The importance of solvation in the design of ligands targeting membrane proteins†
Angel
González
ab,
Marta
Murcia
c,
Bellinda
Benhamú
c,
Mercedes
Campillo
a,
María L.
López-Rodríguez
*c and
Leonardo
Pardo
*a
aLaboratori de Medicina Computacional, Unitat de Bioestadística, Facultat de Medicina, Universitat Autònoma de Barcelona, E-08193, Bellaterra, Barcelona, Spain
bUniversidad Andrés Bello, Facultad de Ciencias Biológicas, República 252, Santiago, Chile
cDepartamento de Química Orgánica I, Facultad de Ciencias Químicas, Universidad Complutense, E-28040, Madrid, Spain
First published on 7th February 2011
Abstract
A crucial contribution to the ligand-receptor binding affinity is, in addition to their electrostatic and van der Waals interactions, the desolvation of the ligand. This is of special relevance in membrane proteins because the ligand has to be transferred from the aqueous environment to the transmembrane binding site crevice. Herein we report the synthesis of new serotonin 5-HT4receptor antagonists that replace a key carbonyl group by the thiocarbonyl bioisoster. This modification enhances experimental 5-HT4 receptor binding affinities by as much as 91 times. Free energy perturbation calculations have shown that the significant decrease of the penalty of desolvation, facilitating the entrance of the ligands into the binding site crevice, compensates for the weaker ligand-receptor interaction.
Introduction
Ligand-receptor binding affinities are usually judged as a function of the chemical and geometrical complementarity between the small ligand and the macromolecular receptor.1,2 These comprise electrostatic interactions like salt bridges, hydrogen bonds, and aromatic-aromatic interactions, among others, and mutual spatial complementarity in van der Waals interactions. A crucial contribution to the binding affinity, frequently under-appreciated, comes from desolvation of the ligand.1,2 This is of special relevance in membrane proteins because, in contrast to water-soluble proteins, the ligand has to be transferred from the extracellular aqueous environment to the binding site crevice in the transmembrane domain, frequently apart from bulk water. Thus, ligands targeting membrane proteins must be, in most of the cases, entirely desolvated to bind the receptor. This is significant because membrane proteins like G protein-coupled receptors (GPCRs), transport proteins, or ion channels are among the most prominent target families for drug design.3 For instance, GPCRs are only a small subset of the human genome (2%–3%) but it is estimated that around 40% of prescribed drugs act through these receptors.4Herein, we have synthesized new serotonin 5-HT4receptor (5-HT4R) antagonists that replace a key carbonyl group by the thiocarbonyl bioisoster. We have previously shown that this carbonyl group together with a protonated nitrogen atom, an aromatic moiety, and a voluminous substituent are the essential determinants for the recognition of 5-HT4R antagonists by the receptor.5 Thus, the compound with the thiocarbonyl group interacts more weakly with the receptor than the compound with the carbonyl group. However, we have found that the carbonyl to thiocarbonyl modification enhances the ligand-receptor binding affinity. Free energy perturbation (FEP) calculations suggest that this effect is due to the significant decrease of the penalty of ligand desolvation.
