Metabolic response to low-level toxicant exposure in a novel renal tubule epithelial cell system†
James Keith
Ellis
a,
Toby James
Athersuch
ac,
Rachel
Cavill
a,
Robert
Radford
b,
Craig
Slattery
b,
Paul
Jennings
d,
Tara
McMorrow
b,
Michael P.
Ryan
b,
Timothy Mark David
Ebbels
*a and
Hector Charles
Keun
*a
aBiomolecular Medicine, Department of Surgery and Cancer, Faculty of Medicine, Imperial College London, Sir Alexander Fleming Building, South Kensington, London, SW7 2AZ, UK. E-mail: h.keun@imperial.ac.uk, t.ebbels@imperial.ac.uk
bUCD School of Biomolecular & Biomedical Sciences, UCD Conway Institute, University College Dublin, Dublin, Ireland
cMRC-HPA Centre for Environment & Health, Department of Epidemiology and Biostatistics, School of Public Health, Faculty of Medicine, Imperial College London, London, UK
dDivision of Physiology, Department of Physiology and Medical Physics, Innsbruck Medical University, Innsbruck, Austria
First published on 19th November 2010
Abstract
Toxicity testing is vital to protect human health from exposure to toxic chemicals in the environment. Furthermore, combining novel cellular models with molecular profiling technologies, such as metabolomics can add new insight into the molecular basis of toxicity and provide a rich source of biomarkers that are urgently required in a 21st Century approach to toxicology. We have used an NMR-based metabolic profiling approach to characterise for the first time the metabolome of the RPTEC/TERT1 cell line, an immortalised non-tumour human renal epithelial cell line that recapitulates phenotypic characteristics that are absent in other in vitro renal cell models. RPTEC/TERT1 cells were cultured with either the dosing vehicle (DMSO) or with exposure to one of six compounds (nifedipine, potassium bromate, monuron, D-mannitol, ochratoxin A and sodium diclofenac), several of which are known to cause renal effects. Aqueous intracellular and culture media metabolites were profiled by 1H NMR spectroscopy at 6, 24 and 72 hours of exposure to a low effect dose (IC10). We defined the metabolome of the RPTEC/TERT1 cell line and used a principal component analysis approach to derive a panel of key metabolites, which were altered by chemical exposure. By considering only major changes (±1.5 fold change from control) across this metabolite panel we were able to show specific alterations to cellular processes associated with chemical treatment. Our findings suggest that metabolic profiling of RPTEC/TERT1 cells can report on the effect of chemical exposure on multiple cellular pathways at low-level exposure, producing different response profiles for the different compounds tested with a greater number of major metabolic effects observed in the toxin treated cells. Importantly, compounds with established links to chronic renal toxicity produced more diverse and severe perturbations to the cellular metabolome than non-toxic compounds in this model. As these changes can be rationalised with the different pharmacological and toxicity profiles of the chemicals it is suggested that metabolic profiling in the RPTEC/TERT1 model would be useful in investigating the mechanism of action of toxins at a low dose.
Introduction
Toxicity testing is vital to protect human health from exposure to toxic chemicals in both the clinic and the wider environment, however the process is still faced with many significant challenges. The core battery of tests for hazard identification and risk assessment are heavily reliant on extrapolation of observations in animal models to man, a process that is inherently costly, inaccurate and ethically undesirable. Direct assessment of the effects of environmental toxicants in human populations is very difficult as exposure is often at a low concentration and responses are often subtle, early events. Yet there is increasing public and legislative pressure to fill the knowledge gap that exists for many widely used chemicals in terms of possible impacts on human health, for example the Registration, Evaluation, Authorisation and Restriction of Chemicals (REACH) initiative in effect across European Union countries from 2007 (http://ec.europa.eu/enterprise/sectors/chemicals/reach/index_en.htm). In the face of these challenges, there are growing calls to make more use of human-derived cell models coupled to high-throughput molecular profiling platforms that can predict adverse effects by reporting on key cellular pathways that are perturbed by toxicants, i.e. ‘toxicity pathways’ defined at the molecular level (‘Toxicity testing in the 21st century: a vision and a strategy’: National Research Council (U.S.). Committee on Toxicity Testing and Assessment of Environmental Agents).1 Metabolic profiling (metabolomics/metabonomics) is one such strategy for cellular pathway analysis based on the untargeted analysis of the small molecule composition of a biological sample and is a powerful and flexible tool in biomarker discovery.2,3 Metabolic profiling techniques have been widely applied in the field of toxicology,4 mainly in the context of animal studies where urine or plasma have been analysed. NMR-based metabolomic studies have shown that urine analysis can predict if the kidney is a major target organ for potential toxicants5 and can reveal distinct patterns of metabolic perturbations in response to site-specific nephrotoxins.61H NMR spectroscopy has also been used to study human kidney tissue (both carcinoma and normal)7,8 and specific osmolytes have been measured in non-tumour animal renal lines.9 The integration of an in vitro cell model and metabolic profiling techniques allow the study of the intracellular metabolome and its interaction with the culture media in response to a chemical exposure. The utilisation of a human derived cell model would allow chemical screening of potentially toxic chemicals that would not require cross-species correction and may be informative of a chemicals mechanism of toxicity. We have previously demonstrated that our NMR-based approach can be utilised to study the endogenous metabolome of an in vitro cell system and observe changes in response to chemical exposure.10Renal Proximal Tubule Epithelial Cells (RPTECs) are polarised cells that line the tubule of the kidneys and possess a high metabolic activity. They are involved in blood clearance and resorption of essential metabolites and macromolecules from the glomerular ultrafiltrate.11 The study of human RPTEC cells in primary culture is difficult as these cells enter senescence after approximately 20 population doublings.12 However the development of human derived cell lines can over come this issue. The RPTEC/TERT1 cell line is a novel non-cancer derived human cell model, which contains no viral oncogenes but has been immortalised by over expression of human telomerase (hTERT).12 Characterisation of RPTEC/TERT1 cells has already revealed morphological and biochemical properties that show an improved retention of in vivo phenotype e.g. formation of tight junctions and microvilli, and RPTEC specific sodium-dependent phosphate transporters expression.12 In addition, these cells are maintained under serum-free conditions. Thus the cell line has many potential applications in toxicology, tissue engineering, basic cell biology and drug screening.12 In the present study we used an NMR based metabolic profiling approach to characterise the metabolome of the human RPTEC/TERT1 cell line and define the response of the system over a 72 hour period to low-level exposure to a selection of model compounds that affect the kidney. The current study was carried out within the EU funded Framework Program 6 carcinoGENOMICS project,13 which aims to develop omics-based in vitro screens for chemical carcinogens. We show here that the novel RPTEC/TERT1 in vitro cell model possesses metabolomic characteristics, specifically intracellular accumulation of renal osmolytes, that indicate preserved in vivo functions. Furthermore we observed that exposure to renal toxicants with different pharmacological and toxicity profiles, at levels that are not grossly cytotoxic, produced contrasting perturbations to metabolism that reflect specific physiological effects—a pre-requisite for the use of this approach in toxicity testing.
