Evaluation of FR901464 analogues in vitro and in vivo
Sami
Osman
a,
William R.
Waud
b,
Gregory S.
Gorman
c,
Billy W.
Day
ad and
Kazunori
Koide
*a
aDepartment of Chemistry, University of Pittsburgh, 219 Parkman Avenue, Pittsburgh, Pennsylvania 15260. E-mail: koide@pitt.edu; Fax: +1 412 624 8611; Tel: +1 412 624 8767
bDepartment of Cancer Therapeutics and Immunology, Southern Research Institute, Birmingham, Alabama
cDepartment of Toxicology and Bioanalytical Science, Southern Research Institute, Birmingham, Alabama
dDepartment of Pharmaceutical Sciences, University of Pittsburgh, 3501 Fifth Avenue, Pittsburgh, Pennsylvania 15213
First published on 2nd November 2010
Abstract
This study was designed to determine the in vitro and in vivo antitumor behaviour of three analogues of the natural product FR901464. All analogues demonstrated effective inhibition of cell proliferation. Minimal in vivo antitumor activity was observed, warranting further PK and antitumor efficacy studies.
Introduction
FR901464 (Fig. 1) is an antitumor natural product that was isolated from the Pseudomonas sp. No. 2663 by Nakajima et al.1–3 It activates a reporter gene driven by the SV40 promoter in MCF-7 cells.2 It suppresses the mRNA levels of p53, p21 Cip-1, E2F-1, and c-Myc while not influencing the expression of a housekeeping gene.1FR901464 inhibits the growth of human lung cancer A549 cells, human breast cancer MCF-7 cells, human colon cancer HCT-116 cells, and murine leukemia P-388 cells with GI50 values of 1–2 nM.1 Its in vivo toxicity was observed in mice at 0.32 mg kg−1 (i.p. Q1D × 4) and 1.0 mg kg−1 (i.p. Q4D × 3).1 The preclinical studies with mice showed that FR901464 was effective against murine leukemia P-388 (at 0.056–0.18 mg kg−1, i.p., Q1D × 4) and human lung cancer A549 (at 0.1–0.56 mg kg−1, i.p., Q4D × 3) xenografts.1 It should be noted that FR901464 was injected into the mice only one day after the introduction of the tumor fragments to the mice.1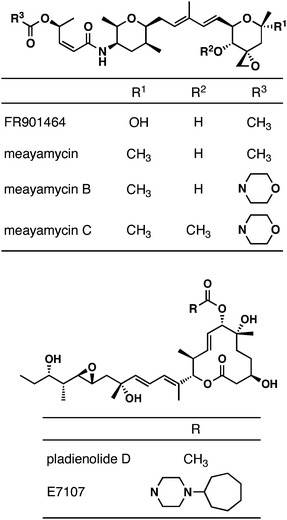 | ||
| Fig. 1 Structures of FR901464, meayamycins and pladienolides. | ||
The structural and biological activities of FR901464 inspired other groups to discover novel natural products with similar biological activity. One such notable class of natural products is the pladienolides.4–7 Recently, it was found that both FR9014648 and the pladienolides9 bind to a subunit of the human spliceosome, splicing factor 3b (SF3b). Structure–activity relationship (SAR) and pharmacokinetic studies of the pladienolides led to the development of E7107 for its anticancer activity.9 E7107 showed remarkable antitumor activity in mouse xenograft models.9 It was then examined in Phase I clinical trials for solid tumors.10 This was presumably the first pre-mRNA splicing inhibitor to be tested in humans. In that study, E7107 was shown to reversibly inhibit pre-mRNA processing in humans.10 This clinical study has now been suspended.11
Although FR901464 was discovered and tested in xenograft models prior to the pladienolides, its progress in terms of drug development lags behind. Studies of FR901464 and its analogues through total chemical syntheses and biological analyses have provided substantial insights into the SAR of FR901464. The structural similarities between FR901464 and the pladienolides enabled the design and synthesis of simplified analogues in large quantities for animal studies.12 A preliminary study shows somewhat promising antitumor activity with one of the simpler analogues.12
We too have been engaged in the chemical and biological studies of FR901464 and its analogues. After our total synthesis of FR901464,13,14 we reported the chemically more stable analogue, meayamycin,15 which exhibited GI50 values in the low to middle picomolar range.16 Meayamycin was also shown to inhibit pre-mRNA splicing in a cell-free system and in live cells.16 Meayamycin was more potent against human small lung cancer A549 cells than against nontumorigenic human lung fibroblasts IMR-90.16 Meayamycin was very effective against a multidrug-resistant cell line.16 More recently, meayamycin B was synthesized and found to be even more potent than meayamycin and more stable in serum.17 Here, we wish to report our preliminary studies of meayamycins in vivo.