Results and discussion
We have synthesized benzimidazole-4-carbothioamides 2 and 4 from directly related carboxamides 1 and 3 by treatment with Lawesson's reagent in refluxing toluene (see Experimental section). This modification enhances significantly experimental 5-HT4R binding affinities (see Experimental section) for both unsubstituted [Ki(1) = 13.7 nM vs.Ki(2) = 0.15 nM] and 6-chloro derivatives [Ki(3) = 0.32 nM vs.Ki(4) = 0.081 nM] (Table 1). Importantly, the effect of this modification in the binding affinity is much larger for unsubstituted (91 times, as monitored by the ratio of Ki values; ΔΔGexp = −2.8 kcal mol−1) than for 6-chloro derivatives (4 times; ΔΔGexp = −0.8 kcal mol−1).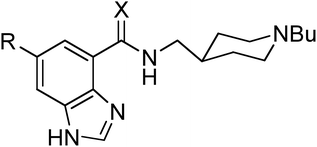
| Compd | X | R | K i a | ΔΔGexpb | ΔΔGFEPc | ΔḠFEP,recd | ΔḠFEP,watd |
|---|---|---|---|---|---|---|---|
| a K i values (nM) are mean values of two to four assays performed in triplicate. b Experimental binding free energy differences (kcal mol−1) between compounds 1 and 2, and 3 and 4, calculated as ΔΔGexp = −0.616 ln(Ki1or3/Ki2or4). c Free energy perturbation (FEP) binding free energy differences (kcal mol−1) between compounds 1 and 2, and 3 and 4, calculated as ΔΔGFEP = ΔḠFEP,rec − ΔḠFEP, wat. d Free energy cost (kcal mol−1) of transforming compound 1 into 2 or 3 into 4 in bulk water (ΔḠFEP,wat) and in complex with the receptor-lipid bilayer system (ΔḠFEP,rec). e Data from ref. 5 All values are shown as mean values ± SEM. | |||||||
| 1 | O | H | 13.7 ± 0.9e | ||||
| 2 | S | H | 0.15 ± 0.04 | −2.8 | −2.7 ± 0.5 | 9.5 ± 0.5 | 12.2 ± 0.3 |
| 3 | O | Cl | 0.32 ± 0.07e | ||||
| 4 | S | Cl | 0.081 ± 0.013 | −0.8 | −1.2 ± 0.3 | 10.5 ± 0.1 | 11.7 ± 0.3 |
In order to understand these effects we constructed three-dimensional models of the complexes between the ligands and a β2-based model of the 5-HT4R (see Experimental section). Site-directed mutagenesis6–8 has shown that 5-HT4R ligands bind side chains of amino acids located within transmembrane helices (TMs) 3, 5, 6 and 7; and in particular 5-HT4R antagonists bind D3.32 and S5.436 (the numbering of the residues corresponds to the generic numbering scheme of Ballesteros & Weinstein that allows easy comparison among residues in the 7TM segments of different receptors9). Thus, the previously reported 5-HT4R ligand 1 (UCM-21195, X = O, R = H) (Table 1) was docked into the receptor model with its protonated piperidine interacting with D3.32, and its carbonyl oxygen hydrogen-bonding the hydroxyl group of S5.43 (Fig. 1D).5 Clearly, binding to S5.43 would be weaker for compound 2 than for compound 1 because the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S⋯H–O hydrogen bond is weaker than the CO⋯H–O hydrogen bond. Similarly, the CS group would also interact more weakly with the extracellular water environment than the CO group. Because binding affinity is a balance of both the stabilization of the ligand-receptor complex and the solvation energy of the ligand we need to evaluate both effects. The free energy cost of transforming compound 1 into 2 in bulk water (ΔḠFEP,wat) and in complex with the receptor-lipid bilayer system (ΔḠFEP,rec) was calculated employing the FEP methodology (see Experimental section). The theoretical simulations show, as expected, that the CS group interacts more weakly with its environment than the CO group in both bulk water and the receptor-lipid system. However, the destabilization of the CS group in water (12.2 kcal mol−1) is larger than in the receptor-lipid bilayer (9.5 kcal mol−1), which results in a theoretical binding free energy difference (ΔΔGFEP) of −2.7 kcal mol−1, which is in very good agreement with the experimental (ΔΔGexp) value of −2.8 kcal mol−1 (Table 1). Thus, the significant decrease of the penalty of desolvation, which facilitates the entrance of the ligands containing CS group into the binding site crevice, compensates for the weaker ligand-receptor interaction.