Methods
Chemicals and reagents
All cell culture plastics were from Costar. All reagents (including nifedipine, D-mannitol, sodium diclofenac, monuron, ochratoxin A and potassium bromate) were of the highest available purity from commercial sources.RPTEC/TERT1 culture and harvesting
The human renal proximal tubular epithelial cell line, RPTEC/TERT1 was originally obtained from the developing laboratory of Dr. Grillari-Voglauer (Institute of Applied Microbiology, BOKU-University of Natural Resources and Applied Life Sciences, Vienna12). Cells were maintained in Dulbecco's modified Eagle's medium Nutrient Mix F12 (1∶1) (Gibco) containing 2 mM L-Glutamine (Gibco), 36 ng mL−1 hydrocortisone (Sigma), 10 ng mL−1 recombinant human epidermal growth factor (EGF, Sigma), 50 U mL−1 penicillin, 50 μg mL−1 streptomycin (Gibco), 5 μg mL−1 insulin, 5 μg mL−1 transferrin and 5 ng mL−1 selenium (ITS(Sigma)). Culture medium was replaced with fresh medium every 48 hours. Cells were maintained in 75 cm2 Costar flasks at 37 °C in a humidified atmosphere containing 95% air and 5% CO2. For compound treatments, cells were grown to confluency and cultured for 10 days prior to experimentation. Control cells were exposed to an appropriate concentration of vehicle control (0.1% v/v dimethyl sulfoxide (DMSO)). Cells were exposed to the indicated test compound (nifedipine (10 μM), D-mannitol (10 mM), sodium diclofenac (30 μM), monuron (200 μM), ochratoxin A (300 nM), potassium bromate (200 μM)) for 6, 24 or 72 hours.For metabolite analysis (at 6, 24 and 72 h), 1 mL of the culture media was transferred to a fresh Eppendorf™ tube, centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 5 min) to remove dead cells and stored at −80 °C for subsequent analysis. The remaining media was aspirated and each well was washed twice with ∼1 mL of Phosphate Buffered Saline (PBS) to remove trace media and the wash solution aspirated. The RPTEC/TERT1 cells were harvested by the addition of 1 mL of methanol to each well (methanol quenching),14 scraping and transferred to an Eppendorf™ tube. Each well was washed again with 1 mL of methanol and the two samples pooled. The pooled methanol quenched sample (2 mL) was dried under a flow of nitrogen gas at room temperature and stored at −80 °C for subsequent extraction.
000 rpm, 5 min) to remove dead cells and stored at −80 °C for subsequent analysis. The remaining media was aspirated and each well was washed twice with ∼1 mL of Phosphate Buffered Saline (PBS) to remove trace media and the wash solution aspirated. The RPTEC/TERT1 cells were harvested by the addition of 1 mL of methanol to each well (methanol quenching),14 scraping and transferred to an Eppendorf™ tube. Each well was washed again with 1 mL of methanol and the two samples pooled. The pooled methanol quenched sample (2 mL) was dried under a flow of nitrogen gas at room temperature and stored at −80 °C for subsequent extraction.
Immunofluorescent microscopy
E-cadherin and Zonula Occludin-1 (ZO-1) protein expression was visualised using indirect immunofluorescence. Briefly, RPTEC/TERT1 cells were grown to confluency on Falcon 8-chamber slides and cultured for 10 days. Cells were fixed using 3.7% formaldehyde/PBS and then permeabilised with 0.2% (v/v) Triton X-100/PBS. For staining cells were incubated with the respective primary antibody (1∶200—ZO-1 (Zymed), 1∶400—E-cadherin (Molecular Probes)) for 2 hours at room temperature. Cells were then incubated with a 1∶200 dilution of the appropriate Alexa488 conjugated secondary antibody (Molecular Probes, Invitrogen). Nuclei were stained using 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI). Slides were mounted and viewed using a Zeiss LSM 510 Meta confocal microscope.
Sample preparation for 1H NMR spectroscopy
Aqueous soluble metabolites were extracted from cultured RPTEC/TERT1 cells using a chloroform/methanol/water extraction method. Briefly, 300 mL of chloroform/methanol (2∶1) solution was added to each Eppendorf™ tube containing the dried methanol quenched sample. The sample was then vortex mixed for 30 seconds, 300 mL of ultrapure water added and vortex mixed again. The sample was centrifuged (16000g, 10 min) and the aqueous and organic layers removed to separate sample tubes (the organic phase was not analyzed in the present study). The extraction process was repeated for each sample and the aqueous sample pooled and left to evaporate at room temperature for ∼12 hours to remove any organic solvent. The aqueous extract was then freeze dried, reconstituted in 600 μL of phosphate buffer (0.2 M Na2HPO4, 0.043 M NaH2PO4, 100 μM TSP, 3 mM NaN3 in 100% D2O) and centrifuged (16000g, 5 min); of which 550 μL was transferred to a standard 5 mm glass NMR tube for spectroscopic analysis.
The culture media was prepared for spectroscopic analysis by the addition of 50 μL of D2O (containing 0.2% TSP (w/v)) to 550 μL of media sample, vortex mixed and 550 μL (TSP: 0.97 mM) transferred to a standard 5 mm glass NMR tube.
All reagents were checked prior to sample preparation by obtaining a 1D 1H NMR spectrum to ensure that they contain no contaminants that may interfere with the downstream spectroscopic analysis.