Materials and methods
Cell culture
HCT-116 cells were obtained as a gift from Professor Andreas Vogt (University of Pittsburgh), the MCF-7 cells were from Dr Marc Lippman, and all other cell lines were purchased from the American Type Culture Collection. All cell culture media were supplemented with 10% fetal bovine serum, 1% L-glutamine and 1% penicillin/streptomycin. HCT-116 cells were grown in McCoy 5A media. MCF-7 and MDA-MB231 cells were grown in RPMI 1640 media. NHDF and NHLF cells were grown in Clonetics® media. HMEC cells were grown in HuMEC Basal medium and HUVEC cells were grown in endothelial cell growth medium. All cell lines were incubated at 37 °C in a humidified atmosphere at 95% air and 5% CO2.Growth inhibition assays
Cell proliferation was evaluated using the MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) dye reduction assay with PMS (phenazine methosulfate) as the electron acceptor or CellTiter-Glo assay performed in triplicate. Briefly, cells were seeded in 96 well plates at 2500 or 4000 cells per well and incubated for 24–72 h at 37 °C. Cells were then exposed to various concentrations (serial three-fold dilutions) of the indicated compounds for 72 h at 37 °C. An MTS or CellTiter-Glo solution was added, and cells were incubated at 37 °C. For an MTS assay, cells were incubated for 2 h, and the absorption signals were measured at 490 and 650 nm. For a CellTiter-Glo assay, cells were incubated at room temperature for 10 min, followed by recording the luminescent signal. Growth inhibition was calculated as defined by the National Cancer Institute [GI50 = 100 × (T − T0)/(C − T0); T0 = cell density at time zero; T = cell density of the test well after period of exposure to test compound; C = cell density of the vehicle treated].18Preparation of dosing solutions
Meayamycin and meayamycin B were dissolved in ethanol and further diluted with sterile water for injection (WFI) to yield a final vehicle composition of 10% ethanol/90% WFI. Paclitaxel was formulated in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 cremophor EL:ethanol (Bristol-Myers Squibb Co., lot no. 6F18936) and further diluted with saline to yield a final vehicle composition of 12.5% cremophor/12.5% ethanol/75% saline. The dosing volume for all agents was 0.1 mL/10 g of body weight.
:1 cremophor EL:ethanol (Bristol-Myers Squibb Co., lot no. 6F18936) and further diluted with saline to yield a final vehicle composition of 12.5% cremophor/12.5% ethanol/75% saline. The dosing volume for all agents was 0.1 mL/10 g of body weight.
Animals and tumor models
Athymic NCr-nu/nu mice, purchased from various commercial suppliers, were housed in microisolator cages and were on a 12-hour light/dark cycle and received filtered Birmingham municipal water and sterilizable rodent diet (Harlan-Teklad TD8656) ad libitum. The two human tumors were obtained from DCTD Tumor Repository/NCI (Frederick, MD, USA) and were maintained in in vivo passages. For the in vivo evaluation of the sensitivity of human tumors to the compounds, male athymic NCr-nu/nu mice were implanted subcutaneously (sc) with 30–40 mg tumor fragments. The day of tumor implantation was designated as Day 0. Tumors were allowed to reach 100–245 mg in weight (100–245 mm3 in size) before the start of treatment. For the dose tolerance experiments, 3 mice were used per group. For the antitumor efficacy experiments, 10 mice were used per group. All procedures were approved by the Southern Research Institutional Animal Care and Use Committee. Animal laboratories of Southern Research are AAALAC accredited.Antitumor activity was assessed on the basis of delay in tumor growth (T-C). The delay in tumor growth is the difference in the median of times post-staging for tumors of the treated and control groups to double in mass two times. Animals whose tumor failed to attain the evaluation size were excluded. Tumors were measured in two dimensions (length and width) twice weekly, and the tumor weight was calculated using the formula (length × width2)/2 and assuming unit density. The mice were also weighed twice weekly. As a positive control, tumor-bearing mice treated with paclitaxel intravenously at 15 mg kg−1/inj on a Q1D × 5 schedule were also examined.