S⋯H–O hydrogen bond is weaker than the CO⋯H–O hydrogen bond. Similarly, the CS group would also interact more weakly with the extracellular water environment than the CO group. Because binding affinity is a balance of both the stabilization of the ligand-receptor complex and the solvation energy of the ligand we need to evaluate both effects. The free energy cost of transforming compound 1 into 2 in bulk water (ΔḠFEP,wat) and in complex with the receptor-lipid bilayer system (ΔḠFEP,rec) was calculated employing the FEP methodology (see Experimental section). The theoretical simulations show, as expected, that the CS group interacts more weakly with its environment than the CO group in both bulk water and the receptor-lipid system. However, the destabilization of the CS group in water (12.2 kcal mol−1) is larger than in the receptor-lipid bilayer (9.5 kcal mol−1), which results in a theoretical binding free energy difference (ΔΔGFEP) of −2.7 kcal mol−1, which is in very good agreement with the experimental (ΔΔGexp) value of −2.8 kcal mol−1 (Table 1). Thus, the significant decrease of the penalty of desolvation, which facilitates the entrance of the ligands containing CS group into the binding site crevice, compensates for the weaker ligand-receptor interaction.
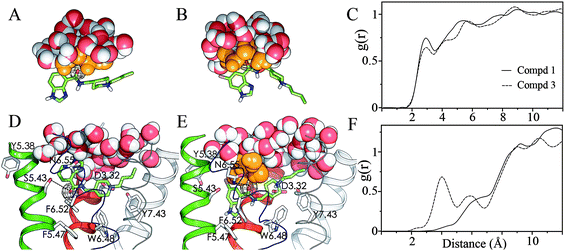 | ||
| Fig. 1 Molecular dynamics snapshots of water distribution around the carbonyl oxygen (black mesh) in bulk water (A, B half sphere) and in the orthosteric binding site of the 5-HT4R (D, E) for compounds 1 (A, D) and 3 (B, E). Ligands 1 and 3 bind the receptor model with their protonated piperidine ring interacting with D3.32 and their carbonyl group interacting with S5.43 and N6.55, respectively (dashed lines in D, E). Waters are drawn in VDW spheres with orange color for the inner solvation shell of the CO group. Compounds 1 and 3 (in sticks) bind within TMs 3, 5 (red and green cartoons) and TMs 6, 7 (blue and cyan thin ribbons), TM 2 is displayed in white cartoon. Only polar hydrogen of the ligands are displayed, the other helices and loops are omitted for clarity. Carbonyl(O)–Water(O) radial distribution functions for compounds 1 (solid line) and 3 (dashed line) in water (C) and the receptor-lipid system (F). | ||
The 6-chloro derivative 3 was also docked into the receptor model similarly to compound 1 (viaS5.43). FEP was also used to estimate the theoretical free energy cost of transforming compound 3 (containing CO) into 4 (containing CS). However, the theoretical ΔΔGFEP value of −3.3 kcal mol−1 obtained for this binding mode (viaS5.43) was in clear disagreement with the experimental ΔΔGexp value of −0.8 kcal mol−1. Because the effect of the CO to CS modification in the binding affinity is much larger for unsubstituted (91 times) than for 6-chloro (4 times) derivatives, we suggest different binding modes to 5-HT4R. Compound 3 was, thus, docked with its protonated piperidine interacting with D3.32 and its carbonyl oxygen hydrogen-bonding N6.55 (Fig. 1E, dashed line). In this model the chlorine atom is located in a small cavity between TMs 3 and 5, in which the halogen atom can interact with Y5.38; and the benzimidazole ring extends between TMs 5 and 6, interacting with the aromatic side chains of F5.47 and F6.52. This mode of binding has been reported for serotonin antagonists.10,11 The theoretical ΔΔGFEP value for transforming compound 3 into 4 calculated for this binding mode (via N6.55) is −1.2 kcal mol−1, which is in very good agreement with the experimental ΔΔGexp value of −0.8 kcal mol−1 (Table 1). As in the case of unsubstituted compounds, the destabilization of the CS group in water (11.7 kcal mol−1) is larger than in the receptor (10.5 kcal mol−1).
