Molecular modeling of the human serotonin1Areceptor: role of membrane cholesterol in ligand binding of the receptor†
Yamuna Devi
Paila‡
*a,
Shrish
Tiwari‡
a,
Durba
Sengupta
b and
Amitabha
Chattopadhyay
*a
aCentre for Cellular and Molecular Biology, Council of Scientific and Industrial Research, Uppal Road, Hyderabad 500 007, India. E-mail: yamuna@ccmb.res.in; amit@ccmb.res.in; Fax: +91-40-2716-0311; Tel: +91-40-2719-2578
bGroningen Biomolecular Sciences and Biotechnology Institute, University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands
First published on 21st October 2010
Abstract
Serotonin1A receptors are important neurotransmitter receptors and belong to the superfamily of G-protein coupled receptors (GPCRs). Although it is an important drug target, the crystal structure of the serotonin1Areceptor has not been solved yet. Earlier homology models of the serotonin1Areceptor were generated using rhodopsin as a template. We have used two recent crystal structures of the human β2-adrenergic receptor, one of which shows specific cholesterol binding site(s), as templates to model the human serotonin1Areceptor. Since the sequence similarity between the serotonin1Areceptor and β2-adrenergic receptor is considerably higher than the similarity between the serotonin1Areceptor and rhodopsin, our model is more reliable. Based on these templates, we generated models of the serotonin1Areceptor in the absence and presence of cholesterol. The receptor model appears more compact in the presence of cholesterol. We validated the stability of ‘compactness’ using coarse-grain MD simulation. Importantly, all ligands exhibit higher binding energies when docked to the receptor in the presence of cholesterol, thereby implying that membrane cholesterol facilitates ligand binding to the serotonin1Areceptor. To the best of our knowledge, this is one of the first reports in which lipid-specific receptor conformations have been modeled by homology modeling.
Introduction
The G-protein coupled receptor (GPCR) superfamily is the largest and most diverse protein family in mammals, involved in signal transduction across membranes.1–3 Cellular signaling by GPCRs involves their activation by ligands present in the extracellular environment and subsequent transduction of signals to cellular interior through concerted changes in their transmembrane domain structure.4 GPCRs are prototypical members of the family of seven transmembrane domain proteins and include >800 members which together constitute ∼5% of the human genome.5 They dictate physiological responses to a diverse array of stimuli that include endogenous ligands such as biogenic amines, peptides, glycoproteins, lipids, nucleotides, Ca2+ ions and various exogenous ligands for sensory perceptions such as odorants, pheromones, and even photons. As a consequence, these receptors mediate multiple physiological processes such as neurotransmission, secretion, cellular differentiation, growth, inflammatory and immune responses. It is therefore only natural that GPCRs have emerged as major targets for the development of novel drug candidates in all clinical areas.6–10 Interestingly, although GPCRs represent 30–50% of current drug targets, only a small fraction of all GPCRs are presently targeted by drugs.11 This raises the exciting possibility that receptors that are not yet recognized could be potential drug targets for diseases that are difficult to treat by currently available drugs. In spite of an enormous interest in GPCR drug design owing to the pharmacological significance of GPCRs, lack of three-dimensional structural information severely limits GPCR drug design.12Membrane proteins are difficult to crystallize in their native conditions due to the intrinsic dependence on surrounding membrane lipids.13 Since a significant portion of any integral membrane protein (such as GPCRs) remains in contact with the membrane lipid environment,14membrane lipids are necessary to maintain their structure and function. Purification and crystallization of membrane proteins therefore continue to be challenging in contemporary biology. Crystallization of GPCRs is even more challenging due to the intrinsic structural flexibility (particularly of the loop regions) giving rise to multiple conformational states (conformational heterogeneity).15 High-resolution structural information of GPCRs is lacking for this reason. Crystal structures of only four GPCRs have been solved with atomic resolution to date.16–19 Due to improved resolution of modern-day crystallography, some of the solved GPCR crystal structures reveal membrane lipids bound in close proximity to the protein, indicating specific interactions of these proteins with surrounding lipids17,20,21 (recently reviewed in ref. 22). Since the crystallization of GPCRs poses considerable challenges, in silico approaches for prediction of GPCR structures are being increasingly used for structure-based drug design.12,23–25 Homology modeling is a widely used approach in which a known structure of a homologous receptor is used as a template for model building. It is based on the concept that three-dimensional structures of proteins are generally more conserved than their amino acid sequences.26 As a result of this, proteins with homologous sequences are expected to show similar three-dimensional structures.
The serotonin1A (5-HT1A) receptor is an important neurotransmitter receptor and belongs to the superfamily of the seven transmembrane domain GPCRs. It is the most extensively studied of the serotonin receptors for a number of reasons.27,28Serotonin receptors have been classified into at least 14 subtypes on the basis of their pharmacological responses to specific ligands, sequence similarities at the gene and amino acid levels, gene organization, and second messenger coupling pathways.29 Serotonin1A receptors are known to play a key role in the generation and modulation of various cognitive, developmental and behavioral functions. Agonists and antagonists of the serotonin1Areceptor have been shown to possess potential therapeutic effects in anxiety- or stress-related disorders.27 As a result, the serotonin1Areceptor serves as an important target in the development of therapeutic agents for neuropsychiatric disorders such as anxiety and depression. Interestingly, mutant (knockout) mice lacking the serotonin1Areceptor exhibit enhanced anxiety-related behavior, and represent an important animal model for genetic vulnerability to complex traits such as anxiety disorders and aggression in higher animals.30,31 Taken together, these results suggest that the serotonin1Areceptor is a central player in a multitude of physiological processes, and represents an important drug target.
