra). This study established that the major and minor diastereomers exhibit (R)- and (S)-configuration, respectively [(R)-9 and (S)-10]. The following steps in Scheme 1 were separately performed on each diastereomer. Tosylation (TsCl, pyridine, 4-DMAP) of the secondary alcohol was followed by SN2 displacement of the corresponding tosylates [(R)-11 and (S)-12)] with NaN3 (DMF, HMPA) to furnish the azides (S)-13 and (R)-14 with inverted configurations. Hydrolysis of the isopropylidene ketal with aqueous acetic acid, followed by benzoylation of the resulting secondary alcohols, provided the benzoates (S)-15 and (R)-16. Our earlier studies on the assembly of the pseudo-trisaccharides 112 and 213 have demonstrated that the desired C5 acceptors are highly unreactive in glycosylation reactions and only trichloroacetimidate donors gave satisfactory results. Therefore, the thioglycosides (S)-15 and (R)-16 were converted to the corresponding trichloroacetimidates (S)-17 and (R)-18 in two successive steps; hydrolysis with NBS in aqueous acetone and treatment of the resulting hemiacetals with CCl3CN in the presence of DBU. The donors (S)-17 and (R)-18 were used in glycosylation reactions without further purification.
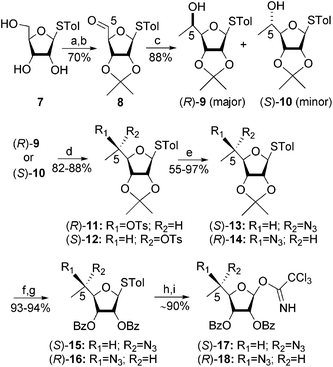 | ||
| Scheme 1 Reagents and conditions: (a) 2,2-dimethoxypropane, CSA, acetone, rt; (b) Dess–Martin periodinane (DMP), DCM, rt; (c) MeMgBr, THF, −30 °C; (d) TsCl, Py, 4-DMAP, rt; (e) NaN3, HMPA, DMF, 70 °C; (f) acetic acid/water (8:2), reflux; (g) BzCl, Py, 4-DMAP, rt; (h) NBS, acetone/water (8:2), −30 °C; (i) CCl3CN, DBU, DCM, 0 °C. | ||
The synthesis of the pseudo-trisaccharides 3–6 was accomplished from the corresponding selectively protected pseudo-disaccharide acceptors 19 and 20, previously reported by us,12,13 and the donors (S)-17 and (R)-18, by using essentially the same chemical transformations as illustrated in Scheme 2. Lewis acid (BF3·Et2O–DCM) promoted glycosylation furnished the protected pseudo-trisaccharides 21–24 in 79–85% isolated yields, exclusively as β-anomers at the newly generated glycosidic linkage. Two sequential deprotection steps: treatment with methylamine to remove all the ester protection, and the Staudinger reaction (Me3P, THF/NaOH) to convert azides to corresponding amines, then afforded the target compounds 3–6 in 79–82% isolated yields for two steps. The structures of all new compounds (3–6) were confirmed by a combination of various 1D and 2D NMR techniques, including 2D 1H–13C HMQC and HMBC, 2D COSY, and 1D selective TOCSY experiments, along with mass spectral analysis (see ESI†).
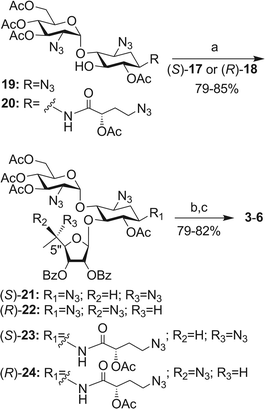 | ||
| Scheme 2 Reagents and conditions: (a) BF3·Et2O, DCM, 4 Å MS, −20 °C; (b) MeNH2–EtOH, rt; (c) PMe3, NaOH, THF, rt. | ||
For the assignment of the stereochemistry at the side-chain C5-alcohols in 9 and 10 (Scheme 1), we separately coupled (DCC, 4-DMAP, CSA) the major product 9 with (R)-2-methoxy-2(1-naphthyl)propanoic acid [(R)-MαNP] and (S)-MαNP of known absolute stereochemistry, to afford the corresponding esters (R,X)-MαNP-27 and (S,X)-MαNP-28 (Scheme 3, panel A), according to a recently reported procedure.16 The absolute configuration at the C5-position (denoted by X) was then determined by using the 1H NMR anisotropy method17 (Scheme 3, panels B and C): the chemical shift difference [Δδ = δ(R, X) − δ(S, X)] for H-3 (−0.30) and H-4 (−0.15) was negative, while that for H-6 (+0.28) was positive. An arrangement of the structures (R,X)-MαNP-27 and (S,X)-MαNP-28 according to the Sector rule16,18 (Scheme 3, panel C: OMαNP and H-5 are positioned on the front and back, respectively, while the Δδ positive part is on the right side of the MαNP plane and the Δδ negative part is on the left side) then confirmed the R configuration (X = R) of the C5 in 9.