Importantly, the enhancement of binding affinity observed upon replacement of the carbonyl with thiocarbonyl is much larger for unsubstituted (1 and 2) than for 6-chloro (3 and 4) derivatives (Table 1). Unsubstituted derivatives 1 and 2 bind 5-HT4R through D3.32 and S5.43 (Fig. 1D). This mode of binding positions these ligands with the carbonyl (Fig. 1D, black mesh) or thiocarbonyl group pointing toward the protein core and the benzimidazole ring toward extracellular loop 2 (ecl2), which fully buries the carbonyl/thiocarbonyl group from the extracellular bulk water. Thus, the destabilization of the CS group, relative to CO, in the binding pocket, is mostly caused by the destabilization of the hydrogen bond interaction with the side chain of S5.43. Clearly, this destabilization in the binding site crevice (9.5 kcal mol−1) is smaller than in bulk water (12.2 kcal mol−1) because in water the CO/CS group can be engaged in multiple hydrogen bond interactions with the water molecules forming the inner solvation shell (orange ball-and-sticks, Fig. 1A).
On the contrary, 6-chloro derivatives 3 and 4 bind 5-HT4R through D3.32 and N6.55, which positions the carbonyl (black mesh) or thiocarbonyl group toward ecl2 and the benzimidazole ring toward the protein core (Fig. 1E). Importantly, ecl2 of rhodopsin (Fig. 2A),12 formed by two β-strands, buries the binding site from the extracellular environment, while ecl2 of the β1- and β2- adrenergic receptors,13,14 formed by a helical segment, partially exposes the binding site to the extracellular environment. Thus, the exposition of the binding site crevice to the extracellular environment will be different (depending on the ecl2 conformation) in the GPCR families. Nevertheless, the 5-HT4R binding site most probably resembles that of the β1- and β2-adrenergic receptors (Fig. 2B). Consequently, the carbonyl/thiocarbonyl groups of the 6-chloro derivatives 3 and 4 are accessible to the extracellular bulk water. Thus, the larger destabilization of the CS group in the receptor-lipid bilayer for 6-chloro (10.5 kcal mol−1) than for unsubstituted (9.5 kcal mol−1) derivatives (Table 1) is attributed i) to the different type of hydrogen bond interaction between the ligand and the receptor (S5.43 or N6.55) and ii) to the larger accessibility of the key carbonyl group by the extracellular water molecules through the channel located between TMs 3, 5–6 (Fig. 2B). The latter point is illustrated by the first peak of the radial distribution function for the distance between the carbonyl group and the water molecules of the receptor-lipid system (Fig. 1F), which is located at ∼6 Å in unsubstituted (solid line) and at ∼4 Å in 6-chloro derivatives (dashed line). This analysis shows that the carbonyl group in 6-chloro derivatives is, in contrast to unsubstituted derivatives, partially solvated. Nevertheless, the destabilization of the CS group in water (11.7 kcal mol−1) remains larger than in the receptor (10.5 kcal mol−1) because the water molecules forming the inner solvation shell (orange ball-and-sticks, Fig. 1B and 1E) can form more optimal hydrogen bond interactions with the carbonyl group in water (the first peak is located at ∼3 Å, dashed line in Fig. 1C) than in the receptor (∼4 Å, dashed line in Fig. 1F). Supplementary Figure 1 shows the radial distribution functions for the distances between S5.43(Oγ) and N6.55(Nδ) and the water molecules of the receptor-lipid system.