1H NMR spectroscopy
High-resolution, 1D, 1H NMR spectra of reconstituted intracellular aqueous extracts and culture media were acquired at 14.1 T (600.13 MHz 1H frequency) using a Bruker AVANCE 600 spectrometer (Bruker Biospin, Rheinstetten, Germany). A 5 mm broadband-inverse tube probehead held at 300 K was used for all NMR experiments. A BACS 60 automated sample changer (Bruker BioSpin) was used for sample management during the analytical run, with acquisitions controlled using Xwin-NMR/Icon-NMR (Bruker Biospin). All spectra were acquired from randomised cellular or media samples. Following sample introduction to the probehead, gradient shimming was used to improve the magnetic field homogeneity. Carr–Purcell–Meiboom–Gill (CPMG) 1H spectra were acquired using the pulse sequence (RD-90°-[τ-180°-τ]n-AQ). The fixed echo time, τ was set to 400 μs, giving a total spin-echo time of 64 ms. During the acquisition period (AQ, 2.73 s), the free induction decay (FID) was recorded into 64 k data-points in the time domain, with a spectral width of 20 ppm. Typically, spectra were recorded as the sum of 128 transients following 16 dummy scans. In both cases, suppression of the water resonance centered at δH = 4.7 ppm was achieved by the application of a presaturation pulse during the relaxation delay (RD, 3 s). Prior to Fourier transformation, all FIDs were multiplied by an exponential function equivalent to a line broadening of 0.3 Hz. Assignment of resonances to specific metabolites was based on the Chenomx profiler in the NMR suite 6.1 software (Chenomx Inc, Alberta, Canada), on the addition of known standards to the biological samples, published literature,15 in-house assignment databases and statistical total correlation spectroscopy16 (STOCSY).NMR data and statistical analysis
Data were imported and manipulated in Matlab (Mathworks) using in-house software written and compiled by Dr T. M. D. Ebbels, Dr H. C. Keun, Dr J. T. Pearce, and Dr R. Cavill. 1H NMR spectra were automatically phased, baseline corrected, and referenced and normalised to the TSP resonance at δ 0. For all subsequent analysis culture media data were normalised to TSP but the aqueous intracellular fraction data were normalised to the median fold change.17 The spectra were digitized using Matlab script developed in-house. Two levels of data resolution were analyzed: full resolution spectra at 32697 data points and reduced resolution spectra that comprised of 166 (intracellular fraction) and 96 (media) data points of varying ppm width. The data points in the reduced resolution data for both the intracellular and media analysis were created using a targeted binning approach. After metabolite identification (see previous section) manually selected regions of the full resolution spectra were integrated, where possible if a metabolite had multiple resonances they were integrated together in a single region, and unassigned resonances were also integrated. Manual integration reduces the signal multiplicity of a metabolite as multiple resonances from the same molecule can be integrated together, chemical shift can be corrected in some cases by integrating a larger spectrum width and almost all the empty regions of the spectrum are removed. Resonances that were identified as a treatment compound were removed from the analysis (both intracellular and media), as were resonances that were only present in a single batch or treatment group, were unable to be identified as a metabolite and demonstrated no time response. D-Mannitol was the only dosing compound which was detectable in the study, in both the intracellular fraction and the culture media. The low concentration (IC10) of the other compounds meant that they were present at levels below the limit of detection of the 1H NMR spectroscopy we performed.
For multivariate analysis the reduced resolution data were exported to SIMCA-P (Umetrics, USA). Principal Component Analysis (PCA) was applied to the reduced resolution data (intracellular and media) to reveal treatment related effects, as well as monitoring RPTEC/TERT1 metabolic response over time (6–72 h) or batch (2 batches) related effects. A batch related effect was observed in both the intracellular fraction and the culture media. To correct for this batch effect all subsequent analysis was carried out separately for each batch, each batch included 3 toxins and a matched control; Batch 1: control 1, nifedipine, potassium bromate, monuron and batch 2: control 2, D-mannitol, ochratoxin A, sodium diclofenac. A time related effect was also observed in both batches over the 72 h time course of the study.
Relative concentrations of media metabolites were converted to a concentration (mM) using the integrated TSP region (0.97 mM). Both the relative intracellular concentrations and media mM concentrations were expressed as change from matched control, where control concentration is equal to one, and the standard error of the mean calculated.
Results
In order to define the metabolome of the RPTEC/TERT1 model and the impact on global metabolic profiles of toxicant exposure, cells were cultured with either the dosing vehicle (DMSO) or with exposure to one of six compounds several of which are known to cause renal effects. These chemicals were selected from the carcinoGENOMICS project phase I model compound list13 and included a mixture of toxicants, primarily human carcinogens (monuron—MON; ochratoxin A—OTA; and potassium bromate—KBrO3), non-toxins with pharmacological activity (sodium diclofenac—DIC; nifedipine—NIF) and one non-toxic control (D-mannitol—MAN). These compounds do not require metabolic activation and the site of action for all is exclusively, or includes, the kidney.13 For this metabolomic study exposures of minimal cytotoxicity (10% of the Inhibitory Concentration (IC10)) were selected on the basis of both resazurin (Alamar Blue™) reduction and lactate dehydrogenase (LDH) release. Where an IC10 could not be accurately determined, as in the case of D-mannitol, a default concentration of 10 mM was used. The IC10 values were determined for the other 5 compounds to be 200 μM (MON), 300 nM (OTA), 1 mM (KBrO3), 30 μM (DIC) and 10 μM (NIF). RPTEC/TERT1 cells (with metabolism quenched by methanol, see Methods) and culture media were harvested at 6, 24 and 72 hours of exposure. Media was centrifuged and the supernatant frozen while cells were extracted post harvest using a chloroform/methanol/water extraction. The aqueous portion of this extract and culture media were profiled using 1D 600 MHz 1H NMR spectroscopy using standard protocols and annotated using a combination of prior literature and currently available metabolome databases.RPTEC/TERT1 morphology has been shown to be similar to primary RPTECs12 by the formation of dome structures and the expression of E-cadherin and Zonula Occluden-1. The RPTEC/TERT1 cells in the present study also demonstrated these characteristics (Fig. 1A–D). Representative 1D spectra (CPMG spin echo) of both the RPTEC/TERT1 intracellular extract and media samples are shown in Fig. 1 (E and F), with resonances from all identified metabolites highlighted. In total 20 metabolites could be identified in each of the two sample types, 8 of which were common to both matrices. Most of the intracellular metabolites detected are common to 1H NMR analysis of other epithelial cell cultures and observed in 1H MAS-NMR analysis of intact renal tissue. Interestingly, the metabolite betaine was observed to be at high intracellular abundance and consistently produced the highest intensity resonance in all spectra from control extract samples. Betaine, together with myo-inositol and glycerophosphocholine are typically observed in high abundance in renal tissue8,18,19 and non-tumour animal renal lines9 by NMR where they play an osmoprotective/osmoregulatory role.