PK blood collection
PK studies were performed in 5-week-old female athymic NCr-nu/nu mice following a single intravenous dose of either 1 or 6 mg kg−1 of meayamycin B and 6 mg kg−1 of meayamycin C. PK blood samples were collected from 3 mice at each time point after compound dosing.Animals were anesthetized with 2:1 CO2/O2 and heparinized microhematocrit tubes were used to collect blood from the retro-orbital sinus. Samples were held on ice until processed. Plasma was separated by centrifugation at 2,400 rpm and stored at −84 °C until assayed.
Measurement of compound in plasma
The residual meayamycin B or C and metabolites were extracted from plasma using CH3CN containing HCO2H (0.5%), followed by protein precipitation and analyzed using broad gradient reverse phase HPLC tandem mass spectrometry. Initially, Q1 mass scans from m/z 300 to 900 were collected on each sample followed by targeted mixed reaction monitoring (MRM) transitions based on the initial Q1 data and predicted metabolites from LightSight (Applied Biosystems, Foster City, CA).Results
Activity in vitro
The antiproliferative activities of meayamycin, meayamycin B and meayamycin C were determined on various human tumor cell lines. Meayamycin was found to inhibit the growth of all of the tumor cell lines tested, with GI50 values ranging from 10 pM to 1230 pM (Table 1).16 The antiproliferative activity of meayamycin was also seen in the nontumorigenic cell lines NHDF, NHFL, HMEC and HUVEC (Table 1). On the other hand, the more biologically stable analogue, meayamycin B, was 3.1 ± 1.9 times more potent than meayamycin against MCF-7, MDA-MB231, A549, DU145, HCT-116, H1299, and PC-3 cell lines.17 Meayamycin C displayed sub- to single-digit nanomolar GI50 values against MCF-7, MDA-MB231 and HCT-116 cancer cell lines. Meayamycin C retained potency to a slightly lesser extent against the normal cell lines NHDF, NHLF, HMEC and HUVEC with GI50 values ranging from 4 to 39 nM. In contrast, doxorubicin inhibited HCT-116 cancer cell proliferation with a GI50 of 70 nM, while in the normal cell lines NHDF, NHLF, HMEC, and HUVEC, its GI50 ranged from 109 to 462 nM (Table 1). Thus, meayamycin C exhibited a two-fold difference between the normal and cancer cell lines. Velcade displayed single digit nanomolar GI50 against these cancer and normal cell lines, except towards the NHDF cell line, which was less sensitive (Table 1). Taken together, the data from these cell lines demonstrated that all of the meayamycin derivatives retained potent antiproliferative activity in tumor cells derived from a variety of organs and showed similar in vitro selectivity to the clinically used anticancer drugs doxorubicin and Velcade.| Cell Line | Tissue | Meayamycin GI50/nM | Meayamycin C GI50/nM | Doxorubicin GI50/nM | Velcade GI50/nM |
|---|---|---|---|---|---|