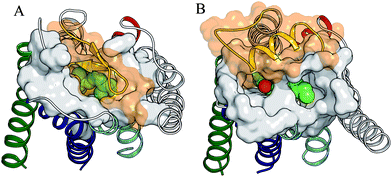 | ||
| Fig. 2 Comparison of solvent accessibilities to the binding site crevice in rhodopsin (A) and in the β2-based homology model of the 5-HT4R (B). 11-cis-retinal (A) and compound 3 (B) are shown in VDW spheres within TMs 3 (red), 5 (green), 6 (blue), and 7 (cyan). TMs 1, 2, 4 and solvent exclusion surface of the residues located at the extracellular region of the helical bundle are drawn in grey. Ecl2 is displayed in yellow with the transparent solvent exclusion surface in orange. The key carbonyl group (red VDW sphere) of compound 3 is accessible from the extracellular water environment through the channel located between TMs 3, 5–6. | ||
Another important difference between unsubstituted and 6-chloro derivatives that favors the former for thiocarbonyl substitution is the penalty of desolvation. The inner solvation shell around the carbonyl group is more closely packed for unsubstituted than for 6-chloro derivatives as shown by the larger area (higher maximum) at ∼3 Å in the radial distribution function (Fig. 1C) for compound 1 (solid line) than 3 (dashed line). The presence of the chlorine atom weakens the interaction between CO and the water environment. Thus, the destabilization of the CS group in water is larger for unsubstituted (12.2 kcal mol−1) than for 6-chloro (11.7 kcal mol−1) derivatives, facilitating the entrance of compound 2, more than 4, into the binding site crevice, with the corresponding improvement of binding affinity.
Experimental section
Alchemical transformations
The free energy cost of transforming ligand 1 into 2 and 3 into 4 in bulk water (ΔḠFEP,wat) and in complex with the receptor-lipid bilayer system (ΔḠFEP,rec) was calculated employing the FEP method15 as depicted in the thermodynamic cycle of Scheme 1. The dual-topology paradigm was implemented for the alchemical transformation of the carbonyl CO group to a thiocarbonyl CS in both, the benzimidazole-4-carboxamides and the 6-chloro derivatives, wherein the initial and final states were defined concomitantly, without interaction. Ligands 1 and 3 were placed in a rectangular box (55 Å × 50 Å × 45 Å in size) containing ∼3700 TIP3P water molecules, which were equilibrated during 1 ns. Modeller v9.516 was used to build a homology model of the 5-HT4R using the crystal structure of the β2-adrenergic receptor (PDB code 2RH1)14 as template. The internal water molecules in the P6.50/D2.50/N7.49/Y7.53 environment that mediate a number of key interhelical interactions were also included in the model.17 Ligands 1 and 3 were docked, by interactive computer graphics, into the receptor model with their protonated piperidine interacting with D3.32 and their carbonyl group interacting with S5.43 and N6.55, respectively. These structures were placed in a rectangular box (approx. 115 Å × 115 Å × 110 Å in size) containing a lipid bilayer (∼280 molecules of palmitoyl-oleoyl-phosphatidylcholine; ∼21500 water molecules and counterions). The ligand-receptor-lipid bilayer systems were equilibrated during 2.5 ns (a positional restraint of 2 kcal mol−1 Å−2 was applied to the Cα atoms of the receptor model, which was gradually turned off to a final value of 0). The reaction path for the FEP calculations was broken into 24 stages of uneven widths. Narrow intermediate states were defined toward the end points of the simulation to circumvent end-point singularities caused by appearing atoms that may clash with their surroundings. The simulations at each value of the coupling parameter λ consist of an equilibration period of 250 ps and a sampling period of 1 ns. Simulations were carried out using NAMD version 2.7,18 the CHARMM27 all-hydrogen force field for protein and lipids,19 and the general Amber force field (GAFF) and HF/6-31G*-derived RESP atomic charges were used for the ligands.20 The molecular dynamics simulations were performed using a 1 fs integration time step, constant pressure, constant temperature of 300 K, and using the particle mesh Ewald method to evaluate electrostatic interactions. The simulations were run forward (transforming the CO group into SO) and backward (SO into CO) to estimate the error in the procedure.