 | ||
| Fig. 1 RPTEC/TERT1 cells were grown to confluency in 6-well plates or Falcon chamber slides, and cultured for 10 days. (A and B) Cell morphology was assessed by phase contrast microscopy at 100× MAG (A) and 200× MAG ((B) arrows indicate fluid-filled domes). E-cadherin (C) and Zonula Occluden-1 (ZO-1(D)) protein expression was visualised using indirect immunofluorescence and a confocal microscope as described in materials and methods (630× MAG). Typical full resolution 600 MHz 1H CPMG spin-echo NMR spectra (median fold change normalised) of aqueous intracellular extract (E) and culture media (F) in a representative spectrum of the RPTEC/TERT1 cell line. Spectra shown are from the control group of batch 1. Metabolites: 1: Isoleucine, 2: Leucine, 3: Valine, 4: EtOH, 5: Threonine, 6: Lactate, 7: Alanine, 8: Lysine, 9: 5-Oxoproline, 10: Glutamine, 11: Pyruvate, 12: DMSO, 13: Tyrosine, 14: Glucose, 15: Myo-inositol, 16: Histidine, 17: 1-methylnicotinamide, 18: NAD+/NADP+, 19: Tryptophan, 20: Contaminant, 21: GSSG, 22: β-alanine, 23: GSH, 24: Aspartate, 25: Phosphocholine, 26: Glutamate, 27: Phenylalanine, 28: Glycerophosphocholine, 29: Betaine, 30: Glycine, 31: MeOH, 32: ATP/ADP/AMP, 33: Formate, 34: Phenol. Aromatic region (5.8–9.5 ppm) in the spectrum of the intracellular fraction (E) is at 3.5 times vertical expansion. Aromatic region (6.8–8.5 ppm) in the spectrum of the media (F) is at 24 times vertical expansion. | ||
To characterise the global impact of compound exposure on the metabolite profile we used a pattern recognition approach based on PCA of normalised peak integrals for all observable and resolvable features (166 and 96 for intracellular and extracellular profiles respectively) in the NMR data. Samples for this study were generated in two separate experiments each containing vehicle controls but with different treatments, hence two models were generated at every time point analysing data from each experiment separately to remove the influence of any ‘batch’ effect on the analysis. Pattern recognition analysis revealed that all of the treatments examined produced consistent perturbations from vehicle controls in terms of the intracellular metabolite profile in at least one time point. As all the toxicants (MON, OTA, KBrO3) produced such effects at 72 h we focussed on models from this time point to assist with the process of selecting key metabolites that could report on potential toxicity in this model (Fig. 2A–D). Loading plots for PCA models from each experimental batch were very similar indicating that the same metabolites were co-varying in both sets of exposures. From the effect on PC1 (the first principal component) scores it appeared that MON decreased levels of betaine while increasing levels of GPC, alanine, glutamate, glutamine and glutathione. OTA appeared to produce an opposite effect to MON, while the non-toxin DIC produced a broadly similar response in this component. KBrO3 caused a separation in an orthogonal component, and thus affected a different set of metabolites, specifically decreasing phosphocholine, and myo-inositol. Neither of the remaining non-toxins produced a consistent perturbation from control at this timepoint. A similar analysis of the extracellular medium at 72 h showed that only MON and DIC produced a consistent perturbation from control, in both cases an apparent increase in lactate production and glucose consumption (Fig. 2E–H). However other metabolites were observed to cause significant variation in the extracellular profiles: 3-hydroxyisovalerate (3-HV), alanine, pyruvate, 5-oxoproline (pyroglutamate), isoleucine, lysine, glutamine, phenylalanine, tyrosine, tryptophan, threonine and leucine.
 | ||
| Fig. 2 The response of the RPTEC/TERT1 cell line to specific toxins at 72 hours in both the aqueous cellular extract and the culture medium. Principal component analysis (PCA) was applied to the intracellular and media metabolic profiles to assess treatment related effects. The reduced resolution data (intracellular = 166 data points/media = 96 data points of varying ppm width) were Pareto scaled in all models. NIF—Nifedipine, KBrO3—Potassium Bromate, MON—Monuron, MAN—D-Mannitol, OTA—Ochratoxin A, DIC—Sodium Diclofenac. PCA scores plots (A, E, C and G) and accompanying loadings plots (B, F, D and H). PC = Principal component. G and H = Phenol resonances removed from analysis. | ||
To define more quantitatively the effect of chemical exposure on this metabolite panel, resonances associated with each metabolite were integrated and the fold change in normalised intensity relative to matched vehicle control was calculated at each time point for all treatments. While the majority (116/180) of treatment effects observed for the intracellular panel were of low magnitude (<|1.2| fold), five metabolites exhibited major changes (>|1.5| fold) in intracellular levels for at least one experimental condition: lactate, alanine, GPC, choline and betaine (Table 1). From the extracellular metabolite panel, six metabolites exhibited major differences in extracellular concentration compared to controls at the end of the experiment (72 h) for at least one condition: lactate, alanine, isoleucine, tryptophan, pyruvate and glucose (Table 2). These major perturbations were unevenly distributed across the different treatments: MON produced the greatest number of effects, followed by two other toxicants OTA and DIC, while KBrO3, NIF and MAN produced the fewest major changes. Furthermore, significant variation was observed between treatments in the direction and magnitude of metabolic perturbations. Hence each chemical exposure appeared to produce a distinct pattern of metabolic responses, reflecting the diversity in the biological properties of the compounds tested in this study. These are described in further detail below and a complete list of all observed perturbations to the targeted metabolite panel for all experimental conditions is available online (ESI†, Table S1).
| Metabolite | Treatment compound | ||||
|---|---|---|---|---|---|
| Nifedipine | Monuron | Mannitol | Ochratoxin A | Diclofenac | |
| Metabolites shown in the above table exhibited major changes (>1.5 fold) in intracellular levels for at least one experimental condition. Control = 1. GPC = glycerophosphocholine. | |||||
| Betaine | — | 0.67 (24 h), 0.25 (72 h) | — | — | 0.53 (72 h) |
| GPC | 0.48 (24 h) | 1.76 (72 h) | 0.67 (72 h) | — | — |
| Lactate | — | 0.32 (72 h) | — | 1.61 (6 h), 0.60 (72 h) | 1.55 (24 h) |
| Choline | — | 0.38 (72 h) | 0.52 (72 h) | — | — |
| Alanine | — | 1.62 (72 h) | — | 0.58 (72 h) | — |
| Metabolite | Treatment compound | |||
|---|---|---|---|---|
| Monuron | Mannitol | Ochratoxin A | Diclofenac | |
| Metabolites shown in the above table exhibited major differences (>1.5 fold) in extracellular concentration compared to controls at the end of the experiment (72 h) for at least one condition. Control = 1. | ||||
| Lactate | 2.01 ± 0.05 | — | — | 2.78 ± 0.03 |
| Alanine | — | — | 0.57 ± 0.03 | — |
| Isoleucine | 0.59 ± 0.29 | — | — | — |
| Tryptophan | — | 0.23 ± 0.11 | — | 2.66 ± 0.22 |
| Pyruvate | — | — | 1.66 ± 0.03 | 2.08 ± 0.15 |
| Glucose | 0.34 ± 0.01 | — | — | 0.33 ± 0.07 |
Perturbations to renal osmolytes and choline metabolism