| HCT-116 | Colon | 0.07 | 1.4 | 70 | 5 |
| MCF-7 | Breast | 0.02 ± 0.0116 | 0.4 | N/A | N/A |
| MDA-MB231 | Breast | 0.07 ± 0.0616 | 1.1 | N/A | N/A |
| NHDF | Skin | 0.44 | 39 | 462 | 334 |
| NHLF | Lung | 0.91 | 12 | 223 | 1.7 |
| HMEC | Mammary | 0.11 | 9 | 276 | 3.5 |
| HUVEC | Vein | 0.06 | 4 | 109 | 1.1 |
Dose and schedule optimization
The tolerability and antitumor activity of various doses and schedules of meayamycin were evaluated in mice. Meayamycin was intravenously administered to non-tumor-bearing male athymic nude mice at dosages of 3.0, 1.0, 0.3, 0.1 and 0.03 mg kg−1/inj once every 4 days for total of three injections (Q4D × 3). Such administrations resulted in no deaths or body weight loss (Fig. 2A). Similarly, intravenous administration of meayamycin B at dosages of 3.0, 1.0, 0.3, 0.1 and 0.03 mg kg−1/inj on a Q4D × 3 schedule was tolerated without deaths. Body weight loss was observed only at a dosage of 3.0 mg kg−1/inj, with a maximum average body weight loss of 11% (3.0 g) (Fig. 2B). Vehicle administration was tolerated without deaths or body weight loss. Meayamycin B was also tolerated at dosages of 0.5 and 1.0 mg kg−1/inj intravenously administered once a day for 5 consecutive days (Q1D × 5) (Fig. 2C). Administration of a dosage of 1.0 mg kg−1/inj resulted in a maximum average weight loss of 10% (2.7 g). Administration of a lower dosage of 0.5 mg kg−1/inj led to the euthanization of one mouse on Day 9 because it was moribund. This treatment resulted in a maximum average weight loss of 6% (1.5 g). Two out of three mice in each group were noted to have swollen tails with skin sloughing off, which prevented us from giving additional injections. Doses of 1.5 mg kg−1/inj and higher administered Q1D × 5 were lethal to all of the treated animals.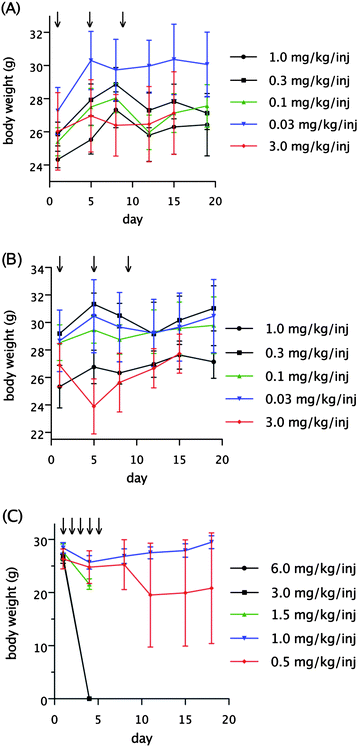 | ||
| Fig. 2 Dose tolerance of meayamycin and meayamycin B in mice (n = 3). (A) Tolerance of meayamycin in male athymic nude mice Q4D × 3. (B) Tolerance of meayamycin B in male athymic nude mice Q4D × 3. (C) Tolerance of meayamycin B in male athymic nude mice Q1D × 5. Arrows denote times of injections. | ||
Following the dose tolerance studies, we proceeded to evaluate the antitumor efficacy of meayamycin. With HCT-116 human colon tumor xenografts, tumor size was between 100 and 245 mm3 (100–245 mg) in size at the initiation of treatment (Day 16) (Fig. 3A). Intravenous administration of meayamycin at doses of 3.0 and 1.5 mg kg−1/inj on a Q4D × 3 treatment schedule lacked antitumor activity. Minimal antitumor activity was observed with a dose of 6 mg kg−1/inj administered Q4D × 3, resulting in a maximum inhibition rate (IR = [1-(median tumor weight treated/median tumor weight control)] × 100%) of 24% on day 41, although this may not be statistically significant (p > 0.05).