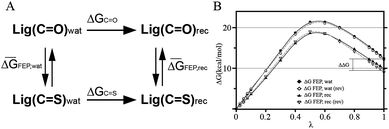 | ||
| Scheme 1 (A) Thermodynamic cycle to estimate the difference in binding free energy between compounds containing CO and CS groups. ΔGCO and ΔGCS are the binding free energies to the 5-HT4R of each ligand, whereas ΔḠFEP,wat and ΔḠFEP,rec are the free energy costs of transforming one ligand into another in the aqueous solution and the membrane-embedded 5-HT4R, respectively. ΔḠFEP,wat and ΔḠFEP,rec are average values calculated from forward and reverse simulations. (B) Free energy change as a function of λ in the forward (solid lines) and reverse (dashed lines) simulations for the transformation of compound 1 into 2, in water (circles) and in the membrane receptor (triangles). The curves in the reverse simulation (rev) were shifted to align the values at λ = 0. Notably, the forward and reverse simulations produce consistent results with relatively small hysteresis. A similar plot was created for the transformation of compound 3 into 4 (not shown) to estimate the standard errors presented in Table 1. | ||
Chemistry
Melting points (uncorrected) were determined on a Stuart Scientific electrothermal apparatus. Infrared (IR) spectra were measured on a Bruker Tensor 27 instrument equipped with a Specac ATR accessory of 5200–650 cm−1 transmission range; frequencies (ν) are expressed in cm−1. Nuclear Magnetic Resonance (NMR) spectra were recorded on a Bruker Avance 300-AM (1H, 300 MHz; 13C, 75 MHz) at the UCM's NMR facilities. Chemical shifts (δ) are expressed in parts per million relative to internal tetramethylsilane; coupling constants (J) are in hertz (Hz). The following abbreviations are used to describe peak patterns when appropriate: s (singlet), d (doublet), t (triplet), br (broad). Elemental analyses (C, H, N, S) were obtained on a LECO CHNS-932 apparatus at the UCM's analysis services and were within 0.5% of the theoretical values, confirming a purity of at least 95% for all tested compounds. Analytical thin-layer chromatography (TLC) was run on Merck silica gel plates (Kieselgel 60 F-254) with detection by UV light (254 nm), ninhydrin solution, or iodine. Flash chromatography was performed on glass column using silica gel type 60 (Merck, particle 230–400 mesh). Unless stated otherwise, starting materials, reagents and solvents were purchased as high-grade commercial products from Sigma-Aldrich, Scharlab or Panreac, and were used without further purification.The following compounds were synthesized according to described procedures: N-[(1-butylpiperidin-4-yl)methyl]-1H-benzimidazole-4-carboxamide (1) and N-[(1-butylpiperidin-4-yl)methyl]-6-chloro-1H-benzimidazole-4-carboxamide (3), and their spectroscopic data are in agreement with those previously reported.21 Spectroscopic data of described compounds were consistent with the proposed structures.
General procedure for the synthesis of benzimidazole-4-carbothioamides (2, 4)
To a solution of benzimidazole-4-carboxamides 1, 3 (1 mmol) in dry toluene (15 mL) under an argon atmosphere was added one portion of Lawesson's reagent (0.3 g, 0.7 mmol) at room temperature, and the reaction mixture was refluxed for 18 h. Then, the solvent was removed under reduced pressure and the crude was taken up in chloroform and washed with 20% aqueous K2CO3. The organic layer was dried (Na2SO4) and evaporated to afford a sticky solid, which was purified by flash chromatography on silica gel (eluent: dichloromethane–ethanol 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and subsequent recrystallization from toluene.
:1) and subsequent recrystallization from toluene.
N-[(1-Butylpiperidin-4-yl)methyl]-1H-benzimidazole-4-carbothioamide (22) was obtained as a yellowish solid in 50% yield (165 mg); mp 153–154 °C. IR (KBr) ν 3450, 3200, 1575, 1480. 1H-NMR (Me2SO-d6) δ 0.88 (t, J = 7.2 Hz, 3H), 1.23–1.41 (m, 6H), 1.80–1.84 (m, 3H), 1.93 (t, J = 10.5 Hz, 2H), 2.28 (t, J = 6.9 Hz, 2H), 2.90 (d, J = 10.5 Hz, 2H), 3.82 (m, 2H), 7.39 (t, J = 7.5 Hz, 1H), 7,80 (d, J = 7.5 Hz, 1H), 8.42 (d, J = 6.0 Hz, 1H), 8.56 (s, 1H), 12.30 (br s, 1H). 13C-NMR (Me2SO-d6) δ 13.9, 20.1, 28.4, 29.7, 34.7, 51.5, 52.9, 57.7, 115.9, 122.2, 125.4, 126.1, 133.9, 138.4, 143.1, 193.0. Anal. (C18H26N4S) C, H, N.