Fig. 3A and B presents treatment-related effects to intracellular betaine and GPC levels across all treatment conditions. While GPC and betaine are both abundant renal osmolytes and accumulate with increases in extracellular tonicity, we observed both increases and decreases in intracellular levels and a lack of coordination in response between these metabolites across treatments, indicating that perturbations to osmoregulation and/or non-specific osmotic stress were unlikely to be primarily responsible for these metabolite changes. Intracellular levels of betaine, likely to reflect a normal, differentiated renal epithelial phenotype, were generally observed to be lower after treatment, with major decreases observed in cells exposed to monuron (up to 4-fold, p = 0.003, 72 h) and DIC (up to 1.9 fold, p = 0.05, 72 h). OTA was the only treatment to cause an increase in betaine (1.29 fold, p = 0.1, 72 h). Levels of choline metabolites are well known to be altered in states of hyperproliferation, e.g. in malignancy, and we have previously reported decreases in intracellular GPC in cultured rat hepatocytes as a result of inhibiting entry into cell cycle.10 In the current study most treatments caused minor reductions in GPC, with a major decrease observed for NIF (up to 2.1-fold, p = 0.02, 24 h). Interestingly, two carcinogens exhibited the opposite response, with major increases in GPC observed after exposure to MON (up to 1.76 fold, p = 0.0008, 72 h) and lesser increases observed in OTA exposed cells at earlier timepoints (up to 1.28 fold, p = 0.04, 6 h). This implied that exposure to these specific compounds at a low, non-toxic dose produced a metabolic state associated with promotion of proliferation and survival. Choline is a precursor to both betaine and GPC which is not directly synthesised but is instead taken up from the culture medium. However perturbations to intracellular choline levels (Fig. 3C) were not generally correlated to either GPC or betaine levels, and did not distinguish clearly toxicants from non-toxicants. Decreases were observed in choline levels for four treatments at 72 h: NIF (1.4 fold, p = 0.005, 72 h), KBrO3 (1.3 fold, p = 0.02, 72 h), MON (2.6 fold, p = 0.01, 72 h) and MAN (1.9 fold, p = 0.6, 72 h). MON exposure produced the greatest fall in both choline and betaine levels, so it was possible that for particular treatments reduced choline uptake and subsequently choline availability contributed to alterations in other metabolites. Generation of betaine from choline involves the mitochondrial enzymes choline oxidase and betaine aldehyde dehydrogenase. Thus variation in betaine may also reflect perturbations to mitochondrial metabolism.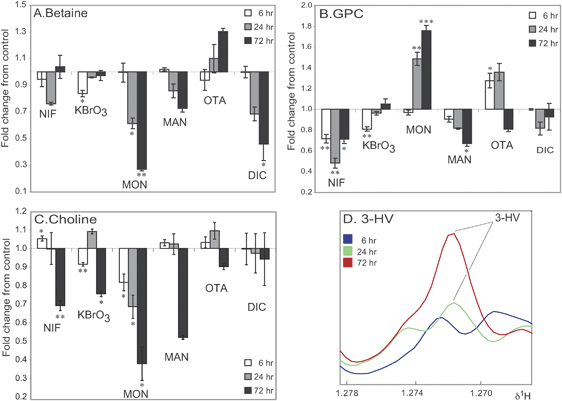 | ||
| Fig. 3 Effect of compound treatment on the concentration of three intracellular metabolites and the effect of ochratoxin A treatment on extracellular 3-HV concentration. (A) Change in relative concentration (fold change relative to control) of intracellular betaine levels in all six treatment groups. (B) Change in relative concentration (fold change relative to control) of intracellular GPC levels in all six treatment groups. (C) Change in relative concentration (fold change relative to control) of intracellular choline levels in all six treatment groups. (D) Averaged 1H CPMG spin-echo NMR spectra (n = 3 per timepoint) showing the increase in 3-HV concentration in the media of OTA treated RPTEC/TERT1 cells. 3-HV = 3-hydroxy-isovalerate. NIF—Nifedipine, KBrO3—Potassium Bromate, MON—Monuron, MAN—D-Mannitol, OTA—Ochratoxin A, DIC—Sodium Diclofenac. Significance compared to control assessed by Welch's t-test. NIF, KBrO3 and MON compared to control 1 and MAN, OTA and DIC compared to control 2. Control = 1. Significance marked as: *p < 0.05, **p < 0.01, ***p < 0.001. Significance values indicated are without multiple testing correction. Upon Bonferroni correction fold changes with a p < 0.001 remain valid (q < 0.05). | ||
Perturbations to glycolysis, the fate of pyruvate and mitochondrial metabolism
Glucose is the primary energy substrate for cultured mammalian cells and after absorption from the extracellular medium is converted to pyruvate during glycolysis with concomitant production of ATP and NADH. We observed major increases in total amount of glucose removed from the media after 72 h exposure to MON (2.9 fold, p = 0.03) and DIC (3.0 fold, p = 0.02). In our study, using low cytotoxic doses, these alterations in glucose uptake from the culture medium are likely to reflect specific perturbations in the rate of glycolysis. In addition to glucose consumption we also observed a net accumulation of extracellular pyruvate, lactate and alanine. Cytosolic pyruvate generated by glycolysis has a number of fates, primarily: transport/diffusion from the cell; anaerobic conversion to lactate to recover NAD+; oxidation via the TCA cycle in mitochondria; or conversion to alanine via transamination with glutamate. Mitochondrial metabolism is the most energy efficient route, generating up to an additional 36 ATP versus 2 ATP from glycolysis alone. Lactate accumulation over 72 h was also markedly increased with both MON (2-fold, p = 0.004) and DIC (2.8 fold, p = 0.007) exposure. The magnitude of effect observed was comparable to the increase in glucose uptake with these treatments, suggesting that both compounds caused an increase in glycolysis without substantially increasing the rate of oxidative metabolism and that much of the excess pyruvate was converted rapidly to lactate.While no other treatment produced significant differences in lactate accumulation by the end of the study, OTA exposure did produce major decreases in extracellular lactate levels within the first 24 h (2-fold, p = 0.09) without a substantial increase in glucose uptake over the same period (1.1 fold, p = 0.9). This indicated that this compound had a very different impact on energy metabolism than DIC or MON, possibly ‘enhancing’ mitochondrial metabolism and increasing the proportion of pyruvate consumed via oxidation. Supporting this hypothesis, we observed that OTA was the only treatment to (a) increase betaine levels, (b) to affect alanine accumulation, reducing this by 1.7 fold (p = 0.01), and (c) was also the only exposure which produced NMR-detectable levels of the metabolite 3-HV (Fig. 3D). 3-HV is the product of irreversible hydrolysis of 3-methylcrotonyl-CoA an intermediate in leucine catabolism produced by the mitochondrial enzyme isovaleryl-CoA dehydrogenase,20,21 and the appearance of this metabolite is consistent with an overall increase in the rates of mitochondrial reactions. Finally, major increases in pyruvate accumulation were observed after 72 h exposure to all three toxicants, DIC (2.1 fold, p = 0.008), OTA (1.7 fold, p = 0.006) and to a lesser extent MON (1.4 fold, p = 0.05). This might result from increased intracellular concentration of pyruvate which is a plausible consequence of both (a) increased glycolysis without an increase in oxidation as apparent for DIC and MON treatment, and (b) suppression of anaerobic conversion of pyruvate to lactate or alanine with a concomitant increase in oxidative metabolism as hypothesised for OTA treatment.