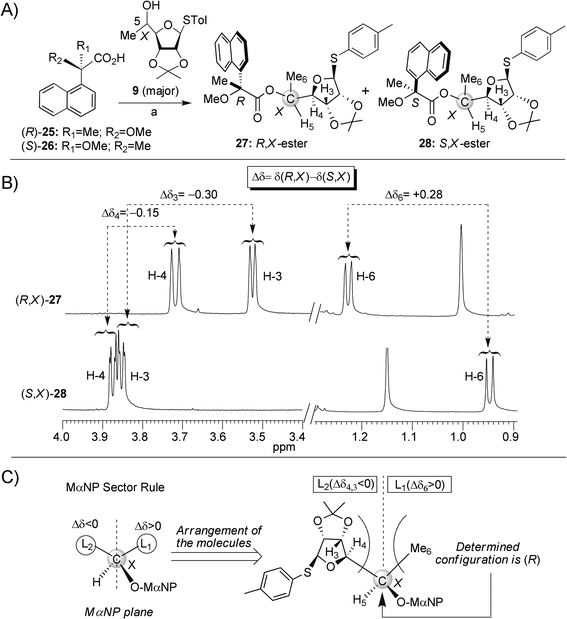 | ||
| Scheme 3 (A) Synthesis of C5-diasteromeric esters (R,X)-27 and (S,X)-28, reagents and conditions: (a) DCC, 4-DMAP, CSA, DCM, rt. (B) 1H NMR spectra of (R,X)-27 and (S,X)-28. The chemical shift differences (Δδ) between particular protons of (R,X)-27 and (S,X)-28 are highlighted. (C) Assignment of absolute configuration at the 5-carbon (denoted by X) of the major alcohol 9 by the Sector rule. | ||
The efficiency of aminoglycoside-induced readthrough is highly dependent on: (i) the identity of the stop codon (UGA > UAG > UAA), (ii) the identity of the first nucleotide immediately downstream from the stop codon (C > U > A ≥ G) and (iii) the local sequence context around the stop codon.19,20 Therefore, in attempts to provide a broad understanding of the structure–activity relationship of the designed structures, we used a variety of constructs containing different sequence contexts around premature stop codons derived from the PCDH15, CFTR, IDUA and Dystrophingenes that underlie USH1, CF, HS and DMD, respectively. The prevalent nonsense mutations of these diseases that we chose were: R3X and R245X for USH1, G542X and W1282X for CF, Q70X for HS and R3381X for DMD (for more details on the sequences see ESI†). Initially, the influence of the chiral C5′′-methyl group on readthrough potential was evaluated on the pseudo-trisaccharides (S)-3 and (R)-4 by using a dual luciferase reporter assay system as described previously by us13 (Fig. 2).