In this work, we have used two recent crystal structures of the human β2-adrenergic receptor17,21 as templates to model the human serotonin1Areceptor. One of these structures shows specific cholesterol binding site(s) in the receptor.21 Based on these templates, we generated molecular models of the serotonin1Areceptor in the absence and presence of cholesterol. The receptor model appears more compact in presence of cholesterol. We validated the stability of ‘compactness’ using coarse-grain MD simulations. We have further used these models to dock agonists and antagonists to the serotonin1Areceptor, in order to identify the role of membrane cholesterol in ligand binding to the receptor. Our results show that the presence of membrane cholesterol facilitates ligand binding to human serotonin1A receptors. To the best of our knowledge, our results represent one of the first reports in which lipid-specific receptor conformations have been modeled by homology modeling.
Results
Sequence alignment and analysis
Human serotonin-1 receptors are divided into five classes which include 1A, 1B, 1D, 1E and 1F subtypes. The serotonin1Areceptor is an important member of the serotonin-1 subfamily and is estimated to have differentiated ∼650 million years ago from the subfamily during the period when vertebrates diverged from invertebrates.32,33 The human gene for the receptor encodes a protein of 422 amino acids. Considering the presence of three consensus sequences for N-linked glycosylation in the amino terminus, and the homology of the receptor with β-adrenergic receptor, it is predicted that the receptor is oriented in the plasma membrane with the amino terminus facing the extracellular region and the carboxy terminus facing the intracellular cytoplasmic region.27,28,34 The transmembrane domains (I–VII) of the receptor are connected by hydrophilic sequences of three extracellular loops (EC1, EC2, EC3) and three intracellular loops (IC1, IC2, IC3). Such an arrangement is typical for the G-protein coupled receptor superfamily.35A striking feature of the serotonin1Areceptor is the length (∼120 residues spanning from 219–343 residues, based on transmembrane helix prediction using TMHMM222) of its third intracellular (IC3) loop. The IC3 loop of the serotonin1Areceptor is considerably longer than the corresponding loop of other members of the serotonin-1 receptor subfamily (see Fig. 1) and is important for coupling to G-proteins. Interestingly, it has been previously reported using site-directed mutagenesis that mutations in IC3 loop of the serotonin1Areceptor alter G-protein coupling from Gi to Gs in a ligand-dependent manner.36 The serotonin1Areceptor was the first of all types of serotonin receptors to be cloned as an intronless genomic clone (G-21) of the human genome, which cross-hybridized with a full length β-adrenergic receptor probe at reduced stringency.27,37 Sequence analysis of this genomic clone (later identified as the serotonin1Areceptor gene) shows ∼48% amino acid similarity with the β2-adrenergic receptor in the transmembrane region. A unique pharmacological feature of the serotonin1Areceptor is its affinity for the class of drugs called β-blockers.38 Interestingly, sequence alignment of all serotonin-1 receptors shows that Asn386 is present only in the serotonin1Areceptor (see Fig. 1), and is also found in β-adrenergic receptors. Importantly, Asn386 is the residue that is involved in the binding of pindolol, a β-blocker (and an antagonist for β-adrenergic receptor).38 The serotonin1Areceptor was initially discovered as an ‘orphan’ receptor and was identified (‘deorphanized’) later.39 The sequence alignment of serotonin1A receptors from vertebrates and invertebrates is shown in Fig. S1 and S2†. These figures show that the sequence of serotonin1A receptors is predominantly conserved from fish to humans, while the conservation among sequences of invertebrates is rather poor.
 | ||
| Fig. 1 Sequence alignment of human serotonin-1 receptors. Human serotonin-1 receptor sequences that include receptors of five subclasses (A, B and D–F) have been used for comparison. The amino acid Asn386, crucial for pindolol binding, is highlighted. See text for more details. | ||
Generation of molecular models of human serotonin1A receptors
As mentioned above, the overall sequence similarity of the human serotonin1Areceptor with the human β2-adrenergic receptor is ∼48% in the transmembrane region. Fig. 2 shows sequences of the template and human serotonin1Areceptor to show that the transmembrane regions are predominantly conserved. The homology model generated by using β2-adrenergic receptor as template would therefore be more reliable in the transmembrane region. Modeling the transmembrane region assumes significance since the ligand binding pockets of serotonin1A receptors are believed to be present in the transmembrane region.27 We therefore chose to focus mainly on the transmembrane region of the receptor for further analysis. We used two recently published structures of the human β2-adrenergic receptor as templates. One of the structures (PDB 2RH1)17 was used to generate the model in the absence of cholesterol. The other structure (PDB 3D4S)21 exhibits bound cholesterol molecules at the Consensus Cholesterol binding Motif (CCM) in between the helices of a monomer. We used the latter structure to generate a model of the serotonin1Areceptor in the presence of cholesterol. The 3D4S structure shows a cholesterol binding site between transmembrane helices I–IV with two cholesterol molecules bound per receptor monomer. The cholesterol binding site appears to be characterized by the presence of a cleft located at the membrane interfacial region.21 Both cholesterol molecules bind in a shallow surface groove formed by segments of the above-mentioned helices (I–IV), thereby providing an increase in the intramolecular occluded surface area, a parameter often correlated to the enhanced thermal stability of proteins.40 Importantly, we have earlier reported that cholesterol binding sites are inherent characteristic features of serotonin1A receptors and are conserved over natural evolution.22 | ||
| Fig. 2 The final alignment used for modeling the human serotonin1Areceptor. Transmembrane helices are highlighted for easy identification. The alignment shows that amino acid residues in transmembrane helices are largely conserved. | ||
The energy-minimized homology models of the human serotonin1Areceptor in the absence and presence of cholesterol are shown in Fig. 3. These models were validated using PROCHECK and WHAT IF servers. The corresponding Ramachandran plots for receptor models in the absence and presence of cholesterol are shown in Fig. 4. Analysis using PROCHECK reveals that >98% of the residues are in overall allowed regions (∼91% in most favored regions and 8% in additional allowed regions) in case of the model generated without cholesterol (see Fig. 4a). Fig. 4b shows that in case of the model in presence of cholesterol, >98% of the residues are in overall allowed regions (∼89% in most favored regions and 9% in additional allowed regions). In addition, models of the serotonin1Areceptor in the absence and presence of cholesterol were validated using the ProSA program (Fig. S3†).