While it was not possible to detect intracellular pyruvate by 1H NMR, we were able to observe major perturbations to intracellular lactate and alanine levels after treatment with DIC, OTA or MON, supporting the interpretation that the pyruvate production is altered with these treatments. However no consistent relationship was apparent between intracellular levels of each metabolite, nor between intracellular levels and extracellular accumulation. One likely contribution to these discrepancies is the fact that the composition of the extracellular media represents a time-average of the metabolic behaviour of the cells whereas extraction of intracellular metabolites provides a ‘snap-shot’ of transient metabolic status. These temporal relationships are further complicated by feedback processes, i.e. reuptake of exported metabolites. For example in our study, lactate accumulation appeared to peak in control cultures within the first 24 h (0.07 mmol h−1), with a small net consumption of lactate occurring during the subsequent 48 h in spite of continuing glucose uptake (−0.06 mmol h−1 between 6–24 h and −0.03 mmol h−1 between 24–72 h). Hence some recycling of extracellular lactate and other sources of pyruvate was likely in all treatment conditions and the net accumulation of these substrates will also be dependent on the capacity for reabsorption via mono-carboxylate transporters.
Perturbations to glutathione metabolism
While glutathione could be detected by 1H NMR of cell extracts, it was not possible to distinguish between oxidised (GSSG) and reduced (GSH) forms of the molecule. Two treatments increased the total glutathione pool, MON (1.32 fold, p = 0.02, 72 h) and OTA (1.23 fold, p = 0.02, 72 h), which may reflect a tendency to increased proliferation. No treatment produced a decrease in total glutathione abundance. Levels of 5-oxoproline, an intermediate in the recycling of cysteine from GSH,22 was also observed to accumulate in the culture medium. 5-Oxoproline is a cyclic lactam form of glutamate that accumulates upon exposure to glutathione depleting toxicants (e.g. bromobenzene)19 and in congenital defects such as glutathione synthase deficiency23 and oxoprolinase deficiency associated with oxidative stress, metabolic acidosis, neurotoxicity.22 While no differences were observed in 5-oxoproline accumulation at the end of the study, a minor increase was detected after 24 h of exposure to nifedipine (1.25 ± 0.02, 24 h) and potassium bromate (1.31 ± 0.02, 24 h). This could indicate that these treatments caused a transient increase in GSH turnover and an increase in oxidative stress. However, 5-oxoproline can also form from spontaneous cyclisation of glutamine in aqueous solution, a reaction made more favourable by non-physiological pH. Hence these effects could reflect a transient shift in acid/base balance during culture.Discussion
Our data show that metabolic profiling of RPTEC/TERT1 cells can report on the effect of chemical exposure on multiple cellular pathways at low-level exposure, producing different response profiles for the different chemicals tested. Importantly, compounds with established links to chronic renal toxicity produced more diverse and severe perturbations to the cellular metabolome than non-toxic compounds in this model. An over view of the major metabolic changes and how they are associated with chemical treatments is shown in Fig. 4. | ||
| Fig. 4 Overview of the intracellular and extracellular metabolic changes in the RPTEC/TERT1 cell line associated with compound exposure. Red indicates that the treatment compound increases the metabolite. Blue indicates that the treatment compound decreases the metabolite. | ||
Many of our observations concur with known biological properties of our chemical panel.
Ochratoxin A is a mycotoxin produced by some fungi and a common contaminant of foodstuffs. It has been classified as a carcinogen in the rodent kidney (reviewed in Mally and Dekant)24 but it's mechanism of action is unclear as there is contrasting empirical evidence. However, it seems unlikely that it is directly genotoxic as there is little evidence for ochratoxin A–DNA adducts25–28 or 8-oxodG29,30 formation but it has been suggested that ochratoxin A mutagenesis is based on oxidative stress.31
At low concentration (nM) OTA has been shown to increase aerobic metabolism or “stimulate” mitochondria.32 A finding that is confirmed in our study where we observe a decrease in extracellular lactate with no accompanying uptake of glucose and increased excretion of pyruvate and 3-HV. Additionally, two studies suggest33,34 that OTA impairs antioxidant defence in the kidney by the down regulation of Nrf-2 dependant gene expression resulting in abasic site formation in DNA both in vitro and in vivo. OTA induced aerobic metabolism, increased according to our findings, and impaired antioxidant defence would increase oxidative stress via the increased production and reduced detoxification of H2O2.
A previous metabonomic study carried out in the rat suggested alteration of energy metabolism and mitochondrial function when the animals were dosed with 0–210 μg kg−1 body weight35 of OTA, where urinary citrate and 2-oxoglutarate levels were observed to decrease. Additionally, urinary 5-oxoproline (glutathione depletion or oxidative stress) concentration was increased, which is an effect we do not observe in the RPTEC/TERT1 in vitro model. Of course, the rodent study by Sieber et al. represents systemic changes at higher doses than the present study and OTA is also active in the liver.
The increase in intracellular GPC levels associated with OTA treatment is suggestive of increased proliferation and survival, both of which are facets of a malignant cancer phenotype. MON treatment was more strongly associated with an increase in cellular proliferation, than OTA, as a greater increase in intracellular GPC levels was observed, which may be expected as both MON and OTA are carcinogenic. MON is a herbicide that causes renal and hepatic carcinogenesis in the male rat through a non-genotoxic mechanism of action.36 Treatment of the RPTEC/TERT1 cell line with MON caused multiple changes to the intracellular and media metabolite profile, suggesting that processes in addition to proliferation, were affected. MON, as well as DIC, both had a negative effect on mitochondrial metabolism. DIC is a widely used analgesic and anti-inflammatory drug and can be hepatotoxic,37 with no genotoxic or carcinogenic action.38 Although MON and DIC are considered as different classes of compound (MON is considered a carcinogen and DIC is non-carcinogenic), both show an increase in glycolysis without an increase in mitochondrial oxidative metabolism. Considered together, these changes and the decreased amounts of betaine (both compounds) and choline (MON only) indicate that mitochondrial metabolism may be perturbed by these treatments.
DIC may also be altering the transportation of lactate and pyruvate across cellular membranes. In a primary epithelial cell line39 (human retinal origin) and immortalised cell line40 (human corneal origin) DIC exposure causes activity changes in the monocarboxylate transporters (SMCT1 and MCT1/MCT4). These effects are of course observed in a very specific cell type but SMCT1 and SMCT2 are expressed in the kidney41 and are involved in lactate and pyruvate transport.