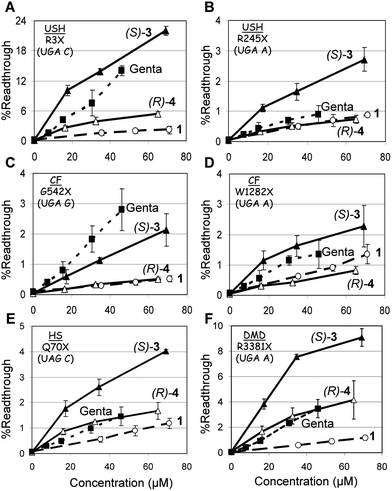 | ||
| Fig. 2 In vitro stop codon suppression levels induced by compounds 1 (○), (S)-3 (▲), (R)-4 (△) and gentamicin (■) in a series of nonsense mutation context constructs representing various genetic diseases (shown in parentheses): (A) R3X (USH1), (B) R245X (USH1), (C) G542X (CF), (D) W1282X (CF), (E) Q70X (HS), and (F) R3381X (DMD). DNA fragments were cloned between BamHI and SacI restriction sites of the p2luc vector and the constructs obtained were transcribed and translated using a TNT quick coupled transcription/translation system.13 The amount of the translated products was evaluated using the dual luciferase reporter assay system and the suppression level was calculated as described previously.21 The results are averages of at least three independent experiments. | ||
As seen from the data in Fig. 2, in all the mutations tested, installation of a (S)-5′′-methyl group (compound (S)-3) on compound 1 dramatically increases its in vitro readthrough activity, whereas that of the (R)-5′′-methyl group (compound (R)-4) is comparatively small. In addition, in all mutations tested (except G542X, Fig. 2C), the readthrough activity of (S)-3 was significantly better than that of the clinical drug gentamicin. Encouraged, we decided to explore the possibility of further potency enhancement. In particular, we were interested to see if the same potency enhancement, due to addition of the (S)-5′′-methyl group, would apply in compound 2 as well. To evaluate the impact of the stereochemistry at the C5′′-position, we constructed and tested both C5′′-diastereomers (S)-5 and (R)-6. Comparative in vitro suppression tests of the pseudo-trisaccharides 2, (S)-5, (R)-6, and gentamicin were performed under the same experimental conditions as for the compounds 1–3 (Fig. 2), and the observed data are shown in Fig. 3.
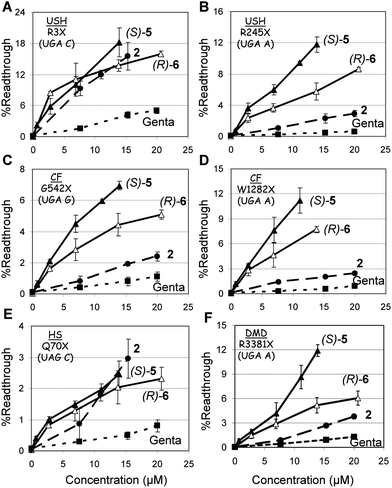 | ||
| Fig. 3 In vitro stop codon suppression levels induced by compounds 2 (●), (S)-5 (▲), (R)-6 (△) and gentamicin (■) in a series of nonsense mutation context constructs representing various genetic diseases (shown in parentheses): (A) R3X (USH1), (B) R245X (USH1), (C) G542X (CF), (D) W1282X (CF), (E) Q70X (HS), and (F) R3381X (DMD). The experiments and data analysis were performed as in the legend of Fig. 2. The results are averages of at least three independent experiments. | ||
From the observed data in Fig. 3 it can be seen that the efficacy of readthrough is substantially different between different constructs and compounds tested, with no obvious dependence of readthrough effectiveness on the type of modification introduced on the aminoglycoside. Nevertheless, in all mutations tested (except R3X and Q70X, Fig. 3A and 3E) compound (S)-5 induced the highest level of readthrough, followed by (R)-6, 2, and gentamicin. The UGA C tetracodon sequence (R3X) showed better translational readthrough than UGA A and UGA G, with the UAG C tetracodon least efficient, in agreement with earlier observations.13,19,20 To further evaluate the readthrough potential of compounds (S)-5 and (R)-6, their activity was assayed in cultured mammalian cells using four different dual luciferase reporter plasmids harboring the PCDH15-R3X and PCDH15-R245X nonsense mutation of USH1, the IDUA-Q70X nonsense mutation of HS, and the CFTR-W1282X nonsense mutation of CF. These reporter constructs were the same as we used above for the in vitro study, and have the distinct advantage of controlling for differences in mRNA levels between normal and nonsense-containing sequences over those of single reporter or direct protein analysis, as previously noted.20,21 The constructs were transfected into a human embryonic kidney cell line (HEK-293) and incubated with varying concentrations of (S)-5, (R)-6, 2 and gentamicin (Fig. 4). In order to ensure suitable cell viability for each of the tested compounds at the concentrations tested, we determined the cell toxicity for each compound by measuring the half-maximal-lethal concentration value (LC50 values) in HEK-293 and HFF (human foreskin fibroblast) cells (Table 1).