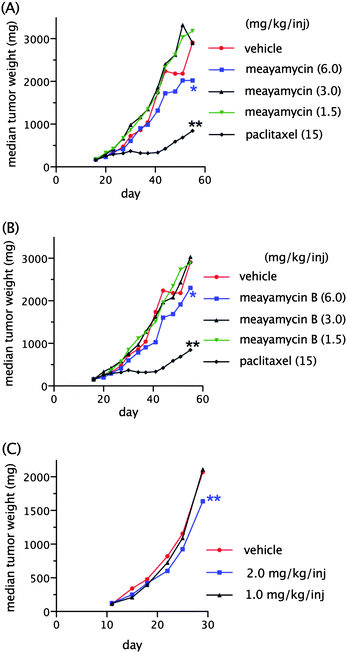 | ||
| Fig. 3 Activity of meayamycin, meayamycin B and paclitaxel in human tumor models (n = 10). (A) Response of HCT-116 tumor implanted sc to treatment with meayamycin (Day 16) and paclitaxel (Day 16). (B) Response of HCT-116 tumor implanted sc to treatment with meayamycin B (Day 16) and paclitaxel (Day 16). (C) Response of PC-3 tumor implanted sc to treatment with meayamycin B (Day 11). Data in (A) and (B) represent the same control group (paclitaxel-treated mice). Statistical significance was evaluated by comparing the time to 2 tumor mass doubling values of vehicle-treated groups to drug-treated groups. p values of less than 0.05 were considered statistically significant. The * signifies 0.05 < p < 0.80 (statistically not significant), and ** signifies p < 0.05. | ||
Next, meayamycin B was evaluated against the same xenograft model (Fig. 3B). Intravenous administration of meayamycin B at doses of 3.0 and 1.5 mg kg−1/inj on a Q4D × 3 schedule was tolerated without deaths and lacked antitumor activity. Administration of meayamycin B at a dose of 6 mg kg−1/inj on a Q4D × 3 schedule led to a maximum IR of 41% on Day 41. It should be noted that this result may be statistically insignificant (p > 0.05) and that one of the ten meayamycin B-treated mice died on Day 20. A vehicle given on the same schedule was tolerated without deaths. Intravenous administration of paclitaxel (positive control) at a dose of 15 mg kg−1/inj on a Q1D × 5 schedule was also tolerated without deaths and showed a maximum IR of 81% on Days 41 and 44.
Meayamycin B's antitumor efficacy was also evaluated in sc-implanted PC-3 human prostate tumor in male mice (Fig. 3C). These mice had tumor sizes of 100–234 mm3 (100–234 mg) at the initiation of treatment. A Q1D × 5 intravenous administration of meayamycin B at a dose of 1 mg kg−1/inj lacked antitumor activity while a dose of 2 mg kg−1/inj led to a maximum IR of 26% on Day 22 and a maximum average bodyweight loss of 15%. Treatment with saline, administered intravenously on a Q1D × 5 treatment schedule, was tolerated without deaths or appreciable bodyweight loss.
Pharmacokinetic studies in mice
Unlike E7107, meayamycin B did not show clear-cut antitumor activity in the xenograft models. We asked whether the compound achieved an adequate plasma concentration in mice. A single dose of meayamycin B (1 mg kg−1) was intravenously administered to female non-tumor-bearing athymic nude mice. A plot of meayamycin B plasma concentration versus time is provided in Fig. 4. If 100% of the injected meayamycin B is in the blood, its concentration should be ∼15 μg mL−1. Meayamycin B was rapidly eliminated from the plasma after the injection, as concentrations reached below 10 ng mL−1 over the first 30 min. Administration of meayamycin B at a higher single dose (6 mg kg−1) to female non-tumor-bearing athymic nude mice also resulted in a rapid plasma clearance (Fig. 4). Concentrations declined by ∼42% from 15 min to 4 h post injection and then were sustained at low percentages. The elimination of meayamycin B showed a prolonged terminal phase in which low plasma concentrations were sustained. This indicates that the elimination of meayamycin B from plasma was unaffected by dose levels.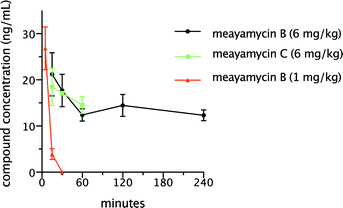 | ||
| Fig. 4 Plasma mean concentration time profiles of meayamycin B and meayamycin C in the plasma of female non-tumor bearing athymic nude mice. n = 3 for each time point. | ||
In order to gain insights into the pharmacokinetics of meayamycin B, we initially attempted to measure meayamycin B and its metabolites in the plasma viaLC-MS. Due to the complexity of the plasma matrix, initial Q1 data were not definitive. Next, targeted mixed reaction monitoring (MRM) transitions were designed based on the predicted metabolites from LightSight (Applied Biosystems, Foster City, CA). The targeted MRM transitions were designed on the basis of the mass shifts of the predicted metabolites. Mass shifts of the parent compound and the fragment ions were monitored. The observed peaks indicated dioxidation, demethylation, and glucuronidation. These metabolites were not quantified because authentic standards were not available. Nonetheless, multiple peaks were observed for the glucuronide mass transition, implying that the glucuronidation of meayamycin B might be a predominant metabolic pathway.