KBrO3 did not cause any “major” changes (<1.5 fold) to the endogenous metabolic profile of the RPTEC/TERT1 cell line in the present experiments. In a previous NMR-based metabonomic study in the rat,42 it was observed that a 2 week dose of KBrO3 (100 and 200 ppm) caused no treatment related changes to the endogenous urinary metabolite profile. A minor increase was detected in extracellular 5-oxoproline levels after 24 h of exposure to NIF and KBrO3. NIF is a dihydropyridine calcium channel blocker and is used as a antihypertensive drug43 and KBrO3 induces tumourigenesis in the kidney of rats and mice,44 and acts at least in part through oxidative DNA damage.45,46 5-Oxoproline is an intermediate in the γ-glutamyl cycle of glutathione metabolism and a reduction of GSH synthesis can result in a raised level of 5-oxoproline.22 5-Oxoprolinuria has been observed in rats that have been dosed with chemicals that induce glutathione or cysteine depletion.19,47 KBrO3 damages DNA and causes the formation of 8-oxodG lesions in the rodent kidney48 and NIF has been shown to be photosensitive and undergo oxidation when exposed to light.49–51 Both treatments could cause a transient increase in GSH turnover, whilst not affecting the concentration of the glutathione pool, and increase oxidative stress.
There are some caveats to the approach used in the current study to examine the metabolic effects of the treatment compounds, specifically the quenching of cellular metabolism. The method used to quench the metabolism of the RPTEC/TERT1 cells has been shown to improve the recovery of some metabolites14 in an epithelial cell line but it is not clear if the quenching efficiency is the same for different cell types. It is clear that certain metabolites, such as the adenosine phosphate pool (ATP/ADP/AMP) or NAD+/NADP+, will be particularly sensitive to quenching and extraction techniques, and it may be necessary to optimise these methods for each cell line. Extracellular metabolite measurements are far less difficult to quench as this is effectively done by removing the media from the cells, for adherent cell lines such as the RPTEC/TERT1, which instantly stops the exchange of metabolites between cells and media. Whereas intracellular metabolite profiles are a snap-shot of the metabolism at the particular time of quenching, the extracellular (media) profiles are an accumulation of the metabolic activity over the period of time up to the point of collection. It is because of this quenching at a single time point that the intracellular metabolite profile is more susceptible to variability due to the timing of sampling. Media (extracellular metabolites) profiles are more stable as they represent an accumulation of metabolites over the time course of the study. Acquisition of a cell culture media NMR spectrum is quicker than the aqueous fraction as it requires less preparation in comparison and it may also be possible to increase the coverage of metabolites detected in culture media by the use of UPLC- and GC-MS.
By combining metabolomics and in vitro human cell models, like the RPTEC/TERT1 line, it may be possible to elucidate the pathways and biological processes that are perturbed by toxins. The National Research Council (NRC) has developed a long-range vision and strategy to advance toxicity testing in the 21st century.1 Here the NRC has referred to “toxicity pathways”, which are the pathways that when sufficiently perturbed can cause adverse health effects in humans. It is in systems such as the one utilised in our study (metabolic profiling and the RPTEC/TERT1 cell system) that these pathways could be defined and the response to toxicants characterised.
We suggest that when perturbations to multiple pathways are observed using metabolic profiling at a low dose a compound is likely to be toxic to the cell type examined. Furthermore a response signature that combines an increased tendency to proliferation and survival, an effect to mitochondrial metabolism and alterations in key renal osmolytes are an indication that a compound may be carcinogenic.
Acknowledgements
This study was funded primarily by the EU 6th Framework grant “carcinoGENOMICS” (PL-037712). This work was also supported in part by the Conway Institute for Biomolecular and Biomedical Research, University College Dublin, under the Programme for Research in Third Level Institutions (PRTLI) administered by the Higher Education Authority of Ireland.References
- S. Gibb, Reprod. Toxicol., 2008, 25, 136–138 CrossRef CAS.
- J. K. Nicholson, J. C. Lindon and E. Holmes, Xenobiotica, 1999, 29, 1181–1189 CrossRef CAS.
- O. Fiehn, Plant Mol. Biol., 2002, 48, 155–171 CrossRef CAS.
- M. Coen, E. Holmes, J. C. Lindon and J. K. Nicholson, Chem. Res. Toxicol., 2008, 21, 9–27 CrossRef CAS.
- T. M. Ebbels, H. C. Keun, O. P. Beckonert, M. E. Bollard, J. C. Lindon, E. Holmes and J. K. Nicholson, J. Proteome Res., 2007, 6, 4407–4422 CrossRef CAS.
- E. Holmes, F. W. Bonner, K. P. Gartland and J. K. Nicholson, J. Pharm. Biomed. Anal., 1990, 8, 959–962 CrossRef CAS.
- D. Moka, R. Vorreuther, H. Schicha, M. Spraul, E. Humpfer, M. Lipinski, P. J. Foxall, J. K. Nicholson and J. C. Lindon, J. Pharm. Biomed. Anal., 1998, 17, 125–132 CrossRef CAS.
- A. R. Tate, P. J. Foxall, E. Holmes, D. Moka, M. Spraul, J. K. Nicholson and J. C. Lindon, NMR Biomed., 2000, 13, 64–71 CrossRef CAS.
- T. Nakanishi, R. S. Balaban and M. B. Burg, Am. J. Physiol., 1988, 255, C181–C191 CAS.
- J. K. Ellis, P. H. Chan, T. Doktorova, T. J. Athersuch, R. Cavill, T. Vanhaecke, V. Rogiers, M. Vinken, J. K. Nicholson and M. D. Ebbels, et al. , J. Proteome Res., 2010, 9, 413–419 CrossRef CAS.
- P. L. Smith, D. A. Buffington and H. D. Humes, Methods Enzymol., 2006, 419, 194–207 CAS.
- M. Wieser, G. Stadler, P. Jennings, B. Streubel, W. Pfaller, P. Ambros, C. Riedl, H. Katinger, J. Grillari and R. Grillari-Voglauer, Am. J. Physiol. Renal Physiol., 2008, 295, F1365–F1375 CrossRef CAS.
- M. Vinken, T. Doktorova, H. Ellinger-Ziegelbauer, H. J. Ahr, E. Lock, P. Carmichael, E. Roggen, D. J. van, J. Kleinjans and J. Castell, et al. , Mutat. Res., 2008, 659, 202–210 CAS.
- Q. Teng, W. Huang, T. W. Collette, D. R. Ekman and C. Tan, Metabolomics, 2009, 5, 199–208 CrossRef CAS.
- T. W. M. Fan and A. N. Lane, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 52, 69–117.
- O. Cloarec, M. E. Dumas, A. Craig, R. H. Barton, J. Trygg, J. Hudson, C. Blancher, D. Gauguier, J. C. Lindon and E. Holmes, et al. , Anal. Chem., 2005, 77, 1282–1289 CrossRef CAS.