 | ||
| Fig. 3 Energy-minimized final models of the human serotonin1Areceptor in the absence (a) and presence (b) of cholesterol. Transmembrane helices I–VII are shown in different colors. Panel (b) shows two cholesterol (yellow) molecules bound to the receptor. The receptor model in presence of cholesterol appears more compact (see text). | ||
 | ||
| Fig. 4 Ramachandran plots calculated for the human serotonin1Areceptor models generated using PROCHECK server. Panels (a) and (b) show Ramachandran plots for models generated in the absence and presence of cholesterol. Fully allowed and partially allowed regions are shown in red and deep yellow, respectively. The analyzed results are shown along with the plots. A vast majority of amino acid residues (>98%) are within most favored and partially allowed regions in both cases. See the Experimental section for other details. | ||
Importantly, Fig. 3 shows that the model of the serotonin1Areceptor in presence of cholesterol is more compact compared to the model generated in absence of cholesterol. The presence of cholesterol appears to increase the packing of the transmembrane helices. In this context, it is worth mentioning here that calculation of packing values of various helices in the β2-adrenergic receptor that are involved in the cholesterol interaction site show that the packing of transmembrane helices II and IV increases upon cholesterol binding. The increased packing would restrict their mobility rendering greater thermal stability to the protein.21 Our results from homology modeling of the serotonin1Areceptor in the presence of cholesterol is in general agreement with these results with the β2-adrenergic receptor. Interestingly, we have recently observed that the thermal stability of the serotonin1Areceptor is enhanced in the presence of cholesterol.41
Validation of the compactness of the receptor model in the presence of cholesterol by MD simulation
Models of the serotonin1Areceptor in the absence and presence of cholesterol, mapped to their coarse-grain representations, were simulated in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and POPC-cholesterol bilayers, respectively. Simulations of 8 μs were performed for both models. To test the integrity of the models, they were compared to the starting structures. The RMSD of the backbone beads of the transmembrane helices of the receptor in the absence and presence of cholesterol is 0.3 nm and 0.5 nm, respectively. Importantly, the volume of transmembrane domains (considering only the backbone) of receptor models in the absence (11.6 ± 0.005 nm3) and presence (11.7 ± 0.01 nm3) of cholesterol is comparable and remains constant during the simulation (see Fig. 5). In addition, the volume of transmembrane domains of the receptor models in the absence (24.3 ± 0.001 nm3) and presence (24.7 ± 0.04 nm3) of cholesterol is similar, even if side chains are included (Fig. 5). | ||
| Fig. 5 Plot of volume of transmembrane domains of serotonin1Areceptor models in the absence (red) and presence (green) of cholesterol with simulation time. Lower two data sets correspond to the volume of only the backbone, while the upper two traces correspond to volume including side chains. | ||
The deviation of the model of the serotonin1Areceptor in the absence of cholesterol from its starting conformation is quite low (indicated by RMSD). The deviation of the model of the serotonin1Areceptor in the presence of cholesterol is relatively high but within the average deviation, for coarse-grain protein structures.42 During the simulation, a small repositioning of helix II was observed when the receptor model was simulated in the presence of cholesterol. We speculate that this deviation is due to the absence of optimal packing of cholesterol molecules in the starting structure since they were placed randomly in the POPC bilayer. Nonetheless, the volume of the receptor models remains constant, thereby ruling out any substantial deviation of the transmembrane helices from their modeled positions. In addition, the volume of both receptor models is comparable, indicating that the ‘compactness’ of the receptor model in the presence of cholesterol is not due to an overall decrease in volume, but rather due to better packing of the side chains resulting in a more compact lumen.
Analysis of molecular docking
Our understanding of the structure of the ligand binding site of GPCRs at the atomic level is still emerging.43 Although the crystal structure of the serotonin1Areceptor has not been worked out yet, mutagenesis studies have provided useful insight in identifying amino acid residues important for ligand binding and G-protein coupling of the receptor (reviewed in ref. 27). In order to identify residues potentially involved in ligand binding, seven ligands (three agonists and four antagonists) were docked to the receptor in the absence and presence of cholesterol. The agonists used were serotonin, 8-hydroxy-2(di-N-propylamino)tetralin (8-OH-DPAT) and 5-carboxamidotryptamine (5-CT) and their chemical structures are shown in Fig. 6. The antagonists used were 4-(2′-methoxy)-phenyl-1-[2′-(N-2′′-pyridinyl)-p-fluorobenzamido]ethyl-piperazine (p-MPPF), N-(2-(1-(4-(2-methoxyphenyl)piperazinyl)-N-(2-pyridinyl)ethyl))cyclohexanecarboxamide (WAY-100635), O-desmethyl derivative of WAY (DWAY) and pyridinylN-oxide derivative of WAY (NOWAY) and their chemical structures are shown in Fig. 7. DWAY and NOWAY have been shown to exhibit high affinity toward the serotonin1Areceptor.44 The selective agonist 8-OH-DPAT was the first compound that did not contain an indole nucleus as part of its structure.44 The availability of 8-OH-DPAT helped in the characterization of the serotonin1Areceptor, thereby making this receptor the most studied amongst serotonin receptors.27Serotonin is the natural ligand of the receptor. 5-CT has been reported to display affinity similar to that of serotonin.44 | ||
| Fig. 6 Chemical structures of agonists for the serotonin1Areceptor. Agonists such as serotonin, 8-OH-DPAT and 5-CT are used to dock to the receptor. | ||
 | ||
| Fig. 7 Chemical structures of antagonists for the serotonin1Areceptor. Antagonists such as p-MPPF, WAY-100635, DWAY and NOWAY are used to dock to the receptor. | ||
Fig. 8 shows the representative docked poses of these ligands to the receptor. The docked poses of ligands to the receptor are the best-energy solutions in the most populated cluster. Clustering analysis with RMSD is shown in Fig. S4†. The docked poses are depicted with the receptor in the absence of cholesterol. Although the docked site remains largely invariant in the presence of cholesterol, the predicted binding energies differ (see later). Docked poses of serotonin and WAY-100635 clearly show that the antagonist binding pocket is relatively shallow compared to the agonist binding pocket. Interestingly, it was previously suggested by us that the antagonist binding site in the receptor could be at a relatively shallow location compared to the agonist binding site.45 This prediction was based on the effect of ethanol on the extent of ligand binding of the receptor.
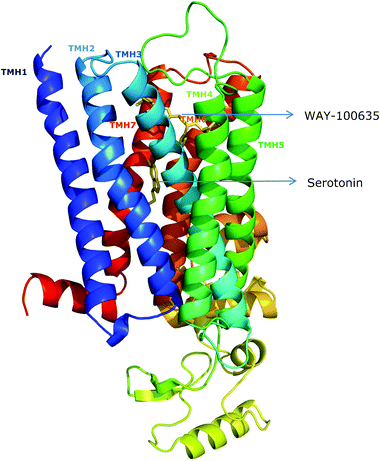 | ||
| Fig. 8 Model of the human serotonin1Areceptor showing docked sites and poses of serotonin (agonist) and WAY-100635 (antagonist). Interestingly, the antagonist binding site is found to be relatively shallow compared to the agonist binding site. Transmembrane helices I–VII are shown in different colors. See the Experimental section for other details. | ||
Although there is no structural data for the serotonin1Areceptor, there is considerable experimental data suggesting possible ligand binding sites in the receptor. Site-directed mutagenesis provides a powerful approach to identify the location of the binding pocket in the receptor. Results obtained from point mutations suggest that Asp82 and Asp116 in transmembrane helices II and III are crucial for binding of serotonin to the receptor.46 It has been suggested that the carboxylate group in Asp82 and/or Asp116 probably acts as a counter-ion to the amine group of serotonin, thereby stabilizing its interaction.27 Interestingly, while Asp82 is conserved in all cloned GPCRs, Asp116 is conserved only in GPCRs that bind biogenic amines. Another possibility for agonist binding is an interaction between a hydrogen bonding group (e.g., hydroxyl group) of serotonin and the hydroxyl group of serine or threonine residues. In addition, agonist binding could be stabilized by aromatic π–π interactions with the surrounding residues.44Fig. 9 and 10 illustrate agonist and antagonist docking poses on the serotonin1Areceptor, and Tables 1 and 2 show amino acid residues of the receptor involved in interactions with ligands. The predicted binding mode of serotonin shows hydrogen bonding with Asp116. Fig. 11 shows the respective predicted binding energy of ligands to the serotonin1Areceptor in the absence and presence of cholesterol. Higher binding energy implies better docking of the ligand to the receptor. The predicted binding energy for docked serotonin is found to be ∼7.25 kcal/mol. Docking of the synthetic specific agonist, 8-OH-DPAT, with the receptor resulted in slightly higher binding energy (∼7.5 kcal/mol). Synthetic ligands appear to bind to the receptor with higher binding energy. In general, it appears that docked antagonists exhibit higher binding energies compared to that of agonists. Importantly, both agonists and antagonist show higher binding energies when docked to the receptor in the presence of cholesterol. Our results therefore demonstrate that membrane cholesterol facilitates ligand binding to the serotonin1Areceptor. Interestingly, we have previously shown that the affinity of specific agonist (8-OH-DPAT)47 and antagonist (p-MPPF)48 to the serotonin1Areceptor is reduced upon cholesterol depletion.
 | ||
| Fig. 9 Representation of docked poses of agonists to the human serotonin1Areceptor. Snapshots of interactions of the serotonin1Areceptor with various agonists from AutoDock are shown. Agonists include serotonin, 8-OH-DPAT and 5-CT. | ||
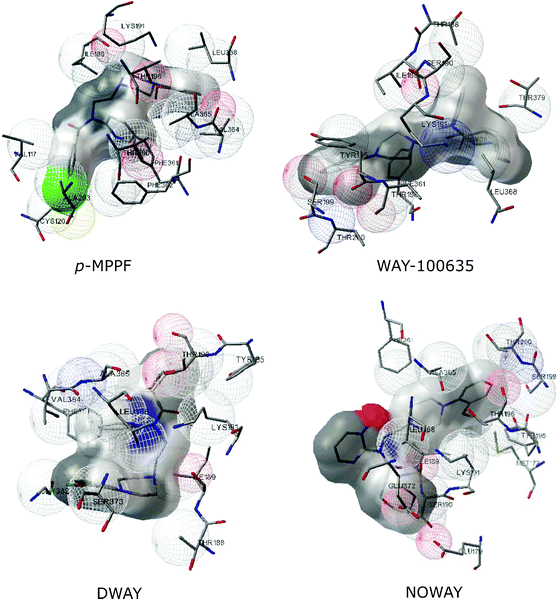 | ||
| Fig. 10 Representation of docked poses of antagonists to the human serotonin1Areceptor. Snapshots of interactions of the serotonin1Areceptor with various antagonists from AutoDock are shown. Antagonists include p-MPPF, WAY-100635, DWAY and NOWAY. | ||
 | ||
| Fig. 11 Predicted binding energies of docked poses of (a) agonists and (b) antagonists to the human serotonin1Areceptor in the absence and presence of cholesterol. Binding energies in the absence and presence of cholesterol are shown as empty and filled bars, respectively. | ||
| Agonist | Interactions involving: | ||
|---|---|---|---|
| Hydrogen bonds | Nonpolar residues | Polar residues | |
| a Residues that are reported to be involved in binding are in bold. | |||
| Serotonin | Asp116 | Leu78 | Asp82 |
| Cys120 | Val85 | ||
| Ser123 | Cys119 | ||
| Gly389 | Phe354 | ||
| Ser393 | Trp358 | ||
| Tyr390 | |||
| Asn392 | |||
| 8-OH-DPAT | Ser190 | Cys109 | Asp110 |
| Met172 | Asp185 | ||
| Trp175 | |||
| Thr177 | |||
| Pro178 | |||
| Pro184 | |||
| Ala186 | |||
| Cys187 | |||
| Thr188 | |||
| Ile189 | |||
| 5-CT | Asp116 | Leu78 | Asp82 |
| Val85 | Ser123 | ||
| Cys119 | Ser393 | ||
| Cys120 | |||
| Phe354 | |||
| Trp358 | |||
| Gly389 | |||
| Tyr390 | |||
| Asn392 | |||
| Antagonist | Interactions involving: | |
|---|---|---|
| Nonpolar residues | Polar residues | |
| p-MPPF | Val117 | Lys191 |
| Cys120 | ||
| Ile189 | ||
| Thr196 | ||
| Thr200 | ||
| Ala203 | ||
| Phe361 | ||
| Phe362 | ||
| Val364 | ||
| Ala365 | ||
| Leu368 | ||
| WAY-100635 | Thr188 | Ser190 |
| Ile189 | Lys191 | |
| Tyr195 | Ser199 | |
| Thr196 | ||
| Thr200 | ||
| Phe361 | ||
| Leu368 | ||
| Thr379 | ||
| Gly382 | ||
| DWAY | Ile189 | Lys191 |
| Thr188 | Ser373 | |
| Tyr195 | ||
| Thr196 | ||
| Ala365 | ||
| Leu368 | ||
| Phe361 | ||
| Val364 | ||
| Gly382 | ||
| NOWAY | Ile189 | Glu372 |
| Met172 | Lys191 | |
| Phe361 | Ser199 | |
| Ala365 | Ser190 | |
| Thr200 | Glu179 | |
| Tyr195 | ||
| Thr196 | ||
| Leu368 | ||
Discussion