N-[(1-Butylpiperidin-4-yl)methyl]-6-chloro-1H-benzimidazole-4-carbothioamide (44) was obtained as a yellow solid in 47% yield (171 mg); mp 154–156 °C (d). IR (KBr) ν 3220, 1600, 1490, 1450. 1H-NMR (Me2SO-d6) δ 0.85 (t, J = 7.3 Hz, 3H), 1.21–1.39 (m, 6H), 1.75–1.78 (m, 3H), 1.90 (t, J = 10.6 Hz, 2H), 2.26 (t, J = 7.7 Hz, 2H), 2.88 (d, J = 10.3 Hz, 2H), 3.76 (m, 2H), 7.83 (s, 1H), 8.25 (s, 1H), 8.54 (s, 1H), 12.25 (br s, 1H). 13C-NMR (Me2SO-d6) δ 13.8, 19.9, 27.2, 28.5, 33.7, 51.1, 52.1, 56.8, 115.4, 125.3, 126.4, 126.6, 135.5, 137.0, 144.5, 191.6. Anal. (C18H25ClN4S) C, H, N.
Radioligand binding assays at 5-HT4R
Tissues were stored at −80 °C for subsequent use and homogenized on a Polytron PT-10 homogenizer. Membrane suspensions were centrifuged on a Beckman J2-HS instrument. Binding assays were performed according to the procedure previously described.5 The rat striatum was homogenized in 15 volumes of ice-cold 50 mM HEPES buffer (pH 7.4 at 4 °C) and centrifuged at 48000 g for 10 min. The pellet was re-suspended in 4.5 mL of assay buffer (50 mM HEPES, pH 7.4 at 4 °C). Fractions of 100 μL of the final membrane suspension were incubated at 37 °C for 30 min with 0.1 nM [3H]GR113808 (85 Ci/mmol), in the presence or absence of six concentrations of the competing drug, in a final volume of 1 mL of assay buffer. Nonspecific binding was determined with 30 μM 5-HT and represented less than 30% of the total binding. Competing drug, nonspecific, total, and radioligand bindings were defined in triplicate. Incubation was terminated by rapid vacuum filtration through Whatman GF/B filters presoaked in 0.05% poly(ethylenimine), using a Brandel cell harvester. The filters were then washed once with 4 mL of ice-cold 50 mM HEPES, pH 7.4, at 4 °C and dried. The filters were placed in poly(ethylene) vials to which were added 4 mL of a scintillation cocktail (Aquasol), and the radioactivity bound to the filters was measured by liquid scintillation spectrometry. The data were analyzed by an iterative curve-fitting procedure (program Prism, Graph Pad), which provided IC50, Ki, and r2 values for test compounds.Conclusions
Herein we report the synthesis of new serotonin 5-HT4R antagonists that replace a key carbonyl group of previously reported compounds 1 and 35 by the thiocarbonyl bioisoster to obtain compounds 2 and 4. Notably, this modification enhances experimental 5-HT4R binding affinities (Table 1). FEP calculations have shown that the significant decrease of the penalty of desolvation, facilitating the entrance of the ligands containing thiocarbonyl into the binding site crevice, compensates for the weaker ligand-receptor interaction.We want to emphasize that these effects cannot be generalized to all targets and circumstances of drug design. The penalty of ligand desolvation is a significant factor in the binding affinity only in the cases where the ligand must be entirely desolvated to elicit its function (ligands targeting proteins with the binding site crevice apart from bulk water or ligands that interact with the protein macromolecule through the membrane bilayer). As shown before, this effect of ligand desolvation can improve binding affinity by as much as two orders of magnitude (91 times). Thus, for these particular cases, we intend to design compounds that interact stronger with the receptor than with water.
