Kinetic mechanism determination and analysis of metal requirement of dehydroquinate synthase from Mycobacterium tuberculosisH37Rv: an essential step in the function-based rational design of anti-TB drugs
Jordana Dutra de
Mendonça
ab,
Osao
Adachi
c,
Leonardo Astolfi
Rosado
ad,
Rodrigo Gay
Ducati
a,
Diogenes Santiago
Santos
*a and
Luiz Augusto
Basso
*a
aCentro de Pesquisas em Biologia Molecular e Funcional (CPBMF), Instituto Nacional de Ciência e Tecnologia em Tuberculose (INCT-TB), Pontifícia Universidade Católica do Rio Grande do Sul (PUCRS), 6681/92-A Av. Ipiranga, 90619-900, Porto Alegre, RS, Brazil. E-mail: luiz.basso@pucrs.br; diogenes@pucrs.br; Tel: +55-51-33203629
bPrograma de Pós-Graduação em Ciências Biológicas: Bioquímica, Universidade Federal do Rio Grande do Sul (UFRGS), Porto Alegre, RS, Brazil
cDepartment of Biological Chemistry, Faculty of Agriculture, Yamaguchi University, Yamaguchi 753-8515, Japan
dPrograma de Pós-Graduação em Medicina e Ciências da Saúde (PUCRS), Porto Alegre, RS, Brazil
First published on 26th October 2010
Abstract
The number of new cases of tuberculosis (TB) arising each year is increasing globally. Migration, socio-economic deprivation, HIV co-infection and the emergence of drug-resistant strains of Mycobacterium tuberculosis, the main causative agent of TB in humans, have all contributed to the increasing number of TB cases worldwide. Proteins that are essential to the pathogen survival and absent in the host, such as enzymes of the shikimate pathway, are attractive targets to the development of new anti-TB drugs. Here we describe the metal requirement and kinetic mechanism determination of M. tuberculosisdehydroquinate synthase (MtDHQS). True steady-state kinetic parameters determination and ligand binding data suggested that the MtDHQS-catalyzed chemical reaction follows a rapid-equilibrium random mechanism. Treatment with EDTA abolished completely the activity of MtDHQS, and addition of Co2+ and Zn2+ led to, respectively, full and partial recovery of the enzyme activity. Excess Zn2+ inhibited the MtDHQS activity, and isotitration microcalorimetry data revealed two sequential binding sites, which is consistent with the existence of a secondary inhibitory site. We also report measurements of metal concentrations by inductively coupled plasma atomic emission spectrometry. The constants of the cyclic reduction and oxidation of NAD+ and NADH, respectively, during the reaction of MtDHQS was monitored by a stopped-flow instrument, under single-turnover experimental conditions. These results provide a better understanding of the mode of action of MtDHQS that should be useful to guide the rational (function-based) design of inhibitors of this enzyme that can be further evaluated as anti-TB drugs.
Introduction
Although the estimated per capita tuberculosis (TB) incidence was stable in 2005, the number of new cases arising each year is still increasing globally.1 Among the 15 countries with the highest estimated TB incidence rates, 13 are located in Africa, a phenomenon linked to high rates of Mycobacterium tuberculosis, the causative agent of TB in humans, and HIV co-infection, making TB the major cause of death in HIV-positive patients, which represent 23% of the estimated 2 million HIV deaths in 2007. According to the World Health Organization, there were an estimated 9.4 million new TB cases in 2008, from which 1.4 million were HIV-positive, with 1.8 million deaths total—equal to 4500 deaths a day.2 Migration, socio-economic deprivation, HIV co-infection and the greater use of immunosuppressive agents in healthcare have all contributed to the increasing number of TB cases worldwide, mainly in countries where it was considered eradicated.3 Furthermore, the recent report of the occurrence of at least one case of extensively-resistant drug TB—resistant to isoniazid and rifampicin plus resistance to any fluoroquinolone and at least one of three injectable second line drugs used in TB treatment—in 57 countries worldwide, has characterized a global spread and alarmed the public health authorities.4 Even more recently, totally drug-resistant TB strains (TDR-TB) have been identified that are resistant to all first- and second-line drugs tested (i.e.aminoglycosides, cyclic polypeptides, fluoroquinolones, thioamides, serine analogues and salicylic acid derivatives).5Although TB can be cured with the current short-course therapy, this six-month-long treatment and its host toxicity lead to patients’ non adherence.6 There is thus an urgent need for the development of both better vaccines and new and more efficient antimycobacterial agents with novel mechanisms of action. Future drugs should present selective toxicity, be active against drug-resistant and non-resistant strains, shorten the duration of TB treatment to improve patients' compliance and, ideally, do not have pharmacological interactions with antiretroviral drugs commonly used to treat HIV.7,8
The complete sequencing of the M. tuberculosisH37Rv genome has provided a better understanding of the biology of this pathogen and has also identified potential targets and biochemical pathways that may be used in prophylactic and therapeutic interventions.9Proteins that are essential to the pathogen survival and absent from the host, such as enzymes of the shikimate pathway, are attractive targets for the development of new antitubercular drugs. In 2002, Parish and Stoker showed that this pathway is essential to the viability of the bacilli, thereby validating the shikimate pathway enzymes as potential targets for the design of inhibitors with potential anti-TB activity.10
The aroB-encoded dehydroquinate synthase (DHQS, EC 4.6.1.3) catalyzes the conversion of 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) to dehydroquinate (DHQ) in the shikimate pathway (Fig. 1). This pathway leads to the biosynthesis of chorismate, the precursor of aromatic compounds such as phenylalanine, tyrosine and tryptophan, and a range of other metabolites.11DHQS has been characterized in other microorganisms (E. coli,12B. circulans,13N. crassa,14A. nidulans15) and higher plants (P. mungo16 and Sorghum17) as a metalloenzyme with preference for Co2+ and Zn2+ that requires catalytic amounts of NAD+. Pathogenic bacteria harboring mutations in the aroBgene sequence are attenuated for virulence.18 The mechanism of the DHQS reaction appears to closely resemble that of 2-deoxy-scyllo-inosose synthase (DOIS) in the 2-deoxystreptamine biosynthesis, as well as the protein sequence.19,20 The TDR targets database has ranked M. tuberculosisDHQS (MtDHQS) as one of the top 50 druggable protein targets for anti-TB drug development,21 and Target-TB identified MtDHQS as a protein target belonging to intermediary metabolism and respiration class.22
 | ||
| Fig. 1 Chemical reaction catalyzed by DHQS. | ||
The rational design of chemotherapeutic agents may be divided into function-based and structure-based design. Knowledge of the molecular structure of the active site and of the mode of action of an enzyme should thus aid the design of inhibitors that may be used as antimycobacterial agents. Here we describe the kinetic mechanism of the MtDHQS-catalyzed chemical reaction assessed by steady-state and pre-steady-state kinetics, fluorescence spectroscopy, and isothermal titration calorimetry. We also present the metal requirements of this enzyme studied by enzyme activity measurements and atomic absorption analysis. These results represent an important step for the rational design of potent MtDHQS inhibitors that can further be used as anti-TB drugs.
Results
Steady-state kinetic parameters and enzyme mechanism
To determine the true steady-state kinetic parameters and the MtDHQS enzyme mechanism, initial velocity as a function of substrate concentration was plotted as a linear function of reciprocal of initial velocity against the reciprocal of substrate concentration (double-reciprocal or Lineweaver–Burk plot). The double-reciprocal plots showed a family of lines intersecting to the left of the y-axis (Fig. 2), which is consistent with ternary complex formation and a sequential mechanism.23 Data were plotted in reciprocal form and fitted to the equation for a sequential initial velocity pattern (eqn (1)), yielding the following values for the true steady-state kinetic parameters: kcat = 0.63 (± 0.03) s−1, KDAHP = 6.3 (± 1.1) μM, KNAD+ = 70 (± 12) μM, kcat/KDAHP = 1.0 (± 0.2) × 105 M−1s−1, and kcat/KNAD+ = 9.0 (± 1.6) × 103 M−1s−1, and KD(DAHP) = 9.9 (± 3.4) μM. | ||
| Fig. 2 Double-reciprocal plots for MtDHQS with either DAHP (A) or NAD+ (B) as the variable substrate. Each curve represents fixed-varying levels of the co-substrate, ranging from 40 to 400 μM for NAD+ and 3 to 50 μM for DAHP. | ||
Metal requirement analysis
As divalent metal has been proposed as an important cofactor for DHQS catalysis, analysis of metal requirement for MtDHQS enzyme activity was carried out. No increase in the maximum velocity of MtDHQS was observed at saturating concentrations of DAHP and NAD+ when the non-chelated enzyme was assayed in the presence of different divalent metals, thereby justifying the absence of these compounds in the assay mixture (data not shown).Isothermal titration calorimetry
To determine the relative affinities of the metal binding site of the two divalent metals that showed the best effect on restoring MtDHQS enzyme activity, Co2+ and Zn2+ were titrated to chelated enzyme (56 μM). Fig. 3 shows the isotherm generated by isothermal titration calorimetry (ITC) and the data after peak integration, subtraction of blank titration data (not shown), concentration normalization (heat normalized to the molar ratio), and analysis by Origin 7.0 suite programs. | ||
| Fig. 3 ITC analysis of Co2+ (A) and Zn2+ (B) binding to MtDHQS. The top graphs show raw data of the heat pulses resulting from a titration of metal-free MtDHQS (56 μM) in the calorimetric cell with a 0.5 μL injection of 1.5 mM of each metal followed by 19 subsequent 2 μL injections. The bottom graphs show the integrated heat pulses, normalized per mol of injectant as a function of the molar ratio (metal concentration/MtDHQS concentration). These binding curves were best fitted to a two-site sequential model equation. | ||
Data from Co2+ titration analysis indicated the presence of two sequential binding sites (Fig. 3A, where the primary binding site has a KD of 16 μM and the secondary site has a KD of 781 μM (exothermic processes). The overall binding isotherm for the interaction of MtDHQS with Zn2+ is biphasic (Fig. 3B) and it is best fitted to a model of two sequential binding sites. Binding to the first binding site is accompanied by a negative enthalpy change (KD of 167 μM; exothermic process), whereas binding to the second site (KD of 0.3 μM) is accompanied by a positive enthalpy change (endothermic process).
Equilibrium ligand binding to MtDHQS
MtDHQS intrinsic protein fluorescence measurements were carried out to both determine the order of substrate/product addition/dissociation on/from the catalytic site and distinguish the enzyme kinetic mechanism. DAHP binding to free MtDHQS enzyme resulted in a decrease of intrinsic protein fluorescence. The quench in protein fluorescence upon MtDHQS-DAHP binary complex formation plotted as a function of substrate concentration (Fig. 4A) was hyperbolic yielding an equilibrium dissociation constant value of 73 (± 7) μM.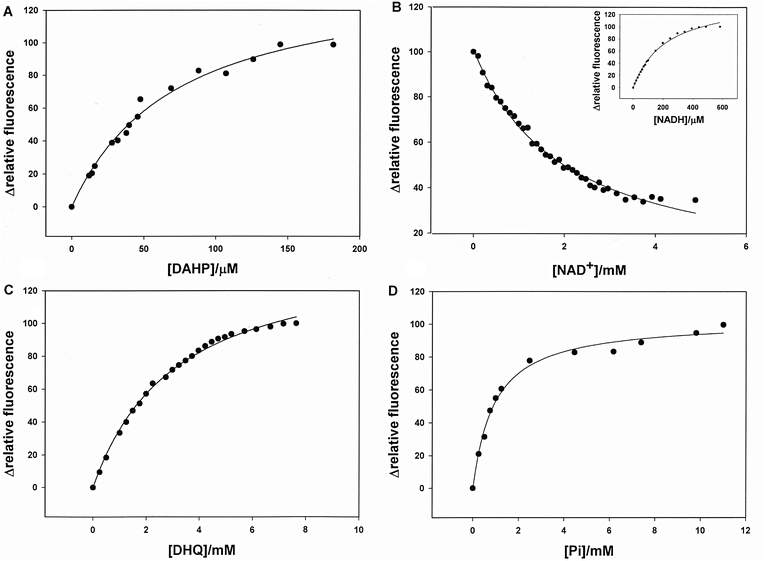 | ||
| Fig. 4 Fluorescence spectroscopy of equilibrium ligand binding to MtDHQS. Dependence of MtDHQS relative protein fluorescence change upon binding of DAHP substrate (A), and the products DHQ (C) and Pi (D). The dependence of the change in MtDHQS-NADH fluorescence in the competitive assay as a function of increasing NAD+ concentration is presented in (B). The inset shows the dependence of the enhancement of nicotinamide fluorescence (460 nm) due to resonance energy transfer from the enhancement in protein fluorescence (350 nm) upon NADH binding to free enzyme. | ||
No change in intrinsic protein fluorescence could be detected upon binding of NAD+ to free MtDHQS. However, the absence of protein fluorescence change upon NAD+ binding to free enzyme cannot firmly be interpreted as absence of MtDHQS-NAD+ binary complex formation. Accordingly, a competitive assay with NADH (an inhibitor of DHQS) was performed.14,24 The titration of NADH in MtDHQS solution causes a hyperbolic increase in nucleotide fluorescence (460 nm) upon excitation of intrinsic protein fluorescence at 300 nm, indicating the binding of NADH to free MtDHQS, with KD = 215 (± 13) μM (Fig. 4B-inset). These results show that there is resonance energy transfer (RET) in which the energy of protein fluorescence (340 nm) is transferred to NADH resulting in nicotinamide fluorescence at 460 nm. In the presence of NADH at concentration near its KD value, NAD+ was added and the RET nicotinamide fluorescence was measured. Plots of NAD+ concentration versus relative nicotinamide fluorescence variation were hyperbolic, and the data were fitted to an equation for competitive binding (eqn (2)). This analysis yielded a value of 895 (± 50) μM for the equilibrium dissociation constant of NAD+ (Fig. 4B). Binding of both products resulted in intrinsic protein fluorescence changes, and the hyperbolic plots yielded equilibrium dissociation constant values of 3100 (± 135) μM for DHQ (Fig. 4C) and 920 (± 80) μM for Pi (Fig. 4D).
Pre-steady-state kinetic analysis
NADH transient formation in the oxidation of DAHP by NAD+ was monitored by pre-steady-state kinetics in a stopped-flow instrument (Fig. 5). Single-turnover experimental conditions (20 μM MtDHQS, 2.5 mM NAD+ and 15 μM DAHP; mixing-chamber concentrations) were employed in which a large NAD+ concentration was pre-incubated with MtDHQS enzyme. These experimental conditions were chosen to try to both ensure that most of MtDHQS is in the form of binary complex with NAD+ and to simplify the reaction model with a substrate, an intermediate, and a product (Fig. 6A and B). A scheme has been proposed by Hirayama et al.25 that involves a reversible binding step (Fig. 6A). In this scheme, k1 is the rate of oxidation of NAD+ and concomitant formation of the first intermediate (Fig. 1), k−1 is the rate constant for the back reaction, and k2 is the rate constant containing the phosphate elimination step and the reduction step of the enone intermediate to form the cyclic enol ether intermediate and oxidation of NADH to NAD+ (Fig. 1 and 6A). On the other hand, an even simpler case of two consecutive irreversible reactions (Fig. 6B) can be considered for analysis of pre-steady-state kinetics data under single turnover conditions.26,27 In this scheme (Fig. 6B), k1 is an apparent first-order rate constant comprising the reduction of NAD+ to NADH, formation of the first intermediate, and the phosphate β-elimination step (including abstraction of a proton at C-5) with concomitant formation of the enone intermediate (Fig. 1), whereas k2 is an apparent first-order rate constant containing the reduction of the enone intermediate by NADH to form the cyclic enol ether with concomitant NAD+ formation (Fig. 1 and 6B). It should be pointed out that this analysis implies that the rate of DAHP binding to MtDHQS is much faster than the remaining rate constants (not rate limiting). Moreover, assuming the two steps (Fig. 6B) as irreversible is justifiable because these reactions occur in the absence of products. Moreover, in this scheme it is implicit that measurements of absorbance at 340 nm detect the rate of formation of E:NADH:S′ intermediate (k1) and the rate of decay of this intermediate to form E:NAD+:S′′. This simplified analysis allows fitting the results to eqn (3), yielding values of k1 = 47 (± 2) s−1 and k2 = 4.36 (± 0.06) s−1 (Fig. 5). | ||
| Fig. 5 Pre-steady-state kinetic analysis of MtDHQS reaction. The NADH production (M) was monitored by a stopped-flow instrument under single turnover conditions (20 μM MtDHQS was pre-incubated with 2.5 mM NAD+ and mixed with 15 μM DAHP; mixing-chamber concentrations). Experimental data are presented by black-filled circles, and fitting the experimental data to eqn (3) yielded the predicted values plotted as a solid line. | ||
 | ||
| Fig. 6 Proposed pre-steady-state kinetic model of the MtDHQS reaction. The rate constant k1 is the substrate oxidation rate by NAD+, and k2 is the rate containing the elimination step of phosphate and reduction step by NADH. Absorbance at 340 nm for NADH formed in the enzyme reaction was monitored and fitted to eqn (3) Scheme A is for a mechanism that involves a reversible binding step followed by an irreversible reaction, whereas scheme B is for a mechanism of two consecutive irreversible reactions. | ||
Discussion
The rational-based development of MtDHQS inhibitors requires characterization of its catalytic mechanism. Because of the complex mechanism in which DHQS is involved, the enzyme has been speculated as a spectator of its own reaction and suggest its catalysis as a simple oxidoreductase with several reactions occurring spontaneously.28 However, the arrangement of the active site of DHQS indicates that the enzyme is not just a spectator in catalysis but stabilizes intermediates and prevents side reactions through its entire reaction pathway.29 The elimination of phosphate from DAHP to generate DHQ catalyzed by DHQS requires catalytic amounts of NAD+ for activity, even though the enzyme-catalyzed chemical reaction is redox neutral. DHQS has attracted considerable mechanistic interest and has long been regarded as a catalytic marvel due to its ability to perform several consecutive chemical reactions in one active site during each catalyzed turnover of substrate into product. In this sequence (Fig. 1), which is mechanistically unusually diverse for a single enzyme, it appears to mediate five sequential transformations:12 (i) the oxidation of the secondary alcohol at C-5; (ii) the β-elimination of inorganic phosphate across C-6 and C-7; (iii) the reduction of the resulting enone at C-5; (iv) the ring opening of the enol pyranose; and (v) the final intramolecular aldol-like reaction to yield DHQ (resonance stabilization of enolate ion, and nucleophilic addition of C-7—as a carbanion—to C-2 carbonyl, production of an alkoxide ion, and protonation of the latter). It has been shown that the β-elimination of Pi across C-6 and C-7 occurs in a syn fashion and the transition state for the subsequent intramolecular aldol reaction has a chairlike geometry.30The mechanism of the DHQS reaction is similar to the mechanism described for DOIS in the 2-deoxystreptamine biosynthesis, including the divalent metal ion requirement for activity, and Co2+ as the most effective to enzyme activity, and the cyclic reduction and reoxidation of NAD+ and NADH, respectively, during the enzyme-catalyzed reaction prior to release of DHQ from the active site.19 The Bacillus circulans btrCgene shows significant sequence similarity to various DHQS and the catalytic domain, and the metal binding residues are conserved between DOIS and DHQSs.20 However, it has been pointed out that there exist dissimilarities between B. circulans DOIS and DHQSs, particularly in the stereochemistry of overall reactions.31 We have previously reported that amino acid sequence comparison between MtDHQS and DHQSs from other organisms showed highly conserved residues,32 which are likely involved in protein function and activity. Multiple sequence alignment between MtDHQS (Gene bank access code: NP_217054.1), DHQS domain of Aspergillus nidulans AROM protein (AnDHQS, for which there is structural data; PDB access code: 1DQS), and Bacillus circulans DOIS (BcDOIS; PDB access code: 2GRU) amino acid sequences show conservation of key residues (Fig. 8).33 Note that the numbering given here is for the actual position of a particular amino acid in the polypeptide chain of each enzyme, and the numbering given in Fig. 8 is to show sequence comparison results after introduction of gaps. The similarity of the mode of action of MtDHQS and BcDOIS is borne out by the conservation of key amino acid residues (Fig. 8). However, as there is structural data on AnDHQS, emphasis of the likely roles played by MtDHQS side chains will be placed on conservation of amino acid residues in AnDHQS and in MtDHQS polypeptide sequences. The crystal structure of AnDHQS has been solved at 1.8 Å resolution29 with Zn2+, NAD+ and carbaphosphonate. The latter is a sub-nanomolar slow-binding inhibitor of E. coliDHQS.34 The active site is located in a cleft between two domains of homodimeric AnDHQS.29 The pentacoordinated Zn2+ interacts with Glu194, His271 and His287 and two carbaphosphonate inhibitor hydroxyls in AnDHQS.29 The corresponding amino acid residues in MtDHQS are Glu186, His249 and His265 (Fig. 8), which are all conserved. In concert with hydride transfer of C5 of DAHP to C4 of the NAD+ nicotinamide moiety, a proton from C5 hydroxyl group is transferred to a water molecule and relayed (proton-shuffling system) to His275 of AnDHQS. A similar role may be played by the conserved His253 in MtDHQS (Fig. 8). It has been suggested that a possible role for Zn2+ is to facilitate hydride transfer and proton loss by polarizing the C5 hydroxyl group of DAHP.29 The β-elimination step appears to involve interactions between the oxygens of the phosphate group with Lys152, Asn162, Asn268 and Lys356 of one subunit, and Arg130 from the other monomer. The corresponding conserved residues in MtDHQS are Lys144, Asn154, Asn246 and Lys323 of one subunit, and Arg122 from the other monomer. NADH hydride transfer to C5 ketone of the enone intermediate (Fig. 1) may be followed by proton transfer to C5 hydroxyl group from MtDHQS His253 conserved side chain involved in the proton-shuffling system. After ring opening, there is a rotation about the C5–C6 bond that is followed by aldol condensation and protonation of the alkoxide intermediate (Fig. 1). To prevent epimerization at C2 in DHQS-catalyzed chemical reaction, the carboxylic acid group on C2 of the cyclic enol ether (Fig. 1) is held in place by interactions with a Lys152, Lys250 and Arg264 in AnDHQS.29 The corresponding residues in MtDHQS are Lys144, Lys228 and Arg242, which are all conserved (Fig. 8).
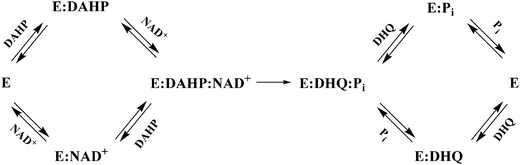 | ||
| Fig. 7 Proposed kinetic mechanism for MtDHQS. | ||
 | ||
| Fig. 8 Comparison of amino acid sequences. Shown are DHQS sequences for M. tuberculosis (Mt_DHQS; 362 amino acids; Gene bank access code: NP_217054.1), Aspergillus nidulansDHQS (An_DHQS; 402 amino acids; Gene bank access code: NT_107015.1), and Bacillus circulans DOIS (Bc_DOIS; 368 amino acids; Gene bank access code: BAE07067.1). Multiple amino acid sequence alignment was performed by ClustalW,33 using the Gonnet matrix for amino acids substitutions and considering gap penalties. | ||
The divalent metal cofactor has been proposed as an important factor during the catalysis of DHQSs. To elucidate this role, analysis of the metal requirement of MtDHQS was performed. The need for no addition of divalent metal in the standard assay with non-chelated enzyme can be explained by the general conditions of the expression system that provided appropriate amounts of metal for MtDHQS enzyme activity. The treatment with EDTA results in rapid formation of inactive apoenzyme, and DAHP binding prevents the removal of the metal from the active site, which suggests that the metal is less accessible to EDTA when DAHP is bound to the enzyme active site. Conversely, NAD+ has no protective effect. This protective behavior of DAHP was previously described for E. coli12 and N. crassa14 enzymes. As discussed above, the pentacoordinated Zn2+ interacts with one glutamate and two histidine amino acid side chains and two hydroxyl groups of carbaphosphonate inhibitor,29 which would account for the protective effect of DAHP. On the other hand, the lack of protective effect by NAD+ suggests that there is no interaction between this cofactor and Zn2+ ion.
In the reactivation studies, the addition of Co2+ in the reaction mixture containing the chelating agent restored completely the MtDHQS activity, similar to B. subtilis13 and E. coli12 enzymes, but differing from N. crassa that rapidly recovered the enzyme's activity with Zn2+ rather than Co2+.14 In contrast, the presence of excess of Zn2+ leads to decreased rates for MtDHQS at concentration values larger than 200 μM (data not shown), which suggests that zinc may also bind to a second inhibitory site. This is similar to DHQS from E. coli12 and A. nidulans35 and was confirmed by the ITC assays of metal binding to the mycobacterial enzyme, where the titration of Co2+ and Zn2+ of MtDHQS in solution indicated the existence of two sequential binding sites. There appears to be positive cooperativity between the two MtDHQS binding sites since there is a decrease in the equilibrium dissociation constant for binding to the second site (0.3 μM) as compared to binding to the first site (167 μM). We have recently been able to obtain crystal of the MtDHQS enzyme, and, hopefully, these structural efforts will shed further light on the role of a metal in catalysis and/or binding (ongoing experiments).
The atomic absorption analysis showed the absence of any significant quantity of cobalt and the presence of a small, but significant, amount of zinc. The zinc content of the final preparation is 0.2 mol of Zn2+ per mol of MtDHQS, indicating that the metal sites of the enzyme were only partially saturated in crude extract. This substoichiometric value obtained may represent the loss of the metal during the purification process, similar to that described for other enzymes, like DAHP(Phe) synthase from E. coli.36 It is interesting to note that the purified recombinant MtDHQS enzyme did not contain significant amount of cobalt associated, even though the metal reactivation assays here described demonstrated preference for Co2+ However, based on the higher bioavailability of Zn2+, it seems likely that DHQS is naturally a zinc-dependent metalloenzyme.14 Taken together, these results demonstrate a central role for the metal ion in the catalytic mechanism, since each chemical transformation mediated by dehydroquinate synthase has, in simpler enzymatic systems, been shown to involve metal cofactors.12
The pattern of the double-reciprocal plots was consistent with ternary complex formation and a sequential mechanism. The mechanisms of ping-pong and rapid equilibrium ordered could be discarded, since these mechanisms display parallel lines and intersecting lines at the y-axis, respectively.23 The initial velocity data and equilibrium binding analysis are consistent with either a steady-state or a rapid-equilibrium random kinetic mechanism, in which both DAHP and NAD+ bind to the free enzyme, and there is no preferential order for dissociation of the products, DHQ and Pi, from the active site (Fig. 7). However, steady-state random order mechanisms usually display non-linear Lineweaver–Burk plots due to squared terms in its rate equation.23,37 Accordingly, the results here presented suggest that the MtDHQS enzyme mechanism is rapid-equilibrium random order. The true kinetic parameters showed that MtDHQS has a lower catalytic efficiency when compared to others DHQSs. The turnover numbers of 24 s−1 for E. coli,38 19 s−1 for N. crassa14 and 6.8 s−1 for A. nidulans,35 are significantly higher when compared with 0.63 s−1 for MtDHQS. The KDAHP values are, however, similar as compared to E. coli (5.5 μM),12N. crassa (1.4 μM),14 and somewhat lower than the value for A. nidulans monofunctional DHQS domain (21 μM).35 Notwithstanding, the DAHP substrate specificity constant value of 1.0 × 105 M−1s−1 (kcat/KDAHP = apparent second-order rate constant) for MtDHQS is more than 250-fold lower than the E. colienzyme value (2.5 × 107 M−1s−1).12 On the other hand, the NAD+ Michaelis–Menten constant value for MtDHQS (KNAD+ = 70 μM) is approximately 900-fold larger than for E. coli (KNAD+ = 80 nM),12 350-fold larger than N. crassa (KNAD+ < 0.2 μM)14 and 37-fold larger than A. nidulans monofunctional DHQS domain (KNAD+ = 1.9 μM).35
DHQS is mechanistically distinguished by its catalytic use of NAD+. The reduction of NAD+ to NADH during the first step of the enzyme-catalyzed reaction is followed by reoxidation of NADH to NAD+ at a later enzyme-catalyzed step prior to release of DHQ from the enzyme active site. Pre-steady-state kinetics assay of a single enzyme turnover was thus carried out to determine the rate constants of the conversion NAD+ → NADH → NAD+ (Fig. 5). Experimental conditions were chosen so that only a single turnover of MtDHQS-catalyzed chemical reaction is possible and the transient changes in absorbance at 340 nm were measured. The single turnover data were analysed using a simple case for two consecutive reactions as described by Fersht26 and Hiromi,27 and fitting the data to eqn (3) yielded apparent rate constant values of 47 s−1 for k1 and 4.36 s−1 for k2 (Fig. 5). It is noteworthy that detection of part of the pre-steady-state absorbance signal due to NAD+ to NADH conversion was not possible because it occurred in the dead time of the apparatus (≤1.5 ms) (Fig. 5). It should be pointed out that we do not wish to imply that NADH to NAD+ conversion may be associated with product release (Pi) and/or conversion of the first intermediate to the enone intermediate (Fig. 1). Importantly, the pre-steady-state kinetic data demonstrate that there is a transient increase in absorbance at 340 nm associated with NADH formation followed by its oxidation back to NAD+ that occurs only in the presence of MtDAHP-catalyzed conversion of DAHP to DHQ. In addition, the rates of NAD+ → NADH → NAD+ conversions are not rate limiting because they are larger than the kcat value (0.63 s−1).
Conclusions
Rational inhibitor design relies on mechanistic and structural information on the target enzyme. Enzyme inhibitors make up roughly 25% of the drugs marketed in the United States.39 Enzymes catalyze multistep chemical reactions to achieve rate accelerations by stabilization of transition state structure(s).40 Accordingly, mechanistic analysis should always be a top priority for enzyme-targeted drug programs aiming at the rational design of potent enzyme inhibitors. Moreover, ITC has been used as an important technique for the direct determination of thermodynamic and kinetic parameters of enzymatic reactions.41 It has recently been pointed out that recognition of the limitations of high-throughput screening approaches in the discovery of candidate drugs has rekindled interest in rational design methods.42 Understanding the mode of action of MtDHQS will inform us on how to better design inhibitors targeting this enzyme with potential therapeutic application in TB chemotherapy. Accordingly, it is hoped that the results here described may be useful to the rational design of anti-TB agents and that they may contribute to our understanding of the biology of M. tuberculosis.Experimental
Homogeneous MtDHQS production
The homogeneous solution of recombinant MtDHQS was obtained as we have previously described.32Protein concentration was determined by the Bradford Protein Assay Kit (Bio-Rad Laboratories), using bovine serum albumin as standard.43Enzyme assay and initial velocity
MtDHQS activity was measured by coupling the conversion of 3-deoxy-D-arabino-heptulosonate 7-phosphate (DAHP) into dehydroquinate (DHQ), and monitoring the dehydroquinate dehydratase-catalyzed conversion of DHQ to dehydroshikimate at 234 nm (ε234nm = 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1) in an UV-2550 UV/Visible spectrophotometer (Shimadzu).38,44 The standard assay was performed in 5-mm pathlength quartz cuvettes at 25 °C in a total volume of 1.5 mL containing 50 mM Tris-HCl pH 7.5, 120 μM DAHP (Toronto Chemical Research), 600 μM NAD+ (Sigma), and 1 unit of dehydroquinate dehydratase (DHase) from M. tuberculosis. The true steady-state kinetic parameters were determined by varying concentrations of DAHP (3–50 μM) against varied-fixed concentrations of NAD+ (40–400 μM). One unit of enzyme activity was defined as the amount of protein that catalyzes the consumption of 1 μmol of DAHP/min at 25 °C. The family of lines intersecting to the left of the y-axis in double-reciprocal plots were fitted to eqn (1) for a mechanism involving ternary complex formation and a sequential substrate binding:
000 M−1 cm−1) in an UV-2550 UV/Visible spectrophotometer (Shimadzu).38,44 The standard assay was performed in 5-mm pathlength quartz cuvettes at 25 °C in a total volume of 1.5 mL containing 50 mM Tris-HCl pH 7.5, 120 μM DAHP (Toronto Chemical Research), 600 μM NAD+ (Sigma), and 1 unit of dehydroquinate dehydratase (DHase) from M. tuberculosis. The true steady-state kinetic parameters were determined by varying concentrations of DAHP (3–50 μM) against varied-fixed concentrations of NAD+ (40–400 μM). One unit of enzyme activity was defined as the amount of protein that catalyzes the consumption of 1 μmol of DAHP/min at 25 °C. The family of lines intersecting to the left of the y-axis in double-reciprocal plots were fitted to eqn (1) for a mechanism involving ternary complex formation and a sequential substrate binding: | (1) |
Metal requirement analysis
Divalent metal ions were removed from all solutions by stirring with Chelex resin (BioRad), followed by filtration through a Milipore 0.22 μM sterilizing membrane.135.70 Da).
Isothermal titration calorimetry (ITC)
The protein binding constants of protein-divalent metal binary complex formation were determined by ITC using a MicroCal ITC-200 microcalorimeter (Thermo). ITC measurements were carried out at 25 °C, and titrations were carried out using a 40 μL-syringe and with stirring at 500 rpm. Each titration consisted of a preliminary injection of 0.5 μL, followed by 19 injections of 2 μL into a cell containing 200 μL of protein sample of 56 μM. To correct for dilution and mixture effects, a series of baselines were performed, in which injections of metal were carried out into buffer, and subtracted from data to obtain accurate heat exchanges. The integrated heat changes (area under each peak in μcal) were plotted as kcal/mole of injectant versus the molar ratio ligand/macromolecule (Fig. 3, lower panels). The binding curve data were best fitted to standard equations using a model for two-site sequential model as implemented in Microcal ORIGIN 7.0 software package (Thermo-MicroCal Software).Equilibrium ligand binding to MtDHQS
Fluorescence measurements were carried out in a RF-5301 PC Spectrofluorophotometer (Shimadzu) at 25 °C. Measurements of intrinsic MtDHQS protein fluorescence were carried out using excitation wavelength at 300 nm in each binding experiment, and the emission wavelength ranged from 320 to 450 nm (maximum emission wavelength at 340 nm). For competitive assays of NADH and NAD+, the nicotinamide fluorescence was monitored, with excitation wavelength at 300 nm and the emission wavelength ranging from 320 to 500 nm (with maximum emission wavelength at 460 nm due to RET). The slits for excitation and emission were 1.5 and 15 nm, respectively. Fluorescence titrations of binary complex formation were carried out by making microlitre additions of the following compounds to 2 mL containing 2 μM MtDHQS: 4 mM DAHP stock solution (1.999–181.34 μM final concentration); 200 mM NAD+ stock solution (0.0989–4.877 mM final concentration); 500 mM DHQ solution (0.249–7.634 mM final concentration); 500 mM Pi stock solution (0.249–11.00 mM final concentration); 20 mM NADH stock solution (9.995–582.52 μM final concentration). Control experiments were employed to both determine the maximum ligand concentrations to be used with no significant inner filter effect and to account for any dilution effect on fluorescence. Data were fitted to a hyperbolic equation for DAHP, NADH, DHQ, and Pi. For competitive binding to determine the equilibrium dissociation constant of NAD+, the data of relative fluorescence (f) as a function of NAD+ concentration were fitted to eqn (2), in which F represents the maximum relative fluorescence change upon ligand binding, A the NAD+ concentration, Ka the equilibrium dissociation constant for NAD+, Ki is the equilibrium dissociation constant for NADH (215 μM), and I the fixed NADH concentration (215 μM). | (2) |
Pre-steady-state kinetic analysis
Absorption of NADH (340 nm) produced in the catalytic cycle was analyzed in an Applied-Photophysics SX-18MV-R (Leatherhead, UK) stopped-flow instrument. Data acquisition was carried out using a split time base (0.2 and 1 s), and a 1-cm pathlength mixing chamber. After pre-incubation of 20 μM MtDHQS with 2.5 mM NAD+ at 25 °C for 10 min, 15 μM DAHP was mixed to start the single-turnover enzyme reaction (mixed-chamber or final concentration). A large NAD+ concentration was employed to ascertain that most of MtDHQS is in the form of binary complex and to simplify the reaction model (Fig. 6A and B). Considering the simplest case for two consecutive reactions of pre-steady-state data under single turnover conditions,26,27eqn (3) can be employed to yield estimates for k1 and k2. Accordingly, pre-steady-state kinetics data were fitted to eqn (3) by regression analysis using SigmaPlot 9.1 package software (Systat Software, Inc.). | (3) |
Acknowledgements
This work was supported by funds of Millennium Initiative Program and National Institute of Science and Technology on Tuberculosis (INCT-TB), MCT-CNPq, Ministry of Health - Department of Science and Technology (DECIT) - Secretary of Health Policy (Brazil) to L.A.B. and D.S.S. L.A.B. (CNPq, 520182/99-5) and D.S.S. (CNPq, 304051/1975-06) are Research Career Awardees of the National Research Council of Brazil (CNPq). J.D.M. acknowledges an MSc scholarship awarded by CNPq.References
- WHO, 2010 http://www.who.int/mediacentre/factsheets/fs104/en/index.html. Accessed on 01-15-2010.
- WHO, 2009 Global tuberculosis control: a short update to the 2009 report. WHO/HTM/TB/2009.426.
- A. Goodman and M. Lipman, Clin. Med., 2008, 8, 531–534 Search PubMed.
- Centers for Disease Control and Prevention, MMWR., 2006, 55, 301–305; WHO, 2010. http://www.who.int/tb/challenges/xdr/xdr_map_sep09.pdf. Accessed on 01-21-2010.
- A. A. Velayati, P. Farnia, M. R. Masjedi, T. A. Ibrahim, P. Tabarsi, R. Z. Haroun, H. O. Kuan, J. Ghanavi, P. Farnia and M. Varahram, Eur. Respir. J., 2009, 34, 1202–1203 CrossRef CAS.
- Y. Zhang, Annu. Rev. Pharmacol. Toxicol., 2005, 45, 529–564 CrossRef CAS.
- R. Ducati, L. A. Basso and D. S. Santos, Curr. Drug Targets, 2007, 8, 423–435 Search PubMed.
- M. Jassal and W. R. Bishai, Lancet Infect. Dis., 2009, 9, 19–30 CrossRef.
- S. T. Cole, R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V. Gordon, K. Eiglmeier, S. Gas, C. E. Barry, 3rd, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, A. Krogh, J. McLean, S. Moule, L. Murphy, K. Oliver, J. Osborne, M. A. Quail, M. A. Rajandream, J. Rogers, S. Rutter, K. Seeger, J. Skelton, R. Squares, S. Squares, J. E. Sulston, K. Taylor, S. Whitehead and B. G. Barrell, Nature, 1998, 393, 537–544 CrossRef CAS.
- T. Parish and N. G. Stoker, Microbiology, 2002, 148, 3069–3077 CAS.
- R. Bentley, Crit. Rev. Biochem. Mol. Biol., 1990, 25, 307–384 CrossRef CAS.
- S. L. Bender, S. Mehdi and J. R. Knowles, Biochemistry, 1989, 28, 7555–7560 CrossRef CAS.
- N. Hasan and E. W. Nester, J. Biol. Chem., 1978, 253, 4999–5004 CAS.
- J. M. Lambert, M. R. Boocock and J. R. Coggins, Biochem. J., 1985, 226, 817–829 CAS.
- J. D. Moore, J. R. Coggins, R. Virden and A. R. Hawkins, Biochem J., 1994, 301, 297–304 CAS.
- E. Yamamoto, Phytochemistry, 1980, 19, 779–781 CrossRef CAS.
- R. Saijo and T. Kosuge, Phytochemistry, 1978, 17, 223–225 CrossRef CAS.
- A. Gunel-Ozcan, K. A. Brown, A. G. Allen and D. J. Maskel, Microb. Pathog., 1997, 23, 311–316 CrossRef CAS.
- F. Kudo, Y. Hosomi, H. Tamegai and K. Kakinuma, J. Antibiot., 1999, 52, 81–88 CAS.
- F. Kudo, H. Tamegai, T. Fujiwara, U. Tagami, K. Hirayama and K. Kakinuma, J. Antibiot., 1999, 52, 559–571 CAS.
- F. Agüero, B. Al-Lazikani, M. Aslett, M. Berriman, F. S. Buckner, R. K. Campbell, S. Carmona, I. M. Carruthers, A. W. Chan, F. Chen, G. J. Crowther, M. A. Doyle, C. Hertz-Fowler, A. L. Hopkins, G. McAllister, S. Nwaka, J. P. Overington, A. Pain, G. V. Paolini, U. Pieper, S. A. Ralph, A. Riechers, D. S. Roos, A. Sali, D. Shanmugam, T. Suzuki, W. C. Van Voorhis and C. L. Verlinde, Nat. Rev. Drug Discovery, 2008, 7, 900–907 CrossRef.
- K. Raman, K. Yeturu and N. Chandra, BMC Syst. Biol., 2008, 2, 10 CrossRef.
- I. H. Segel, in Enzyme Kinetics, Behavior and Analysis of Rapid Equilibrium and Steady-state Enzyme Systems, John Wiley & Sons, Inc., New York, 1975, 957 pp Search PubMed.
- P. R. Srinivasan, J. Rothschild and B. D. Sprinson, J. Biol. Chem., 1963, 238, 3176–3182.
- T. Hirayama, F. Kudo, Z. Huang and T. Egushi, Bioorg. Med. Chem., 2007, 15, 418–423 CrossRef CAS.
- A. Fersht, in Enzyme Structure and Mechanism, W. H. Freeman and Company, 2nd edn, 1985, ch. 4, pp. 121–154 Search PubMed.
- K. Hiromi, in Kinetics of Fast Enzyme Reactions: Theory and Practice, Kodansha Scientific Books, Tokyo, 1979, ch. 4, pp. 188–253 Search PubMed.
- P. A. Barlett and K. Satake, J. Am. Chem. Soc., 1988, 110, 1628–1630 CrossRef CAS.
- E. P. Carpenter, A. R. Hawkins, J. W Frost and K. A. Brown, Nature, 1998, 394, 299–302 CrossRef CAS.
- T. Widlanski, S. L. Bender and J. R. Knowles, Biochemistry, 1989, 28, 7572–7582 CrossRef CAS.
- E. Nango, T. Eguchi and K. Kakinuma, J. Org. Chem., 2004, 69, 593–600 CrossRef CAS.
- J. D. Mendonça, F. Ely, M. S. Palma, J. Frazzon, L. A. Basso and D. S. Santos, J. Bacteriol., 2007, 189, 6246–6252 CrossRef.
- J. D. Thompson, D. G. Higgins and T. J. Gibson, Nucleic Acids Res., 1994, 22, 4673–4680 CrossRef CAS.
- S. L. Bender, T. Widlanski and J. R. Knowles, Biochemistry, 1989, 28, 7560–7572 CrossRef CAS.
- A. Park, H. K. Lamb, C. Nichols, J. D. Moore, K. A. Brown, A. Cooper, I. G. Charles, D. K. Stammers and A. R. Hawkins, Protein Sci., 2006, 13, 2108–2119.
- C. M. Stephens and R. Bauerle, J. Biol. Chem., 1991, 266, 20810–20817 CAS.
- P. C. Engel, in Enzyme Kinetics: The Steady-state Approach, John Wiley & Sons, Inc., New York, 1977, ch. 5, pp. 45–73 Search PubMed.
- U. S. Maitra and D. B. Sprinson, J. Biol. Chem., 1978, 253, 5426–30 CAS.
- J. G. Robertson, Biochemistry, 2005, 44, 5561–5571 CrossRef CAS.
- J. G. Robertson, Curr. Opin. Struct. Biol., 2007, 17, 674–679 CrossRef CAS.
- M. L. Bianconi, Biophys. Chem., 2007, 126, 59–64 CrossRef CAS.
- J. E. Ladbury, G. K. Klebe and E. Freire, Nat. Rev. Drug Discovery, 2010, 9, 23–27 CrossRef CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- S. Mituhashi and B. D. Davis, Biochim. Biophys. Acta, 1954, 15, 54–61 CAS.
| This journal is © The Royal Society of Chemistry 2011 |