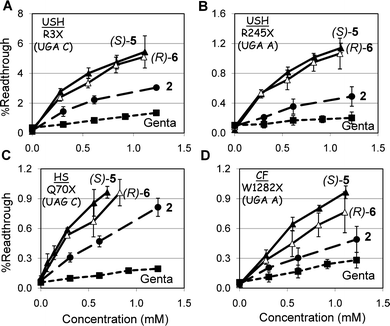 | ||
| Fig. 4 Ex vivo suppression of the (A) PCDH15-R3X, (B) PCDH15-R245X, (C) IDUA-Q70X, and (D) CFTR-W1282X nonsense mutations by compounds 2 (●), (S)-5 (▲), (R)-6 (△) and gentamicin (■). The constructs of p2luc plasmid harboring the R3X, R245X, Q70X and W1282X mutations were transfected to HEK-293 cells using lipofectamine 2000 and the tested compounds were added 6 h post transfection. Cells were harvested after 16 h incubation and luciferase activity was determined using the dual luciferase reporter assay system (Promega™). Stop codon readthrough was calculated as described previously.21 The results are averages of at least three independent experiments. | ||
| Aminoglycoside | Cell toxicity LC50 (mM)b | Translation inhibitionc | Antibacterial activity MIC (μM)d | |||
|---|---|---|---|---|---|---|
| HEK-293 | HFF | Eukaryotic system IC50 (μM) | Prokaryotic system IC50 (nM) | E. coli R477/100 | B. subtilis ATCC6633 | |
| a In all biological tests, all tested aminoglycosides were in their sulfate salt forms. The concentrations reported refer to that of the free amine form of each aminoglycoside. b Aminoglycoside-induced cell toxicity was measured in human embryonic kidney cells, HEK-293, and human foreskin fibroblast cells, HFF, and calculated as a ratio between the number of living cells in cultures grown in the presence of the compound tested, versus cultures grown without compound. The half-maximal lethal concentration (LC50) values were obtained from fitting concentration-response curves to the data of at least three independent experiments, using GraFit 5 software. c Prokaryotic and eukaryotic translation inhibition was quantified in coupled transcription/translation assays as previously described by us.11 The half-maximal concentration (IC50) values were obtained from fitting concentration response curves to the data of at least three independent experiments, using GraFit 5 software. d The MIC values were determined by using the double-microdilution method, with two different starting concentrations of each tested compound (384 μg mL−1 and 6144 μg mL−1). All the experiments were performed in duplicates and analogous results were obtained in three different experiments. | ||||||
| Gentamicin | 2.5 ± 0.3 | 3.2 ± 0.3 | 62 ± 9 | 28 ± 4 | 6 | <0.75 |
| Paromomycin | 4.1 ± 0.5 | 3.1 ± 0.4 | 57 ± 4 | 51 ± 5 | 22 | 1.2 |
| Compound 1 | 21.4 ± 3.9 | 21.8 ± 0.9 | 31 ± 4 | 460 ± 50 | 790 | 100 |
| Compound 2 | 6.1 ± 0.6 | 7.8 ± 0.4 | 24 ± 1 | 160 ± 20 | 588 | 70 |
| Compound (S)-3 | 23.5 ± 0.6 | 21.8 ± 0.5 | 16 ± 1.3 | 1960 ± 206 | 2659 | 83 |
| Compound (R)-4 | 19.8 ± 0.4 | 20.1 ± 0.6 | 28 ± 1.1 | 2132 ± 478 | 4989 | 78 |
| Compound (S)-5 | 10.1 ± 0.8 | 8.1 ± 1.4 | 5.2 ± 0.7 | 2266 ± 196 | 1067 | 33 |
| Compound (R)-6 | 13.9 ± 1.3 | 19.3 ± 1.5 | 4.6 ± 0.6 | 811 ± 59 | 1057 | 33 |
In all the mutations tested, the observed efficacy of aminoglycoside-induced readthrough was in the order (S)-5 ≥ (R)-6 > 2 > gentamicin. This trend for (S)-5 and (R)-6 (Fig. 4) was similar to that observed for the suppression of the same stop mutations in vitro (Fig. 3), even though the gap of potency difference between (S)-5 and (R)-6 was smaller than that observed for the suppression of the same mutations in cell-free extracts. Interestingly, however, the observed significantly higher readthrough potencies of both (S)-5 and (R)-6, over