Acknowledgements
This work was supported by grants from MICINN (SAF2007-67008-C02, SAF2010-22198-C02), CAM (S-SAL/0249/2006), and ISCIII (RD07/0067/0008).References
- H. Gohlke and G. Klebe, Angew. Chem., Int. Ed., 2002, 41, 2644–2676 CrossRef CAS.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS.
- P. Imming, C. Sinning and A. Meyer, Nat. Rev. Drug Discovery, 2006, 5, 821–834 CrossRef CAS.
- M. C. Lagerstrom and H. B. Schioth, Nat. Rev. Drug Discovery, 2008, 7, 339–357 CrossRef.
- M. L. Lopez-Rodriguez, M. Murcia, B. Benhamu, A. Viso, M. Campillo and L. Pardo, J. Med. Chem., 2002, 45, 4806–4815 CrossRef CAS.
- J. Mialet, Y. Dahmoune, F. Lezoualc'h, I. Berque-Bestel, P. Eftekhari, J. Hoebeke, S. Sicsic, M. Langlois and R. Fischmeister, Br. J. Pharmacol., 2000, 130, 527–538 CrossRef CAS.
- L. Rivail, M. Giner, M. Gastineau, M. Berthouze, J. L. Soulier, R. Fischmeister, F. Lezoualc'h, B. Maigret, S. Sicsic and I. Berque-Bestel, Br. J. Pharmacol., 2004, 143, 361–370 CrossRef CAS.
- L. Pellissier, J. Sallander, M. Campillo, F. Gaven, E. Queffeulou, M. Pillot, A. Dumuis, S. Claeysen, J. Bockaert and L. Pardo, Mol. Pharmacol., 2009, 75, 982–990 CrossRef CAS.
- J. A. Ballesteros and H. Weinstein, Methods in Neurosciences, 25, 366–428 Search PubMed.
- M. J. Smit, H. F. Vischer, R. A. Bakker, A. Jongejan, H. Timmerman, L. Pardo and R. Leurs, Annu. Rev. Pharmacol., 2007, 47, 53–87 Search PubMed.
- T. de la Fuente, M. Martin-Fontecha, J. Sallander, B. Benhamu, M. Campillo, R. A. Medina, L. P. Pellissier, S. Claeysen, A. Dumuis, L. Pardo and M. L. Lopez-Rodriguez, J. Med. Chem., 2010, 53, 1357–1369 CrossRef CAS.
- K. Palczewski, T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. L. Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto and M. Miyano, Science, 2000, 289, 739–745 CrossRef CAS.
- T. Warne, M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov, P. C. Edwards, R. Henderson, A. G. Leslie, C. G. Tate and G. F. Schertler, Nature, 2008, 454, 486–491 CrossRef CAS.
- D. M. Rosenbaum, V. Cherezov, M. A. Hanson, S. G. Rasmussen, F. S. Thian, T. S. Kobilka, H. J. Choi, X. J. Yao, W. I. Weis, R. C. Stevens and B. K. Kobilka, Science, 2007, 318, 1266–1273 CrossRef CAS.
- R. W. Zwanzig, J. Chem. Phys., 1954, 22, 783–1426 CrossRef CAS.
- M. A. Marti-Renom, A. C. Stuart, A. Fiser, R. Sanchez, F. Melo and A. Sali, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 291–325 CrossRef CAS.
- L. Pardo, X. Deupi, N. Dolker, M. L. Lopez-Rodriguez and M. Campillo, ChemBioChem, 2007, 8, 19–24 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS.
- A. D. MacKerell Jr., D. Bashford, M. Bellott, R. L. Dunbrack Jr., J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher III, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef.
- M. L. Lopez-Rodriguez, B. Benhamu, A. Viso, M. J. Morcillo, M. Murcia, L. Orensanz, M. J. Alfaro and M. I. Martin, Bioorg. Med. Chem., 1999, 7, 2271–2281 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c0md00258e |
| This journal is © The Royal Society of Chemistry 2011 |