We hypothesized that meayamycin C, which has no hydroxy group, might be resistant to glucuronidation, thereby prolonging its half-life in plasma. To test this hypothesis, meayamycin C was synthesized17 and intravenously administered to female non-tumor-bearing athymic nude mice at a single dose of 6 mg kg−1. As shown in Fig. 4, meayamycin C plasma concentration fell quickly within 1 h after the injection and then remained below 10 ng ml−1 for the next 4 h, suggesting that the glucuronidation of meayamycin B was not the major reason for its poor pharmacokinetics.
To ensure that the measured levels in plasma were accurate and not affected by poor extraction of meayamycin B caused by covalent binding to proteins, we evaluated the extraction efficiency after incubation. Samples were prepared in triplicate by spiking mouse plasma at a concentration of 10 μM of meayamycin B and incubating them in a shaking water bath for 15 min at 37 °C. This is the same sample generation procedure that would be used to determine the plasma protein binding properties of drugs.19,20 After incubation, the samples were extracted using acetonitrile and analyzed using tandem mass spectrometry with the identical methodology as employed for the previous samples described above. The amount of meayamycin B extracted from the incubated plasma samples was compared to a 10 μM solvent standard prepared in a starting mobile phase. The analysis showed that ∼ 95% of meayamycin B spiked into the incubated plasma samples was recovered. This result suggested it was not likely that strong irreversible protein binding occurred.
Discussion
The spliceosome is an emerging molecular target for the treatment of cancer.21,22FR901464 and its various derivatives have been reported to have potent anticancer activity against several mammalian cancer cell lines, presumably through the inhibition of the spliceosome.8,16,23 Their in vivo applications are, however, at an earlier stage compared to E7107.This study shows an attempt to evaluate the anticancer activities of novel synthetic FR901464 analogues in in vitro and in vivo cancer models. The antiproliferative activity of meayamycin and derivatives was first determined by measuring their GI50 values with various cancer cell lines. Our results, together with previously reported data, showed that all of the FR901464 derivatives used in this study exhibited strong antiproliferative activity against these cell lines. The order of potency was meayamycin B > meayamycin > meayamycin C. Despite the potency of meayamycin and meayamycin C in cancer and non-cancerous cells, the therapeutic index increased from meayamycin to meayamycin C (if, e.g., we compare GI50 in the HCT-116 cell line and the non-cancerous cell lines in Table 1). The therapeutic index for meayamcyin is 6 (NHDF cell line), 2.7 (NHLF cell line), 1.6 (HMEC cell line), and 1 (HUVEC cell line). On the other hand, the therapeutic index for meayamycin C is 15 (NHDF cell line), 4.6 (NHLF cell line), 3.5 (HMEC cell line), and 1.5 (HUVEC cell line). This indicates that there is some degree of selectivity of meamycin C towards the HCT-116 cancer cell line. This quantitative analysis should be met with caution because the margin of error could be greater when the growth inhibition of nontumorigenic cells was measured.
Subsequently, we initiated an in vivo study to determine whether the compounds could inhibit tumor growth in HCT-116 and PC-3 xenograft models. First, we determined a maximum tolerated dose of meayamycin and meayamycin B. Doses for both compounds ranged from 0.03 to 3 mg kg−1/inj administered intravenously on a Q4D × 3 schedule. The treated animals were monitored for weight loss and mortality. No considerable weight loss or deaths were observed in any of the mice during or after treatment, except that a maximum average body weight loss of 11% was observed with meayamycin B at a dose of 3 mg kg−1/inj. Meayamycin B was also tolerated at a different dosing schedule, Q1D × 5, at doses of 0.5 and 1.0 mg kg−1/inj. A dose of 1.0 mg kg−1/inj led to only a maximum average body weight loss of 10%. Higher doses (1.5–6.0 mg kg−1/inj) were lethal for mice. Webb et al. designed structurally simpler analogues that were tolerated at much higher doses (≤50 mg kg−1 at a Q1D × 5 schedule), but that may be attributed to the lower antiproliferative activity (GI50 were at high nM to single-digit μM).23 These data show that doses of meayamcyin and meayamycin B can be administered safely by the intravenous route on Q4D × 3 and Q1D × 5 schedules.