that of 2 in R3X and Q70X (Fig. 4A and 4C), was considerably different from those of the same mutations in vitro (Fig. 3A and 3E). While these data may point to a better cell permeability of both (S)-5 and (R)-6 over that of 2, due to the presence of the 5′′-methyl group, more experiments are needed to understand this issue satisfactorily. Nevertheless, comparison of the observed cell toxicity data in Table 1 with the readthrough activity data in Fig. 2–4 demonstrates that the installation of an (S)-5′′-methyl group either on compound 1 to give compound (S)-3, or on compound 2 to give compound (S)-5, does not significantly affect the cytotoxicity (LC50 values of 21.4 and 23.5 mM for 1 and (S)-3, and 6.1 and 10.1 mM for 2 and (S)-5, respectively in HEK-293), while it greatly increases the observed stop codon suppression activity (1 < (S)-3 and 2 < (S)-5). This observation is one of the key outcomes of the present work. Thus, the observed similar cell toxicity of the compounds (S)-5 and 2 in HEK-293 and HFF cells (Table 1), together with substantially elevated suppression activity of (S)-5 over that of 2 both in vitro and ex vivo in cultured cells, indicate that the compound (S)-5 may represent a more superior choice than compound 2 in suppression therapy.
It is noteworthy, however, that the comparative exvivo suppression data in Fig. 4 show only a small preference of (S)-5 over that of (R)-6, while the cell toxicity data in Table 1 indicate a small (HEK-293 cells) to significantly (HFF cells) better cell toxicity profile of (R)-6 over that of (S)-5. Therefore, one may argue that the in vivo performance of the (R)-6 diastereomer might be even better than that of (S)-5. In addition, a very recent study on gentamicin demonstrated that the inversion of an absolute configuration at a single carbon atom, from (S)-6′-gentamicin C2 to (R)-6′-gentamicin C2, significantly reduces cell toxicity and apparent nephrotoxicity of the (R)-diastereomer in comparison to that of the (S)-diastereomer, as determined in cell culture and animal studies, while the bactericidal efficacy is not affected.22 Based on these observations it is clear that additional toxicity tests, including nephrotoxicity and ototoxicity, the major drawbacks of aminoglycosides, can resolve this issue satisfactorily and validate the observed benefit of either (S)-5 or (R)-6 over that of 2 and gentamicin. These experiments are currently under way and will be reported in due course.
The impact of the (S)-5′′-methyl group on the elevated readthrough activities of (S)-3 and (S)-5 is further supported by the observed eukaryotic translation inhibition data (Table 1). The efficacy with which (S)-5 (half-maximal inhibitory concentration value IC50 = 5.2 μM) inhibits eukaryotic translation is greater than that of (S)-3 (IC50 = 16.0 μM) and 2 (IC50 = 24.0 μM), a similar trend to that observed for readthrough activity: (S)-5 > (S)-3 > 2 (Fig. 2–4). In addition, the comparison of IC50 values of (S)-3 and (S)-5 to those of their parent structures 1 and 2 (IC50 values of 31 and 24 μM, respectively) reveals that (S)-3 and (S)-5 are 1.9-fold and 4.6-fold more specific to the eukaryotic ribosome than their parents 1 and 2, indicating that the observed impact of the (S)-5′′-methyl group on the elevated readthrough activities of (S)-3 and (S)-5 is associated with their increased specificity to the eukaryotic ribosome.