- F. Dieterle, A. Ross, G. Schlotterbeck and H. Senn, Anal. Chem., 2006, 78, 4281–4290 CrossRef CAS.
- Y. Wang, M. E. Bollard, J. K. Nicholson and E. Holmes, J. Pharm. Biomed. Anal., 2006, 40, 375–381 CrossRef CAS.
- N. J. Waters, C. J. Waterfield, R. D. Farrant, E. Holmes and J. K. Nicholson, J. Proteome Res., 2006, 5, 1448–1459 CrossRef CAS.
- K. Daschner, I. Couee and S. Binder, Plant Physiol., 2001, 126, 601–612 CrossRef CAS.
- J. Vockley and R. Ensenauer, Am. J. Med. Genet., Part C, 2006, 142C, 95–103 Search PubMed.
- S. C. Lu, Mol. Aspects Med., 2009, 30, 42–59 CrossRef CAS.
- Z. Z. Shi, G. M. Habib, W. J. Rhead, W. A. Gahl, X. He, S. Sazer and M. W. Lieberman, Nat. Genet., 1996, 14, 361–365 CrossRef CAS.
- A. Mally and W. Dekant, Mol. Nutr. Food Res., 2009, 53, 467–478 CrossRef CAS.
- C. Schlatter, J. Studer-Rohr and T. Rasonyi, Food Addit. Contam., 1996, 13(Suppl), 43–44 CAS.
- J. Gautier, J. Richoz, D. H. Welti, J. Markovic, E. Gremaud, F. P. Guengerich and R. J. Turesky, Chem. Res. Toxicol., 2001, 14, 34–45 CrossRef CAS.
- A. Mally, H. Zepnik, P. Wanek, E. Eder, K. Dingley, H. Ihmels, W. Volkel and W. Dekant, Chem. Res. Toxicol., 2004, 17, 234–242 CrossRef CAS.
- A. Mally, G. Pepe, S. Ravoori, M. Fiore, R. C. Gupta, W. Dekant and P. Mosesso, Chem. Res. Toxicol., 2005, 18, 1253–1261 CrossRef CAS.
- J. C. Gautier, D. Holzhaeuser, J. Markovic, E. Gremaud, B. Schilter and R. J. Turesky, Free Radical Biol. Med., 2001, 30, 1089–1098 CrossRef CAS.
- A. Mally, W. Volkel, A. Amberg, M. Kurz, P. Wanek, E. Eder, G. Hard and W. Dekant, Chem. Res. Toxicol., 2005, 18, 1242–1252 CrossRef CAS.
- N. Palma, S. Cinelli, O. Sapora, S. H. Wilson and E. Dogliotti, Chem. Res. Toxicol., 2007, 20, 1031–1037 CrossRef CAS.
- M. Gekle, C. Sauvant and G. Schwerdt, Mol. Nutr. Food Res., 2005, 49, 118–130 CrossRef CAS.
- M. Marin-Kuan, S. Nestler, C. Verguet, C. Bezencon, D. Piguet, R. Mansourian, J. Holzwarth, M. Grigorov, T. Delatour and P. Mantle, et al. , Toxicol. Sci., 2006, 89, 120–134 CAS.
- C. Cavin, T. Delatour, M. Marin-Kuan, D. Holzhauser, L. Higgins, C. Bezencon, G. Guignard, S. Junod, J. Richoz-Payot and E. Gremaud, et al. , Toxicol. Sci., 2007, 96, 30–39 CAS.
- M. Sieber, S. Wagner, E. Rached, A. Amberg, A. Mally and W. Dekant, Chem. Res. Toxicol., 2009, 22, 1221–1231 CrossRef CAS.
- C. R. Elcombe, J. Odum, J. R. Foster, S. Stone, S. Hasmall, A. R. Soames, I. Kimber and J. Ashby, Environ. Health Perspect., 2002, 110, 363–375 CrossRef CAS.
- U. A. Boelsterli, Toxicol. Appl. Pharmacol., 2003, 192, 307–322 CrossRef CAS.
- R. D. Snyder and J. W. Green, Mutat. Res., 2001, 488, 151–169 CAS.
- S. Ananth, L. Zhuang, E. Gopal, S. Itagaki, B. Ellappan, S. B. Smith, V. Ganapathy and P. Martin, Biochem. Biophys. Res. Commun., 2010, 394, 75–80 CrossRef CAS.
- K. S. Vellonen, M. Hakli, N. Merezhinskaya, T. Tervo, P. Honkakoski and A. Urtti, Eur. J. Pharm. Sci., 2010, 39, 241–247 CrossRef CAS.
- E. Gopal, N. S. Umapathy, P. M. Martin, S. Ananth, J. P. Gnana-Prakasam, H. Becker, C. A. Wagner, V. Ganapathy and P. D. Prasad, Biochim. Biophys. Acta, 2007, 1768, 2690–2697 CAS.
- A. Mally, A. Amberg, G. C. Hard and W. Dekant, Toxicology, 2007, 230, 244–255 CrossRef CAS.
- R. Pontremoli, G. Leoncini and A. Parodi, Expert. Rev. Cardiovasc. Ther., 2005, 3, 43–50 Search PubMed.
- A. B. DeAngelo, M. H. George, S. R. Kilburn, T. M. Moore and D. C. Wolf, Toxicol. Pathol., 1998, 26, 587–594 CAS.
- S. Kawanishi and M. Murata, Toxicology, 2006, 221, 172–178 CrossRef CAS.
- X. Zhang, S. D. De, B. Sun, J. Fisher, R. J. Bull, J. A. Cotruvo and B. S. Cummings, Toxicology, 2010, 269, 13–23 CrossRef CAS.
- F. Y. Ghauri, A. E. McLean, D. Beales, I. D. Wilson and J. K. Nicholson, Biochem. Pharmacol., 1993, 46, 953–957 CrossRef CAS.
- T. Umemura, A. Takagi, K. Sai, R. Hasegawa and Y. Kurokawa, Arch. Toxicol., 1998, 72, 264–269 CrossRef CAS.
- W. A. Al-Turk, I. A. Majeed, W. J. Murray, D. W. Newton and S. Othman, Int. J. Pharm., 1988, 41, 227–230 CrossRef CAS.
- I. A. Majeed, W. J. Murray, D. W. Newton, S. Othman and W. A. Al-Turk, J. Pharm. Pharmacol., 1987, 39, 1044–1046 CAS.
- R. Testa, E. Dolfini, C. Reschiotto, C. Secchi and P. A. Biondi, Farmaco, Ed. Prat., 1979, 34, 463–473 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the effect of toxin treatment on metabolite concentration and full assignments of all identified metabolite resonances in spectra. See DOI: 10.1039/c0mb00146e |
| This journal is © The Royal Society of Chemistry 2011 |