The in vivo antitumor activity of meayamycin and meayamycin B was initially evaluated in the HCT-116 xenograft model in mice. Dosing schedule studies revealed that intravenously administered meayamycin and meayamycin B achieved IR values of 24% (Day 41) and 41% (Day 41), respectively, using a Q4D × 3 dosing schedule. With the low IR values of both meayamycin and meayamycin B, we decided to increase the frequency of injections. We chose meayamycin B for this experiment because meayamycin B was more stable than meayamycin in mouse serum (t1/2 = 13 h vs. 2 h).17 Due to the availability of xenograft models at the time, we switched from HCT-116 to PC-3 in the next study. Against the PC-3 tumor xenograft, meayamycin B elicited an IR of 26% on Day 22 using a Q1D × 5 dosing schedule. A higher dose was lethal to mice at this injection frequency, which limited our window of optimizations. Although a positive control was omitted in this experiment, the xenograft model might be considered valid because the tumor growth (200 mg to 400 mg) of the vehicle-treated group occurred over 3.1 days, a rate within the range we previously observed (1.5–3.9 days).24 In HCT-116 tumor xenografts, paclitaxel exhibited an IR of 81% on Days 41 and 44 using a Q1D × 5 dosing schedule. The interpretation of the available preclinical data is limited by the lack of information of plasma FR901464-like PK parameters that are associated with antitumor activity in animal models.
Our study illustrated that less than 1% of the injected meayamycin B was found in plasma within 30 min after administration. A glucuronidated meayamycin B metabolite exhibited the strongest signal in the mass spectrometric evaluations when compared to other detected metabolites. This result led us to hypothesize that meayamycin C might be resistant to glucuronidation. To test this hypothesis, meayamycin C was synthesized and its plasma concentrations were evaluated in female non-tumor-bearing mice after intravenous administration. The plasma concentrations of meayamycin C fell quickly within 1 h after the injection and remained below 10 ng ml−1 for the next 4 h, suggesting that the glucuronidation of meayamycin B was not the major reason for its poor pharmacokinetics. Although meayamycin B rapidly diminished in mouse plasma, the compound showed some antitumor activity, IR = 41%. It is possible that some of the metabolites of this compound, formed by the action of metabolizing enzymes, are the active form(s) of the agent.
Conclusions
In summary, the meayamycins displayed broad-spectrum antiproliferative activity, and meayamycin B exhibited minimal antitumor efficacy with Q4D × 3 and Q1D × 5 dosing schedules and no noticeable side effects in the HCT-116 and PC-3 xenograft models. The minimal antitumor efficacy of meayamycin B may be attributed to the poor PK profiles of this compound in mice. The mechanism underlying this observation is unclear. Further pharmacokinetics, antitumor efficacy, and toxicologic studies are needed to evaluate this promising new avenue in cancer treatment. We are developing various other meayamycin derivatives that would have more desirable pharmacological properties.Acknowledgements
S.O. is a recipient of a Graduate Excellence Fellowship from the University of Pittsburgh. The synthesis of FR901464 analogues was supported by the National Cancer Institute (R01 CA120792).Notes and references
- H. Nakajima, Y. Hori, H. Terano, M. Okuhara, T. Manda, S. Matsumoto and K. Shimomura, J. Antibiot., 1996, 49, 1204 CAS.
- H. Nakajima, B. Sato, T. Fujita, S. Takase, H. Terano and M. Okuhara, J. Antibiot., 1996, 49, 1196 CAS.
- H. Nakajima, S. Takase, H. Terano and H. Tanaka, J. Antibiot., 1997, 50, 96 CAS.