Finally, the designed structures 3–6 were investigated as antibacterial agents against both Gram-negative (Escherichia coli) and Gram-positive (Bacillus subtilis) bacteria, together with their prokaryotic translation inhibition activities (Table 1). In our previous studies,11–13 we have shown that compounds 1 and 2 are about 10-fold weaker inhibitors of prokaryotic translation than gentamicin and paromomycin and exhibit almost no bactericidal activity against both Gram-negative and Gram-positive bacteria. Therefore, we were interested whether the new set of compounds 3–6 retain similar properties or not. The measured IC50 values in Table 1 show that the efficacy with which 3–6 inhibit the prokaryotic ribosome is significantly lower than that of paromomycin and gentamicin, in accordance with the observed antibacterial data of this set of compounds; while gentamicin and paromomycin exhibit significant antibacterial activities against both E. coli and B. subtilis, compounds 3–6 lack considerable antibacterial activity. The observed data with 3–6 are similar to those observed for 1 and 2 and further support the previously reported correlation in aminoglycosides between prokaryotic translation inhibition activity and MIC values: decreased inhibition of prokaryotic translation is associated with the decrease in antibacterial activity.23
Furthermore, the observed continued inability of our previous leads, 112 and 2,13 along with current compounds 3–6, to show significant antibacterial activity in conjunction with their decreased prokaryotic ribosome specificity, suggest that by reducing the specificity to prokaryotic ribosome (and as such wiping away from aminoglycosides their ‘natural’ antibacterial activity) we may reduce their action on eukaryotic mitochondrial protein synthesis machinery and as such significantly reduce their toxic effects on humans. This view is supported by the fact that the mammalian mitochondrial protein synthesis machinery is very similar to the prokaryotic machinery24 and that the aminoglycoside-induced toxicity may, at least in part, be connected to drug-mediated dysfunction of the mitochondrial ribosome.25,26 The observed significantly increased eukaryotic translation inhibition activity (which actually measures only the inhibition of cytoplasmic protein synthesis and not that of mitochondrial protein synthesis) together with the significantly reduced cytotoxicity (Table 1) of the developed compounds 1–6 in comparison to those of gentamicin and paromomycin, further support this opinion.
In summary, here we discovered a new pharmacophore, the (S)-5′′-methyl group, as a valuable structural element of the ribosamine ring (ring III) that significantly affects suppression activity and has no significant influence on cell toxicity. The observed high impact of the (S)-5′′-methyl group on the elevated readthrough activity and specificity to the eukaryotic ribosome, along with no significant influence on the cell toxicity, support the feasibility of testing this novel pharmacophore for the generation of novel structures that may act as drugs for the treatment of genetic diseases. Finally, the observed low toxicity and high readthrough activity of (S)-3 and (S)-5 relative to the clinical drug gentamicin, in targeting all six different nonsense constructs underlying USH1, CF, DMD and HS, support the feasibility of testing these novel aminoglycosides in treating these diseases in animal and human subjects.
Acknowledgements
We thank John F. Atkins (University of Utah) for providing the p2luc plasmid, and Varvara Pokrovskaya of our group for antibacterial tests. This work was supported by research grants from the US-Israel Binational Science Foundation (grant no. 2006/301) and by the Mitchel Fund (grant no. 2012386). J.K. acknowledges the Schulich Postdoctoral Fellowship; D.A.-G. acknowledges the Miriam and Aaron Gutwirth Memorial Fellowship; V.B. acknowledges the financial support by the Center of Absorption in Science, the Ministry of Immigration Absorption and the Ministry of Science and Technology, Israel (Kamea Program).References
- K. M. Keeling and D. M. Bedwell, Curr. Pharmacogenomics, 2005, 3, 259 Search PubMed.
- L. V. Zingman, S. Park, T. M. Olson, A. E. Alekseev and A. Terzic, Clin. Pharmacol. Ther., 2007, 81, 99 CrossRef CAS.
- M. Mort, D. Ivanov, D. N. Cooper and N. A. Chuzhanova, Hum. Mutat., 2008, 29, 1037 CrossRef CAS.
- R. Kellermayer, Eur. J. Med. Genet., 2006, 49, 445 CrossRef.
- J. F. Burke and A. E. Mogg, Nucleic Acids Res., 1985, 13, 6265 CrossRef CAS.
- M. Hainrichson, I. Nudelman and T. Baasov, Org. Biomol. Chem., 2008, 6, 227 RSC.
- J. A. Holbrook, G. Neu-Yilik, M. W. Hentze and A. E. Kulozik, Nat. Genet., 2004, 36, 801 CrossRef CAS.
- G. Karpati and H. Lochmuller, Ann. Neurol., 2001, 49, 693 CrossRef CAS.
- A. P. Carter, W. M. Clemons, D. E. Brodersen, R. J. Morgan-Warren, B. T. Wimberly and V. Ramakrishnan, Nature, 2000, 407, 340 CrossRef CAS.
- M. A. Borovinskaya, R. D. Pai, W. Zhang, B. S. Schuwirth, J. M. Holton, G. Hirokawa, H. Kaji, A. Kaji and J. H. Cate, Nat. Struct. Mol. Biol., 2007, 14, 727 CrossRef CAS.
- J. Kondo, M. Hainrichson, I. Nudelman, D. Shallom-Shezifi, C. M. Barbieri, D. S. Pilch, E. Westhof and T. Baasov, ChemBioChem, 2007, 8, 1700 CrossRef.
- I. Nudelman, A. Rebibo-Sabbah, D. Shallom-Shezifi, M. Hainrichson, I. Stahl, T. Ben-Yosef and T. Baasov, Bioorg. Med. Chem. Lett., 2006, 16, 6310 CrossRef CAS.
- I. Nudelman, A. Rebibo-Sabbah, M. Cherniavsky, V. Belakhov, M. Hainrichson, F. Chen, J. Schacht, D. S. Pilch, T. Ben-Yosef and T. Baasov, J. Med. Chem., 2009, 52, 2836 CrossRef CAS.
- A. Rebibo-Sabbah, I. Nudelman, Z. M. Ahmed, T. Baasov and T. Ben-Yosef, Hum. Genet., 2007, 122, 373 CrossRef CAS.
- I. Nudelman, D. Glikin, B. Smolkin, M. Hainrichson, V. Belakhov and T. Baasov, Bioorg. Med. Chem., 2010, 18, 3735 CrossRef CAS.
- Y. Kasai, H. Taji, T. Fujita, Y. Yamamoto, M. Akagi, A. Sugio, S. Kuwahara, M. Watanabe, N. Harada, A. Ichikawa and V. Schurig, Chirality, 2004, 16, 569 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512 CrossRef CAS.
- B. M. Trost, J. L. Belletire, S. Godleski, P. G. Mcdougal, J. M. Balkovec, J. J. Baldwin, M. E. Christy, G. S. Ponticello, S. L. Varga and J. P. Springer, J. Org. Chem., 1986, 51, 2370 CrossRef CAS.
- M. Manuvakhova, K. Keeling and D. M. Bedwell, RNA, 2000, 6, 1044 CrossRef CAS.
- M. T. Howard, B. H. Shirts, L. M. Petros, K. M. Flanigan, R. F. Gesteland and J. F. Atkins, Ann. Neurol., 2000, 48, 164 CrossRef CAS.
- G. Grentzmann, J. A. Ingram, P. J. Kelly, R. F. Gesteland and J. F. Atkins, RNA, 1998, 4, 479 CAS.
- R. M. Sandoval, J. P. Reilly, W. Running, S. B. Campos, J. R. Santos, C. L. Phillips and B. A. Molitoris, J. Am. Soc. Nephrol., 2006, 17, 2697 CrossRef CAS.
- W. A. Greenberg, E. S. Priestley, P. S. Sears, P. B. Alper, C. Rosenbohm, M. Hendrix, S. C. Hung and C. H. Wong, J. Am. Chem. Soc., 1999, 121, 6527 CrossRef CAS.
- J. Kondo and E. Westhof, Nucleic Acids Res., 2008, 36, 2654 CrossRef CAS.
- E. C. Bottger, B. Springer, T. Prammananan, Y. Kidan and P. Sander, EMBO Rep., 2001, 2, 318 CrossRef CAS.
- S. N. Hobbie, C. M. Bruell, S. Akshay, S. K. Kalapala, D. Shcherbakov and E. C. Bottger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3244 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of 3–6, 27 and 28 with full spectral data including 1H and 13C NMR spectra, along with the in vitrotranslation inhibition and cytotoxicity data. See DOI: 10.1039/c0md00195c |
| ‡ Equal contribution. |
| This journal is © The Royal Society of Chemistry 2011 |