Lipid–protein interactions play a crucial role in maintaining the structure and function of integral membrane proteins and receptors.49–52 A large portion of any given transmembrane receptor remains in contact with the lipid environment. This raises the obvious possibility that membrane lipids could be important modulators of receptor structure and function. Work from a number of groups has contributed to our understanding of the requirement of specific lipids for maintaining the proper topology, structure and function of membrane proteins.49,50,53,54Cholesterol is an essential and representative lipid in higher eukaryotic cellular membranes.55,56 Importantly, cholesterol plays a vital role in the function and organization of GPCRs.51,57 These effects of cholesterol on GPCRs have been attributed either to specific interactions of lipids with amino acids in proteins or to bulk properties of membranes.58In addition to the recently reported cholesterol-bound crystal structure of the β2-adrenergic receptor,17,21 the crystal structure of a photo-stationary state, highly enriched in metarhodopsin I, showed a cholesterol molecule between two rhodopsin monomers.20 These authors also reported that cholesterol could improve the reliability and yield of crystallization. In this structure, cholesterol is shown to be oriented with its tetracyclic ring aligned normal to the membrane bilayer. These authors proposed that some of the tryptophans in transmembrane helices would be able to interact with the tetracyclic ring of cholesterol. Specific cholesterol interaction (i.e., through a direct interaction that could induce a conformational change in the receptor) has been shown for GPCRs such as oxytocin57,59,60 and galanin61 receptors. In addition, new information on lipid-mediated dimerization of cannabinoid receptors has been obtained by homology modeling using the crystal structure of β2-adrenergic receptor.62 Interestingly, recent molecular dynamics simulations have shown that membrane cholesterol specifically interacts with transmembrane domains of GPCRs such as rhodopsin63 and human A2A adenosine receptor,64 thereby stabilizing helix II of the human A2A adenosine receptor. A consequence of this interaction is that the receptor couples to G-proteins only in the presence of cholesterol.
The overall objective of the present work is to generate and validate a reliable homology model of the serotonin1Areceptor and use this model to dock well-characterized ligands to the receptor and explore the role of cholesterol in ligand binding. Although homology models of the serotonin1Areceptor were previously generated using rhodopsin as a template,65,66 our work represents the first comprehensive model of the human serotonin1Areceptor generated using the human β2-adrenergic receptor as a template. The success of homology modeling often depends on the similarity of structural and functional relationships between the template and the modeled protein.43,67 The quality and refinement of the model would improve with availability of better templates. It is in this context that our present model is more reliable since the sequence similarity (∼48%) between the human serotonin1Areceptor and human β2-adrenergic receptor in the transmembrane region is considerably higher compared to the similarity (∼38%) between serotonin1Areceptor and rhodopsin sequences.
Previous work from our laboratory has comprehensively demonstrated the requirement of membrane cholesterol in the function and organization of the serotonin1Areceptor.47,48,51,52,68,69 We have recently proposed that membrane cholesterol could occupy ‘nonannular’ binding site(s) on the receptor.22 Nonannular sites are characterized by lack of accessibility to annular lipids, i.e., these sites cannot be displaced by competition with annular lipids. In the present work, we combined homology modeling with cholesterol-specific conformations of the serotonin1Areceptor. Our results constitute an early attempt to incorporate cholesterol specificity of GPCRs in homology modeling. In the context of increasing pharmacological relevance of the serotonin1Areceptor, interaction of this transmembrane protein with surrounding lipids (such as cholesterol) assumes greater significance in its structure and function in healthy and diseased states.
GPCRs are involved in a multitude of physiological functions and represent important drug targets. Although the pharmacological and signaling features of GPCRs have been extensively studied, aspects related to their interaction with membrane lipids have been addressed in very few cases. In this context, the realization that lipids such as cholesterol could influence the structure and function of GPCRs has remarkably transformed our idea regarding the function of this important class of membrane proteins. With progress in deciphering molecular details on the nature of this interaction, our overall understanding of GPCR function in health and disease would improve significantly, thereby enhancing our ability to design better therapeutic strategies to combat diseases related to malfunctioning of these receptors. A comprehensive understanding of GPCR function in relation to the membrane lipid environment is important, in view of the enormous implications of GPCR function in human health,7,8 and the observation that several diagnosed diseases are attributed to altered lipid–protein interactions.70,71 We conclude that homology modeling,12 in combination with emerging knowledge of lipid specificities of GPCRs,52 represent a promising approach to use in future drug discovery research.
Experimental section
Sequence analysis
All sequences used for the analysis, including the sequence (BBA94488) of the human serotonin1Areceptor, were obtained from the NCBI (http://www.ncbi.nlm.nih.gov/) database. Sequence alignment of the human serotonin1Areceptor and human β2-adrenergic receptor was carried out with ClustalX (version 2.0.11). Sequence alignments were performed on sets of receptor sequences to identify conserved residues that could have structural and or functional implications. Sequence alignments were carried out with all subtypes of the serotonin-1 receptor (A, B; D–F) and the serotonin1Areceptor from invertebrates and vertebrates. Alignments were constructed using ClustalX and were manually-edited and viewed using Jalview (version 2.4) to retain high equivalence of conserved regions. Residues are numbered according to the human serotonin1Areceptor sequence throughout the text.Homology modeling
The serotonin1Areceptor and β2-adrenergic receptor are both biogenic amine GPCRs, and enjoy a sequence similarity of ∼48%. The human serotonin1A (BBA94488) and human β2-adrenergic receptor (2RH1) sequences were aligned using ClustalX and viewed and edited using Jalview. Recently reported crystal structures of the human β2-adrenergic receptor were obtained from RCSB (http://www.rcsb.org/pdb) and used as templates. Accession numbers of the crystal structures of human β2-adrenergic receptors in the absence and presence of cholesterol are 2RH117 and 3D4S,21 respectively. We chose 3D4S as a template for modeling the human serotonin1Areceptor due to the presence of two cholesterol molecules in the structure. The coordinates corresponding to 1–28 and 343–365 segments were not available in 2RH1, and coordinates corresponding to 1–31 and 343–365 segments were not available in 3D4S crystal structures due to poor electron density, and therefore these residues were removed from the query sequence prior to alignment. The final alignment was used to construct the model using the software Modeller72 (version 9.7). A set of 200 models was generated, from which the lowest energy structure was used for further processing. The initial low energy model obtained from Modeller was validated by using PROCHECK73 and WHAT IF74 servers. We have also analyzed the side chain orientation of conserved residues (which are conserved at least in all class-A GPCR sequences), by superposing our model against the available crystal structures (2RH1 and 3D4S) in order to analyze the goodness of fit with other structures. We focused mostly on the modeling of the transmembrane helical bundle.Docking study
We carried out blind docking using AutoDock (version 4.0). Blind docking is often used assuming that the ligand binding site is unknown and the structures of the ligand and the receptor are known. In our case, it would be necessary to perform dockings to search the entire surface of the receptor. This was achieved using AutoGrid to create grid maps, with 40 points in each dimension, and creating sets of adjacent grid map volumes that cover the entire transmembrane region of the receptor. Rigid protein-flexible ligand docking of seven different ligands, including agonists and antagonists, was carried out with AutoDock. The agonists include serotonin (natural ligand) and 8-OH-DPAT, and antagonists are p-MPPF and WAY-100635. The AutoDock graphical interface AutoDockTools was used to keep polar hydrogen atoms and add partial charges for protein using the Gasteiger charges. The grid maps were calculated using the auxiliary program Autogrid 4.0. Blind docking was performed using a grid box of 40 × 40 × 40 cells (each cell has dimensions 0.375 × 0.375 × 0.375 Å) by moving it around to cover the entire transmembrane region. The Lamarckian genetic algorithm (LGA) was selected for ligand conformational searching. Docking was carried out with a population size of 150 with 25 × 105 evaluations and a maximum of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 generations. Conformations were analyzed using the MGL AutoDock tools (version 1.5.2). Chemical structures of ligands were obtained from PubChem and three dimensional structures of ligands were generated using Open Babel (version 2.2.3).
000 generations. Conformations were analyzed using the MGL AutoDock tools (version 1.5.2). Chemical structures of ligands were obtained from PubChem and three dimensional structures of ligands were generated using Open Babel (version 2.2.3).
Molecular dynamics simulations of receptor models
Molecular dynamics simulations were performed to validate homology models of the serotonin1Areceptor in the absence and presence of cholesterol. Systems were represented using the MARTINI coarse-grain force-field (version 2.1).75,76 Models were mapped to their coarse-grain representations. The receptor model in the absence of cholesterol was simulated in a pure POPC membrane containing 200 lipid molecules and the receptor model in the presence of cholesterol was simulated in a POPC–cholesterol mixed bilayer containing 80 cholesterol and 80 POPC molecules. Simulations were performed using the GROMACS program package (version 4.0.2),77 with the scheme developed for coarse-grain simulations, under periodic boundary conditions. Temperature was weakly coupled (coupling time 0.1 ps) to a thermostat at 300 K using the Berendsen algorithm.78 Pressure was weakly coupled (coupling time 1.0 ps, compressibility 5 × 10−5 bar) using a semi-isotropic coupling scheme, in which the lateral and perpendicular pressures are coupled independently at 1 bar. Non-bonded interactions were treated with a switch function from 0–1.2 nm for Coulomb interactions and 0.9–1.2 nm for LJ interactions (pair-list update frequency of once per 10 steps). A time step of 25 fs was used.Acknowledgements
This work was supported by the Council of Scientific and Industrial Research (Govt. of India). Y.D.P. was the recipient of a Postdoctoral Fellowship from a CSIR Network project on Nanomaterials and Nanodevices (NWP0035). A.C. is an Adjunct Professor at the Special Centre for Molecular Medicine of Jawaharlal Nehru University (New Delhi, India) and Indian Institute of Science Education and Research (Mohali, India), and Honorary Professor of the Jawaharlal Nehru Centre for Advanced Scientific Research (Bangalore, India). A. C. gratefully acknowledges the J. C. Bose Fellowship (Dept. Science and Technology, Govt. of India). We thank Sandeep Shrivastava for help and members of our laboratory for critically reading the manuscript.References
- K. L. Pierce, R. T. Premont and R. J. Lefkowitz, Nat. Rev. Mol. Cell Biol., 2002, 3, 639–650 CrossRef CAS.
- D. M. Perez, Mol. Pharmacol., 2003, 63, 1202–1205 CrossRef CAS.
- D. M. Rosenbaum, S. G. F. Rasmussen and B. K. Kobilka, Nature, 2009, 459, 356–363 CrossRef CAS.
- U. Gether, Endocr. Rev., 2000, 21, 90–113 CrossRef CAS.
- Y. Zhang, M. E. DeVries and J. Skolnick, PLoS Comput. Biol., 2006, 2, 88–99 CAS.
- Nature reviews drug discovery GPCR questionnaire participants, Nat. Rev. Drug Discov., 3, 577–626 Search PubMed.
- E. Jacoby, R. Bouhelal, M. Gerspacher and K. Seuwen, ChemMedChem, 2006, 1, 760–782 CrossRef CAS.
- S. Schlyer and R. Horuk, Drug Discovery Today, 2006, 11, 481–493 CrossRef CAS.
- P. A. Insel, C. M. Tang, I. Hahntow and M. C. Michel, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 994–1005 CrossRef CAS.
- R. Heilker, M. Wolff, C. S. Tautermann and M. Bieler, Drug Discovery Today, 2009, 14, 231–240 CrossRef CAS.
- S. H. Lin and O. Civelli, Ann. Med., 2004, 36, 204–214 Search PubMed.
- A. Patny, P. V. Desai and M. A. Avery, Curr. Med. Chem., 2006, 13, 1667–1691 CrossRef CAS.
- (a) L. Anson, Nature, 2009, 459, 343 CrossRef CAS; (b) M. Baker, Nat. Methods, 2010, 7, 429–434 CrossRef CAS.
- A. G. Lee, Biochim. Biophys. Acta, Biomembr., 2003, 1612, 1–40 CrossRef CAS.
- B. K. Kobilka and X. Deupi, Trends Pharmacol. Sci., 2007, 28, 397–406 CrossRef CAS.
- K. Palczewski, T. Kumasaka, T. Hori, C. A. Behnke, H. Motoshima, B. A. Fox, I. Le Trong, D. C. Teller, T. Okada, R. E. Stenkamp, M. Yamamoto and M. Miyano, Science, 2000, 289, 739–745 CrossRef CAS.
- V. Cherezov, D. M. Rosenbaum, M. A. Hanson, S. G. F. Rasmussen, F. S. Thian, T. S. Kobilka, H.-J. Choi, P. Kuhn, W. I. Weis, B. K. Kobilka and R. C. Stevens, Science, 2007, 318, 1258–1265 CrossRef CAS.
- T. Warne, M. J. Serrano-Vega, J. G. Baker, R. Moukhametzianov, P. C. Edwards, R. Henderson, A. G. W. Leslie, C. G. Tate and G. F. X. Schertler, Nature, 2008, 454, 486–491 CrossRef CAS.
- V.-P. Jaakola, M. T. Griffith, M. A. Hanson, V. Cherezov, E. Y. T. Chien, J. R. Lane, A. P. Ijzerman and R. C. Stevens, Science, 2008, 322, 1211–1217 CrossRef CAS.
- J. J. Ruprecht, T. Mielke, R. Vogel, C. Villa and G. F. Schertler, EMBO J., 2004, 23, 3609–3620 CrossRef CAS.
- M. A. Hanson, V. Cherezov, M. T. Griffith, C. B. Roth, V.-P. Jaakola, E. Y. T. Chien, J. Velasquez, P. Kuhn and R. C. Stevens, Structure, 2008, 16, 897–905 CrossRef CAS.
- Y. D. Paila, S. Tiwari and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 295–302 CrossRef CAS.
- J. Simms, N. E. Hall, P. H. C. Lam, L. J. Miller, A. Christopoulos, R. Abagyan and P. M. Sexton, in G Protein-Coupled Receptors in Drug Discovery, ed. W. R. Leifert, Humana Press, New York, 2009, vol. 552, pp. 97–113 Search PubMed.
- B. Kneissl, B. Leonhardt, A. Hildebrandt and C. S. Tautermann, J. Med. Chem., 2009, 52, 3166–3173 CrossRef CAS.
- T. Yarnitzky, A. Levit and M. Y. Niv, Curr. Opin. Drug Discov. Devel., 2010, 13, 317–325 Search PubMed.
- A. M. Lesk and C. H. Chothia, Philos. Trans. R. Soc. London, Ser. A, 1986, 317, 345–356 CrossRef CAS.
- T. J. Pucadyil, S. Kalipatnapu and A. Chattopadhyay, Cell. Mol. Neurobiol., 2005, 25, 553–580 CrossRef CAS.
- S. Kalipatnapu and A. Chattopadhyay, Cell. Mol. Neurobiol., 2007, 27, 1097–1116 CrossRef CAS.
- D. Hoyer, J. P. Hannon and G. R. Martin, Pharmacol., Biochem. Behav., 2002, 71, 533–554 CrossRef CAS.
- M. Toth, Eur. J. Pharmacol., 2003, 463, 177–184 CrossRef CAS.
- A. M. Gardier, Behav. Pharmacol., 2009, 20, 18–32 CrossRef CAS.
- S. J. Peroutka and T. A. Howell, Neuropharmacology, 1994, 33, 319–324 CrossRef CAS.
- T. Schöneberg, M. Hofreiter, A. Schulz and H. Römpler, Trends Pharmacol. Sci., 2007, 28, 117–121 CrossRef.
- J. R. Raymond, Y. V. Mukhin, T. W. Gettys and M. N. Garnovskaya, Br. J. Pharmacol., 1999, 127, 1751–1764 CrossRef CAS.
- U. Gether and B. K. Kobilka, J. Biol. Chem., 1998, 273, 17979–17982 CrossRef CAS.
- A. Malmberg and P. G. Strange, J. Neurochem., 2000, 75, 1283–1293 CAS.
- B. K. Kobilka, T. Frielle, S. Collins, T. Yang-Feng, T. S. Kobilka, U. Francke, R. J. Lefkowitz and M. G. Caron, Nature, 1987, 329, 75–79 CrossRef CAS.
- X. M. Guan, S. J. Peroutka and B. K. Kobilka, Mol. Pharmacol., 1992, 41, 695–698 CAS.
- A. Fargin, J. R. Raymond, M. J. Lohse, B. K. Kobilka, M. G. Caron and R. J. Lefkowitz, Nature, 1988, 335, 358–360 CrossRef CAS.
- B. S. DeDecker, R. O'Brien, P. J. Fleming, J. H. Geiger, S. P. Jackson and P. B. Sigler, J. Mol. Biol., 1996, 264, 1072–1084 CrossRef CAS.
- R. Saxena, Y. D. Paila and A. Chattopadhyay, (unpublished observations).
- X. Periole, M. Cavalli, S. J. Marrink and M. A. Ceruso, J. Chem. Theory Comput., 2009, 5, 2531–2543 CrossRef CAS.
- S. R. Jr. Krysteck, S. R. Kimura and A. J. Tebben, J. Comput.-Aided Mol. Des., 2006, 20, 463–470 CrossRef.
- S. L. Kitson, Curr. Pharm. Des., 2007, 13, 2621–2637 CrossRef CAS.
- K. G. Harikumar and A. Chattopadhyay, FEBS Lett., 1998, 438, 96–100 CrossRef CAS.
- B. Y. Ho, A. Karschin, T. Brancheck, N. Davidson and H. A. Lester, FEBS Lett., 1992, 312, 259–262 CrossRef CAS.
- T. J. Pucadyil and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2004, 1663, 188–200 CrossRef CAS.
- T. J. Pucadyil and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2005, 1714, 35–42 CrossRef CAS.
- A. G. Lee, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 62–87 CrossRef CAS.
- H. Palsdottir and C. Hunte, Biochim. Biophys. Acta, Biomembr., 2004, 1666, 2–18 CrossRef CAS.
- T. J. Pucadyil and A. Chattopadhyay, Prog. Lipid Res., 2006, 45, 295–333 CrossRef CAS.
- Y. D. Paila and A. Chattopadhyay, Subcell. Biochem., 2010, 51, 439–466 Search PubMed.
- M. Opekarová and W. Tanner, Biochim. Biophys. Acta, Biomembr., 2003, 1610, 11–22 CrossRef CAS.
- T. K. M. Nyholm, S. Özdirekcan and J. A. Killian, Biochemistry, 2007, 46, 1457–1465 CrossRef CAS.
- K. Simons and E. Ikonen, Science, 2000, 290, 1721–1725 CrossRef CAS.
- O. G. Mouritsen and M. J. Zuckermann, Lipids, 2004, 39, 1101–1113 CrossRef CAS.
- K. Burger, G. Gimpl and F. Fahrenholz, Cell. Mol. Life Sci., 2000, 57, 1577–1592 CrossRef CAS.
- Y. D. Paila and A. Chattopadhyay, Glycoconjugate J., 2009, 26, 711–720 CrossRef CAS.
- G. Gimpl, K. Burger and F. Fahrenholz, Biochemistry, 1997, 36, 10959–10974 CrossRef CAS.
- G. Gimpl, V. Wiegand, K. Burger and F. Fahrenholz, Prog. Brain Res., 2002, 139, 43–55 Search PubMed.
- L. Pang, M. Graziano and S. Wang, Biochemistry, 1999, 38, 12003–12011 CrossRef CAS.
- E. Dainese, S. Oddi and M. Maccarrone, Cell. Mol. Life Sci., 2008, 65, 2277–2279 CrossRef CAS.
- G. Khelashvili, A. Grossfield, S. E. Feller, M. C. Pitman and H. Weinstein, Proteins: Struct., Funct., Bioinf., 2009, 76, 403–417 Search PubMed.
- E. Lyman, C. Higgs, B. Kim, D. Lupyan, J. C. Shelley, R. Farid and G. A. Voth, Structure, 2009, 17, 1660–1668 CrossRef CAS.
- M. Nowak, M. Kołaczkowski, M. Pawłowski and A. J. Bojarski, J. Med. Chem., 2006, 49, 205–214 CrossRef CAS.
- Z.-Y. Yang, W. Lv, R. Tian, H.-W. Jin and H.-H. Zeng, J. Chi. Pharm. Sci., 2009, 18, 151–155 Search PubMed.
- J. C. Mobarec, R. Sanchez and M. Filizola, J. Med. Chem., 2009, 52, 5207–5216 CrossRef CAS.
- Y. D. Paila, M. R. V. S. Murty, M. Vairamani and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 1508–1516 CrossRef CAS.
- T. J. Pucadyil and A. Chattopadhyay, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 655–668 CrossRef CAS.
- P. Pavlidis, M. Ramaswami and M. A. Tanouye, Cell, 1994, 79, 23–33 CrossRef CAS.
- A. Chattopadhyay and Y. D. Paila, Biochem. Biophys. Res. Commun., 2007, 354, 627–633 CrossRef CAS.
- A. Sali and T. L. Blundell, J. Mol. Biol., 1993, 234, 779–815 CrossRef CAS.
- R. A. Laskowski, J. A. Rullmannn, M. W. MacArthur, R. Kaptein and J. M. Thornton, J. Biomol. NMR, 1996, 8, 477–486 CAS.
- R. W. W. Hooft, G. Vriend, C. Sander and E. E. Abola, Nature, 1996, 381, 272 CrossRef CAS.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS.
- L. Monticelli, S. K. Kandasamy, X. Periole, R. G. Larson, D. P. Tieleman and S. J. Marrink, J. Chem. Theory Comput., 2008, 4, 819–834 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. Di Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Multiple sequence alignments, ProSA validation and clustering analysis. See DOI: 10.1039/c0mb00148a |
| ‡ These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2011 |