- Y. Kotake, J. Niijima, M. Nagai, K. Okano, T. Sakai, M. Yoshida, T. Tsuchida, T. Nakashima, K. Dobashi, Y. Mizui, H. Shimizu, T. Uenaka, M. Iwata, M. Asada and K. Yoshimatsu, Clin. Cancer Res., 2003, 9, 6208.
- T. Sakai, N. Asai, A. Okuda, N. Kawamura and Y. Mizui, J. Antibiot., 2004, 57, 180 CAS.
- T. Sakai, T. Sameshima, M. Matsufuji, N. Kawamura, K. Dobashi and Y. Mizui, J. Antibiot., 2004, 57, 173 CAS.
- N. Asai, Y. Kotake, J. Niijima, Y. Fukuda, T. Uehara and T. Sakai, J. Antibiot., 2007, 60, 364 CAS.
- D. Kaida, H. Motoyoshi, E. Tashiro, T. Nojima, M. Hagiwara, K. Ishigami, H. Watanabe, T. Kitahara, T. Yoshida, H. Nakajima, T. Tani, S. Horinouchi and M. Yoshida, Nat. Chem. Biol., 2007, 3, 576 CrossRef CAS.
- Y. Kotake, K. Sagane, T. Owa, Y. Mimori-Kiyosue, H. Shimizu, M. Uesugi, Y. Ishihama, M. Iwata and Y. Mizui, Nat. Chem. Biol., 2007, 3, 570 CrossRef CAS.
- F. A. Eskens, F. J. Ramos, H. Burger, M. J. de Jonge, J. Wanders, A. Lopez-Anaya, J. Baselga and J. Tabernero, J. Clin. Oncol. (Meeting Abstracts), 2009, 27, 3508 Search PubMed.
- http://clinicaltrials.gov/ct2/show/NCT00459823 .
- C. Lagisetti, A. Pourpak, Q. Jiang, X. L. Cui, T. Goronga, S. W. Morris and T. R. Webb, J. Med. Chem., 2008, 51, 6220 CrossRef CAS.
- B. J. Albert and K. Koide, Org. Lett., 2004, 6, 3655 CrossRef CAS.
- B. J. Albert, A. Sivaramakrishnan, T. Naka and K. Koide, J. Am. Chem. Soc., 2006, 128, 2792 CrossRef CAS.
- B. J. Albert, A. Sivaramakrishnan, T. Naka, N. L. Czaicki and K. Koide, J. Am. Chem. Soc., 2007, 129, 2648 CrossRef CAS.
- B. J. Albert, P. A. McPherson, K. O'Brien, N. L. Czaicki, V. DeStefino, S. Osman, M. Li, B. W. Day, P. J. Grabowski, M. J. Moore, A. Vogt and K. Koide, Mol. Cancer Ther., 2009, 8, 2308 CrossRef CAS.
- S. Osman, B. J. Albert, Y. Wang, M. Li, N. C. Czaicki, K. Koide, Eur.–Chem. J., in press DOI:10.1002/chem.201002402.
- http://www.dtp.nci.nih.gov/branches/btb/ivclsp.html .
- H. Fukuda, R. Ohashi, M. Tsuda-Tsukimoto and I. Tamai, Drug Metab. Dispos., 2008, 36, 1275 CrossRef CAS.
- S. X. Peng, C. Henson and L. J. Wilson, J. Chromatogr., B: Biomed. Sci. Appl., 1999, 732, 31 CrossRef CAS.
- J. Tazi, S. Durand and P. Jeanteur, Trends Biochem. Sci., 2005, 30, 469 CrossRef CAS.
- R. J. van Alphen, E. A. C. Wiemer, H. Burger and F. A. L. M. Eskens, Br. J. Cancer, 2009, 100, 228 CrossRef CAS.
- C. Lagisetti, A. Pourpak, T. Goronga, Q. Jiang, X. L. Cui, J. Hyle, J. M. Lahti, S. W. Morris and T. R. Webb, J. Med. Chem., 2009, 52, 6979 CrossRef CAS.
- M. C. Alley, M. G. Hollingshead, D. J. Dykes, W. R. Waud, in Anticancer Drug Development Guide, ed. B. A. Teicher, Humana Press, New Jersey, 2nd edn, 2004, ch. 7, pp. 125–152 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |