DOI:
10.1039/C0MB00125B
(Paper)
Mol. BioSyst., 2011,
7, 129-138
Received
29th July 2010
, Accepted 1st September 2010
First published on 5th November 2010
Abstract
Oleyl 2-acetamido-2-deoxy-α-D-glucopyranoside (1) was previously shown to exhibit antimitotic activity on glioma (C6) and melanoma (A375) cell lines. Preliminary studies about its mechanism of action using 1H MAS NMR suggested that 1 may be altering the metabolism of lipids. We have now studied the effect of 1 on the fatty acid, sphingolipid and ganglioside content in a line of carcinomic human alveolar epithelial cells (A549) using UPLC-MS. Oleic acid and NB-DNJ were used as positive controls for inhibition of fatty acid and ganglioside synthesis, respectively. Compound 1 (10 μM) was more efficient than oleic acid in reducing fatty acid levels of A549 cells, producing a decrease in the range of 40–15%, depending on the acyl chain length and the number of insaturations. In addition, glycoside 1 caused a reduction on ganglioside content of A549 tumor cell line and accumulation of lactosylceramide, the common metabolic precursor for ganglioside biosynthesis. Alteration of ganglioside metabolism was also observed with two galactosylated derivatives of 1, which caused a more pronounced increase in lactosylceramide levels. Compound 1 at higher concentrations (above 30 μM) produced drastic alterations in glycosphingolipid metabolism, leading to cell metabolic profiles very different from those obtained at 10 μM. These biochemical changes were ascribed to activation of endoplasmic reticulum stress pathways.
1. Introduction
Lung cancer is the leading cause of cancer-related human deaths. One of every three cancer-related deaths is attributable to lung cancer with an overall 5-year survival of <15%.1,2 Non-small cell lung cancer accounts for 80% of all lung cancers, including squamous cell carcinomas, adenocarcinomas, and large cell carcinomas.3,4 Therefore, the exploration and development of more effective chemotherapeutic agents that can target the molecules associated with tumor proliferation, angiogenesis, and apoptosis resistance will lead to improved outcomes for lung cancer patients.
Sphingolipids (SLs) are a family of lipids that play essential roles in cell signalling. From a structural standpoint, SLs derive from sphingosine (Sph), a long chain amino alcohol, that is acylated with a long chain fatty acid to give ceramides (Cer), the central core of glycosphingolipids (GSLs) and sphingomyelin (SM) (Scheme 1). Living cells contain several Cer and Cer metabolite species differing in the length and degree of unsaturation of their acyl chain, which appears relevant for their biological activities. Moreover, abnormal SLs metabolism is known to occur in some diseases, such as, certain sphingolipidoses, cancer, diabetes and atherosclerosis.5Gangliosides, a class of sialic acid containing glycosphingolipids, have crucial regulatory roles in tumor formation and progression.6–8 Abnormal ganglioside expression is associated with the malignancy of cancer cells9 and ganglioside content is upregulated in some metastasizing tumor cells, such as lymphoma, fibrosarcoma, and melanoma.10–12 The first step in the biosynthesis of glycosphingolipids is the glycosidation of the lipid ceramide leading to glucosylceramide (GlcCer), and further glycosylation steps leads to complex gangliosides (Scheme 1). Inhibition of the enzyme GlcCer synthase causes a tumorigenicity reduction of transformed cells, mainly due to a decrease in ganglioside levels.7 The imino-sugarN-butyl-deoxynojirimicin (NB-DNJ) (see Chart 1) inhibited melanoma tumor growth by inhibiting GlcCer synthase.13
We recently synthesized a new family of glycolipids with antimitotic activity for cultured glioma and melanoma cells.14 The results indicated that the presence of an oleyl residue linked to the glucosamine backbone increased the inhibitory activity, the oleyl glycoside 1 being the most inhibitory compound (Chart 1). Regarding potential use of 1 for the treatment of brain tumors, we recently reported15,16 the preparation of glycopolymers carrying 1 and their effect on cultured glioma cells. Preliminary studies17 on the mechanism of growth inhibition by 1 using proton magic angle spinning (MAS) NMR, suggested that the compound may be inhibiting the synthesis of tumor fatty acids, probably due to the presence of the oleyl moiety in its structure. In this sense, similar metabolite changes were observed after 24 h incubation of C6 rat glioma cells with 1 or with oleic acid (Chart 1), an inhibitor of fatty acid and cholesterol synthesis.18Fatty acid biosynthesis is upregulated in tumoral cells19 and has been proposed as a new target for cancer treatment.20
The chemical structure of 1 resembles that of GlcCer and their lipid precursors (Scheme 1), and therefore, it seemed reasonable to think that 1 could additionally alter sphingolipid and ganglioside biosynthesis in tumoral cells. Here we present the results of evaluating 1 as inhibitor of GlcCer and SM synthases and the effect on the fatty acids content and sphingolipidome of a human alveolar epithelial cells carcinoma line (A549). We examined also the effect of treatment with 1 and with two galactosylated derivatives on the ganglioside content of A549 cells.
2. Results and discussion
2.1 Effect of 1 on sphingomyelin and GlcCer synthase activity and sphingolipid content
Glycoside
1 inhibited A549 cell proliferation with an IC50 of 20 μM, a value in the concentration range of those obtained14 for rat C6 glioma (IC50, 1.3 μM), human U373 glioma (IC50, 4.3 μM) and A375 melanoma (IC50, 4.8 μM) lines. Compound 1 was previously shown to be more cytotoxic for tumor cells than for normal cells.14,15
For the evaluation of the activity of 1 as inhibitor of SM and GlcCer synthases, A549 cell lysates were used as source of enzymes and fluorescent Cer-C6-NBD as substrate. For GlcCer synthase inhibitory activity, the iminosugar NB-DNJ was used as positive control.13Glycoside 1 was inactive against SM and GlcCer synthases, even at a concentration of 250 μM (Fig. 1). On the other hand, NB-DNJ inhibited GlcCer synthase activity by 65% at 10 μM concentration, without affecting SM synthase.
 |
| | Fig. 1 The effect of compound 1 on sphingomyelin (SM) and glucosylceramide (GlcCer) synthase activity. HPLC-FD profile of fluorescent sphingolipids produced from Cer-C6-NBD in A549 cell lysates, incubated with NB-DNJ (10 μM), compound 1 (250 μM) or ethanol (1 μL), as described in the experimental section. These chromatograms are representative of three different experiments performed in duplicate. | |
To evaluate the effect of glycoside 1 on the sphingolipidome of A549 cells, compound 1 (10 μM) was incubated with the cells for 48 h and then lipids were extracted and analyzed by UPLC-MS, as described in the experimental section. For comparison, A549 cells were also cultured in the presence of 10 μM oleic acid, 50 μM NB-DNJ or vehicle (control).
Among the various sphingolipids present in cells, we focused initially on the Cer and SM content. While no effect was observed with NB-DNJ, the exposure of A549 cells to 1 caused a reduction in the content of both sphingolipids, compared to untreated cells (Fig. 2). Ceramide and sphingomyelin content was reduced 26 and 31%, respectively, the species with C24 acyl chain being that most affected (see ESI†). Oleic acid also caused a reduction on ceramides and sphingomyelins, although to a lesser extent than 1. Concerning the effect on GlcCer content, the treatment with NB-DNJ (50 μM) markedly reduced the GlcCer concentration in the cells, with a total decrease of 65% (Fig. 2). This result is in agreement with previous studies with other cell types,13 and can be ascribed to partial inhibition of GlcCer synthase activity by NB-DNJ. The GlcCer content of cells treated with compound 1 and oleic acid was also reduced, although the effect was less intense than that with NB-DNJ (Fig. 2).
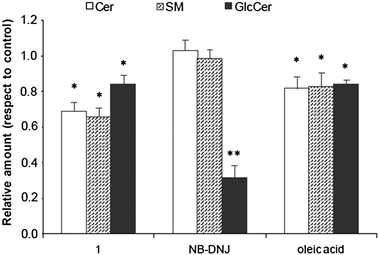 |
| | Fig. 2 Effect of 1, NB-DNJ and oleic acid on ceramide (Cer), sphingomyelin (SM) and glucosylceramide (GlcCer) content of A549 carcinoma cells. Data were normalized to the same number of cells (1 × 106cells). The content in pmol per 1 × 106cells for the control was: 214 ± 43; 3127 ± 277 and 153 ± 16 for Cer, SM and GlcCer respectively. A549 cells were seeded at 250 000 cells per well in 6-well plates. The medium was renewed after 24 h and cells were cultured in medium containing 10% FBS, in the presence of compound 1 (10 μM), NB-DNJ (50 μM), oleic acid (10 μM) or ethanol (1 μL). Two days later, the cells were extracted and analyzed as described in the experimental section. The data are the mean of two experiments performed by triplicate. The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
2.2 Effect of 1 on fatty acid content
Because glycoside 1 is not an inhibitor of the enzymes directly involved in SM and GlcCer biosynthesis, the reduction of these sphingolipids must be related to a decline in the levels of their lipid precursors. As mentioned in the introduction, preliminary studies indicate that glycoside 1 altered lipid metabolism, probably through the inhibition of fatty acid synthesis.17 To confirm this mode of action, fatty acid content was analyzed in cells treated with 1 and with oleic acid, a known inhibitor of fatty acid synthesis.18 As shown in Fig. 3, compound 1 was more effective than oleic acid in reducing fatty acid levels. When the cells were treated with compound 1, a decrease in the range of 15–40%, depending on the acyl chain length and the number of insaturations present, was observed. The decrease in fatty acid content was of the same order as for sphingomyelin and glucosylceramide levels and therefore could account for the drop in the content of those sphingolipids.
 |
| | Fig. 3 Effect of compound 1 on fatty acid production. Black bars: A549 cells treated with 1 (10 μM); empty bars: A549 cells treated with oleic acid (10 μM). The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01; ***p < 0.001. | |
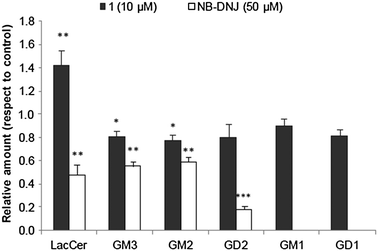 |
| | Fig. 4 The effect of compound 1 and NB-DNJ on the lactosylceramide (LacCer) and ganglioside content of A549 carcinoma cells. The data were normalized to the same number of cells (1 × 106cells). The content in pmol per 1 × 106cells for the control was: 10 ± 3; 554 ± 43; 578 ± 110; 1.3 ± 0.1; 19 ± 2 and 9 ± 1 for LacCer, GM3, GM2, GD2, GM1 and GD1 respectively. Results are referred to samples from cells treated with vehicle for 48 h. Black bars: A549 cells treated with 1 (10 μM); empty bars: A549 cells treated with NB-DNJ (50 μM). The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
To further check the generality of ganglioside depletion by treatment with 1, we tested the effect of the compound on the ganglioside content of other transformed cell lines where 1 had shown antimitotic activity,14i.e. C6 glioma and A375 melanoma cell lines. The cells were cultured in the absence (control) or the presence of 1 (10 μM) for 48 (A375 cells) or 96 h (C6 cells). After this time, gangliosides were extracted and analyzed by HPLC. Based on the elution times of standard gangliosides, and after mass spectrometric analysis of the eluted fractions, the main gangliosides detected in C6 and A375 cell extracts were identified as GM3, GM2, GM1 and GD3. The relative amounts of each ganglioside in compound 1-treated cells were compared to controls (Fig. 5). We observed a decrease in all the gangliosides in both C6 and A375 cells (45 and 20% total ganglioside reduction with respect to control for C6 and A373 cells, respectively). This general ganglioside depletion confirms that 1 must be acting at a step in the biosynthetic pathway prior to the formation of the simplest ganglioside GM3.
 |
| | Fig. 5 Relative amounts of gangliosides in melanoma A375 (black bars) and glioma C6 (empty bars) cells treated with 1 (10 μM) for 96 and 48 h, respectively. Data are means ± SD of two experiments performed in duplicate. The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
2.4 Exposure of tumor cells to higher concentrations of 1
The effect of 1 on lipid and ganglioside content was evaluated at higher concentrations. Incubation of A549 cells with compound 1 at 50 μM led to almost total cell death as observed by the small number of adhering (viable) cells after washing (Fig. 6). A significant ratio of cell death was also observed at 30 μM of 1; the analysis of lipid extracts after treatment with this concentration showed values for sphingolipid and ganglioside levels which are very different from those obtained at 10 μM concentration. As shown in Fig. 7, a significant increase of Cer, GlcCer and LacCer levels was observed in 30 μM 1-treated cells with respect to the control. These results point out that, at concentrations of 30–50 μM, compound 1 caused drastic alterations in the lipid metabolism of A549 cells (some values of sphingolipid and ganglioside levels reversed as compared to those obtained at 10 μM), and leads eventually to cell death. These results are in line with our previous findings in C6 cells,17 which indicated that above a certain concentration range (>40–50 μM), compound 1 causes apoptosis in C6 glioma cells as evidenced by the analysis of DNA-fragmentation. In this cell line, we have now performed preliminary experiments to find the mechanism by which apoptosis is induced. Recently, lipid metabolism has been related to endoplasmic reticulum (ER) stress response pathways and to apoptosis induction. To evaluate whether treatment with compound 1 induce these pathways, we used two reporters, one for the IRE1-XBP1 (inositol-requiring kinase 1) branch pathway (5xUPRE-pGL3) and a second one for the ATF6 (activating transcription factor 6) pathway (ERSE-pGL3) (Fig. 8). As a negative control a related compound previously reported by us,22octyl 6-O-pentaerythrityl-N-acetyl-α-D-glucosaminide (OPG), that was less antimitotic and did not induce apoptosis, was evaluated. As shown in Fig. 8, this compound did not induce any of the two branches. However, treatment of C6 with compound 1 at concentrations ranging from 25 to 100 μM induced ER stress response. Surprisingly, this induction was specific for the IRE1-XBP1 branch pathway (5xUPRE-pGL3), but not for the ATF6 pathways (ERSE-pGL3). Although this is a preliminary study and we have not tested other ER stress pathways (like ATF4 pathway), these data suggest that ER stress activation by compound 1 seems to be specific of the IRE1-XBP1, and not a consequence of a general loss of the ER homeostasis, that should induce ATF6 pathway too. XBP1 has been related to lipogenesis.23 Conditional deletion of XBP1 in the liver of mice reduces lipogenesis in the adult liver. Recently, Goldfinger and coworkers24 have shown that XBP1 increases de novo synthesis of ceramide. Considering the results of our present work, we propose that at low concentrations (<25 μM), compound 1 reduces ceramide and other glycolipids, but does not induce ER stress pathways and cell death. At higher concentration of compound 1 (>25 μM), IRE1-XBP1 branch was induced, increasing ceramide and other glycolipids at the same time. In this scenario, the activation of IRE1-XBP1 branch could result in a feed-back pathway, increasing ceramide and glycolipid accumulation and finally inducing cell death, as observed in A549 cells (Fig. 7).
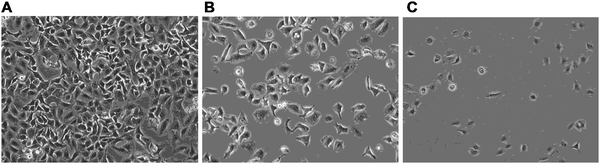 |
| | Fig. 6 Viable A549 cells without treatment (A) and after treatment with 30 and 50 μM compound 1 (B and C, respectively). | |
 |
| | Fig. 7
Ceramide (Cer), sphingomyelin (SM), glucosylceramide (GlcCer), lactosylceramide (LacCer) and gangliosides content in A549 cells treated with 1 (10 and 30 μM). Data were normalized to the same number of cells (1 × 106cells). The content in pmol per 1 × 106cells for the control was: 214 ± 43; 3127 ± 277; 153 ± 16; 10 ± 3; 554 ± 43 and 578 ± 110 for Cer, SM, GlcCer, LacCer, GM3 and GM2 respectively. Data are means ± SD of two experiments performed in duplicate. The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
 |
| | Fig. 8 Compound 1 induced IRE1-XBP1 branch of the ER stress response pathway in C6 cells. A reporter was cotransfected for (A) the ATF6 pathway (ERSE-pGL3) or (B) the IRE1-XBP1 pathway (UPRE-pGL3), and a plasmid with the β-galactosidase gene as control for transfection efficiency. Data are shown as fold induction respect to the control. Tunicamycin (Tm) and octyl 6-O-pentaerythrityl-N-acetyl-α-D-glucosaminide (OPG) were used as positive and negative controls, respectively, for the reporters. Data are means ± SD of three independent experiments performed in triplicate. The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
2.5
Metabolism of 1. Synthesis and evaluation of two galactosylated derivatives of 1
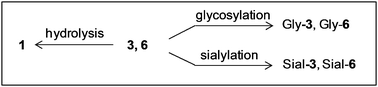
The extracts used for sphingolipidomics with A549 cells were analyzed also for the presence of glycoside 1 and putative metabolites. The presence of 1 was confirmed based on the accurate mass measurement with an error <5 ppm (m/z calculated for C26H50NO6: 472.3638; found: 472.3631, error −1.5 ppm) and its LC retention time, compared to that of an authentic sample (Fig. 9). This observation indicated that the synthetic glycolipid was efficiently taken up by the cells. The presence of oleyl alcohol, that could derived from hydrolysis of 1, was not detected in the analysis by LC-MS. Nevertheless, in order to rule out the possibility that the activity of 1 is due to a small fraction of hydrolysis product, oleyl alcohol was tested as inhibitor of A549 cell division. The IC50 value (>100 μM) was significantly higher than that of 1 and, therefore, the possible formation of oleyl alcohol could not explain the observed inhibitory activity. A new compound was also detected in the lipid extract of cells incubated with 1 that was not present in control samples. Based on its accurate mass measurement (m/z calculated for C32H60NO11: 634.4166; found 634.4164, error −0.5 ppm), its relative retention time compared to that of 1, and relative retention times of C16-LacCer and C16-GlcCer, this compound has been putatively identified as an oleyl disaccharide derived from the glycosylation of 1. We considered the possibility that this compound resulted from galactosylation of 1 by catalysis of LacCer synthase. Such a product could be, in addition to 1, an inhibitor of ganglioside biosynthesis, competing with LacCer for the GM3 synthase. To clarify this question we synthesized two β-galactosylated derivatives of 1 (disaccharides 3 and 6, Scheme 2) and tested their effect on ganglioside content. Comparison of LC retention time and exact mass (see below, Table 1) allowed us to confirm the identity of the metabolite formed from glycosylation of 1 as the β-(1,4)-linked disaccharide 6.
 |
| | Scheme 2 Synthesis of disaccharides 3 and 6: (a) penta-O-acetyl-β-D-galactopyranose, BF3OEt2, CH2Cl2–CH3CN, 0 °C, 2 h; (b) NaOMe/MeOH 0.1 M, rt, 1 h; (c) vinyl acetate, Novozym 435, dioxane, 37 °C, 12 h; (d) galactose peracetate, BF3OEt2, CH2Cl2, 0 °C, 6 h; (e) NaOMe/MeOH 1 M, rt, 30 min; (f) Ac2O, py, rt, 4 h. | |
 |
| | Fig. 9
Metabolism of compound 1 in A549 cells. Selected ion chromatograms generated by LC-MS of lipid extract of cells incubated with 1 (10 μM) or vehicle (control). Traces correspond to compound 1 (retention time 3.31 min, m/z 472.3638), its glycosylated metabolite (3.14 min, m/z 634.4166). For comparison purpose, traces of C16-glucosylceramide (6.90 min, m/z 700.5728) and C16-lactosylceramide (6.62 min, m/z 862.6255) are also shown. | |
| |
rt/mina |
Exact massa (error in ppm) |
Amount (pmol/1 × 106cell) |
Metabolite
|
|
As indicated by UPLC-MS.
|
|
3-treated cells |
3.30 |
472.3638 (1.9) |
2779 ± 297 |
1
|
| |
2.88 |
634.4166 (0.3) |
1148 ± 158 |
3
|
| |
2.70 |
796.4694 (0.0) |
19 ± 3 |
Gly-3 |
| |
2.50 |
925.5120 (−0.8) |
27 ± 2 |
Sial-3 |
|
6-treated cells |
3.30 |
472.3638 (0.6) |
1458 ± 231 |
1
|
| |
3.15 |
634.4166 (1.3) |
809 ± 146 |
6
|
| |
2.90 |
796.4694 (0.6) |
31 ± 11 |
Gly-6 |
| |
2.80 |
925.5120 (−3.0) |
34 ± 7 |
Sial-6 |
The disaccharides were synthesized in a short and straightforward manner from 1 using readily available galactose peracetate as glycosyl donor (Scheme 2). Thus, reaction of galactose peracetate with 1 gave regioselectively β-(1,6)-linked disaccharide 2, which after O-deacetylation afforded target 3. For the synthesis of 6 a highly regioselective acetylation on HO-6 of 1 was carried out by lipase-catalyzed reaction with vinyl acetate in dioxane. The 6-O-acetylated derivative 4 was the only product isolated in 85% yield. Regioselective galactosylation on HO-4 over HO-3 of 4 was obtained using BF3OEt2 as promoter, to give 5 in 31% yield with β-configured glycosidic bond. Deacetylation of 5 furnished unprotected disaccharide 6. The position of the glycosidic bond in 5 was determined from the NMR spectra of its acetylated derivative 7. The 1H NMR spectrum of 7 contained a downfield signal at δ 5.18 ppm (dd, J2,3 = 10.9, J3,4 = 8.9 Hz) for H-3, indicating that acetylation occurred at HO-3 of 6. On the other hand, the signal for H-4 appeared upfield at δ 3.79 ppm, confirming that glycosylation took place at O-4.
The effect of compounds 3 and 6 (15 μM) on sphingolipid and ganglioside content was evaluated in A549 cells. We observed that both disaccharides were partially metabolized and converted into 1 by hydrolysis of the interglycosidic bond (Scheme 3). Compound 3, with the 1,6-linkage, was more susceptible to hydrolysis than 6. We also detected in the extract of cells treated with the disaccharides the presence of two new compounds that based on their accurate mass measurement and relative retention times were assigned to neutral glycohexopyranosyl (Gly) and to sialyl (Sial) derivatives of 3 and 6 (Table 1).
 |
| | Scheme 3
Metabolism of tested disaccharides 3 and 6 in A549 cells. | |
The analysis of sphingolipid content in A549 cells after treatment with 3 and 6 showed minor changes in Cer and SM levels with respect to the control. In the case of GlcCer, the β-(1,4)-linked disaccharide induced an increase of almost 60% of this metabolite as compared to control, while the effect of the β-(1,6)-regioisomer was less intense (Fig. 10). Major changes were observed in the levels of LacCer and gangliosides (Fig. 10). The effect on LacCer levels was more pronounced with the disaccharides than with the monosaccharide 1. The treatment with 3 and 6 caused a 2–3 fold increase in LacCer content, the effect of the 1,6-linked disaccharide was slightly stronger than that of regioisomer 6. Interestingly, while the amounts of monosialo-gangliosides GM1 and GM2 decreased compared to control, the amount of the simple ganglioside GM3 increased in treated cells with respect to control. The latter effect contrasted with the reduction in GM3 level observed after treatment with 1. These results suggest that while 1 may be acting at the step of GM3 formation, the disaccharide derivatives should be interacting with enzymes involved in further steps of the ganglioside biosynthetic pathway. Overall, the results with 3 and 6 support the hypothesis that this family of synthetic compounds alters tumoral cell ganglioside metabolism, leading to significant changes in ganglioside content. In order to confirm this hypothesis, enzymatic assays with glycosyltransferases involved in early steps of ganglioside biosynthesis will have to be done in future studies.
 |
| | Fig. 10 Effect of β-(1,6)- and β-(1,4)-linked disaccharides, 3 and 6 respectively, on sphingolipid and ganglioside content. The data were normalized to the same number of cells (1 × 106cells). The content in pmol per 1 × 106cells for the control was: 214 ± 43; 3127 ± 277; 153 ± 16; 10 ± 3; 554 ± 43; 578 ± 110 and 11.3 ± 0.5 for Cer, SM, GlcCer, LacCer, GM3, GM2 and GM1 respectively. These data are representative of three different experiments performed by duplicate. The diagram includes the ANOVA results for each treatment with respect to control at a significance level of: *p < 0.05; **p < 0.01. | |
Conclusion
It is interesting that a seemingly simple oleyl glycoside exhibits a significant antiproliferative activity in a variety of tumor cell lines. In a recent patent25 describing the use of alkylated iminosugars as anticancer agents, it was found that the compounds bearing an oleyl substituent presented the most potent activity. This finding together with our previous results, in which 1 was the most potent antitumoral compound among those of a series of glucosaminide derivatives, seem to indicate that the activity of 1 as inhibitor of cancer cell growth was likely due to the conjugation of the sugar with the oleyl chain. In the present work, we have investigated the mode of action of 1 under the hypothesis that it could be altering the metabolism of lipids, as indicated in our previous 1H MAS NMR study. We have now found that oleyl glycoside 1 at 10 μM downregulated the synthesis of fatty acids and sphingolipids in A549 cells. In addition, glycoside 1 decreased ganglioside content of human A549, rat C6 and human A375 cells. The intervention on cellular processes important for tumor development may help to understand its antiproliferative activity.
On the other hand, compound 1 at higher concentrations (above 30 μM) caused drastic alterations in glycosphingolipid metabolism, displaying cell metabolic profiles very different from those obtained at 10 μM concentration, and led to cell death. Preliminary results seem to indicate that 1 at concentrations of 25–100 μM activates ER stress response pathways. More precisely, compound 1 induced IRE1-XBP1 branch pathway, but not ATF6 pathway. This induction could explain the drastic changes observed in glycosphingolipid metabolism since transcription factor XBP1 has been identified as a regulator of lipogenesis.
Further work to confirm the interaction of 1 with enzymes involved in ganglioside metabolism and the activation of ER stress response pathways is the aim of our future research.
3. Experimental
3.1 Chemistry
3.1.1 General methods.
Chemicals were purchased puriss p.A. from commercial suppliers or purified by standard techniques. Thin-layer chromatography (TLC) was performed on aluminium sheets 60 F254 Merck silica gel and compounds were visualized by irradiation with UV light and/or by treatment with a solution of Ce2MoO4 or 5% H2SO4 in EtOH, followed by heating. Flash column chromatography was performed using thick walled columns, employing silica gel (Merck 60: 0.040–0.063 mm). The eluent used is indicated, and solvent ratios refer to volume. Melting points are not corrected. Optical rotations were recorded on a Perkin Elmer 241 Polarimeter (λ = 589 nm, 1 dm cell). 1H NMR spectra were registered at 500, 400 or 300 MHz, and 13C NMR were obtained at 125 or 100 MHz using CD3OD as solvent at room temperature. Chemical shift values are reported in parts per million (δ). Coupling constant values (J) are reported in hertz (Hz), and spin multiplicities are indicated by the following symbol: s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet). Mass spectroscopy spectra were registered on a hp series 1100 MSD spectrometer.
3.1.2
Oleyl 2-acetamido-6-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-2-deoxy-α-D-glucopyranoside (2).
To a solution of 1 (360 mg, 0.77 mmol) in anhydrous CH2Cl2–CH3CN (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 40 ml), penta-O-acetyl-β-D-galactopyranose (300 mg, 0.77 mmol) was added and the mixture was cooled to 0 °C. Then, BF3·OEt2 (0.1 ml, 1.3 mmol) was added and the solution was stirred at 0 °C under argon for 2 h. After this time, the reaction mixture was treated with Et3N (0.2 ml), concentrated in vacuo and the residue was purified by silica gel column chromatography (EtOAc) to afford 2 (183 mg, 30%) as a white solid. mp: 67–69 °C; [α]D = +15.3° (C 0.7, MeOH); 1H NMR (400 MHz, CD3OD): δ 5.4–5.3 (m, 3H), 5.2–5.1 (m, 2H), 4.74 (d, 1H, J = 3.6 Hz), 4.67 (dd, 1H, J = 9.0, 4.9 Hz), 4.2–4.1 (m, 4H), 3.84 (dd, 1H, J = 10.7, 3.5 Hz), 3.8–3.6 (m, 4H), 3.4–3.3 (m, 1H), 3.3–3.2 (m, 1H), 2.13 (s, 3H), 2.1–1.9 (m, 16H), 1.5–1.4 (m, 2H), 1.4–1.2 (m, 22H), 0.89 (t, J = 6.7 Hz) ppm; 13C NMR (125 MHz, CD3OD): δ 172.1, 170.6, 170.5, 170.05, 169.9, 129.4, 129.4, 101.3, 96.8, 71.3, 71.1, 70.1, 70.9, 70.3, 69.3, 69.0, 67.4, 67.3, 61.1, 54.0, 31.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.0, 29.0, 28.9, 28.9, 26.7, 26.7, 25.8, 22.3, 21.1, 20.6, 19.4, 19.1, 19.1, 19.1, 13.0 ppm; HRMS (ESI) m/z Anal. calcd. for C40H68NO15: 802.4589, found 802.4589 (M + H)+.
:1, 40 ml), penta-O-acetyl-β-D-galactopyranose (300 mg, 0.77 mmol) was added and the mixture was cooled to 0 °C. Then, BF3·OEt2 (0.1 ml, 1.3 mmol) was added and the solution was stirred at 0 °C under argon for 2 h. After this time, the reaction mixture was treated with Et3N (0.2 ml), concentrated in vacuo and the residue was purified by silica gel column chromatography (EtOAc) to afford 2 (183 mg, 30%) as a white solid. mp: 67–69 °C; [α]D = +15.3° (C 0.7, MeOH); 1H NMR (400 MHz, CD3OD): δ 5.4–5.3 (m, 3H), 5.2–5.1 (m, 2H), 4.74 (d, 1H, J = 3.6 Hz), 4.67 (dd, 1H, J = 9.0, 4.9 Hz), 4.2–4.1 (m, 4H), 3.84 (dd, 1H, J = 10.7, 3.5 Hz), 3.8–3.6 (m, 4H), 3.4–3.3 (m, 1H), 3.3–3.2 (m, 1H), 2.13 (s, 3H), 2.1–1.9 (m, 16H), 1.5–1.4 (m, 2H), 1.4–1.2 (m, 22H), 0.89 (t, J = 6.7 Hz) ppm; 13C NMR (125 MHz, CD3OD): δ 172.1, 170.6, 170.5, 170.05, 169.9, 129.4, 129.4, 101.3, 96.8, 71.3, 71.1, 70.1, 70.9, 70.3, 69.3, 69.0, 67.4, 67.3, 61.1, 54.0, 31.6, 29.5, 29.4, 29.3, 29.3, 29.2, 29.0, 29.0, 28.9, 28.9, 26.7, 26.7, 25.8, 22.3, 21.1, 20.6, 19.4, 19.1, 19.1, 19.1, 13.0 ppm; HRMS (ESI) m/z Anal. calcd. for C40H68NO15: 802.4589, found 802.4589 (M + H)+.
3.1.3
Oleyl 2-acetamido-2-deoxy-6-O-(β-D-galactopyranosyl)-α-D-glucopyranoside (3).
2 (140 mg, 0.17 mmol) was treated with a 0.1 M solution of NaOMe in MeOH (3 ml), and the solution was stirred at room temperature for 1 h. After this time, the mixture was neutralized with Amberlite IR-120 (hydrogen form). The Amberlite was filtered, the solvent was removed in vacuo and the residue was purified by silica gel column chromatography (EtOAc–MeOH 5:1 → 2:1) to give 3 (94 mg, 87%) as a white solid. mp: 172–178 °C; [α]D = +34.4° (C 2.9, MeOH); 1H NMR (400 MHz, CD3OD): δ: 5.4–5.3 (m, 2H), 4.77 (d, 1H, J = 3.6 Hz), 4.30 (d, 1H, J = 7.6 Hz), 4.13 (dd, 1H, J = 10.9, 2.0 Hz), 3.89 (dd, 1H, J = 10.7, 3.6 Hz), 3.9–3.6 (m, 7H), 3.6–3.4 (m, 4H), 3.4–3.3 (m,1H), 2.0–2.0 (m,4H), 1.98 (s, 3H), 1.6–1.5 (m, 2H), 1.4–1.3 (m, 22H), 0.90 (t, 3H, J = 6.9 Hz) ppm; 13C NMR (100 MHz, CD3OD): δ 173.6, 130.8, 130.8, 105.3, 98.5, 76.7, 75.0, 72.7, 72.6, 72.5, 72.2, 70.3, 69.7, 69.1, 62.5, 55.4, 30.9, 30.8, 30.8, 30.7, 30.6, 30.6, 30.5, 30.4, 30.4, 30.3, 28.2, 28.1, 27.3, 23.8, 22.6, 14.5 ppm; HRMS (ESI) m/z Anal. calcd. for C32H60NO11: 634.4166, found: 634.4158 (M + H)+.
3.1.4
Oleyl 2-acetamido-6-O-acetyl-2-deoxy-α-D-glucopyranoside (4).
1 (70.3 mg, 0.15 mmol) was dissolved in dioxane (3 ml) and Novozym 435 lipase (30 mg, 10 mg ml−1) was added. Then, the mixture was treated with vinyl acetate (0.15 mL, 1.6 mmol) and was stirred at 37 °C in an orbitalic shaker (150 rpm) for 12 h. The lipase was filtered, the solvent was removed in vacuo and the residue was purified by silica gel column chromatography (EtOAc) to give 4 (64.9 mg, 85%) as a white solid. mp: 110–111 °C; [α]D = +89.2° (C 0.5, MeOH); 1H NMR (400 MHz, CD3OD): δ: 5.4–5.3 (m, 2H), 4.76 (d, 1H, J = 3.6 Hz), 4.37 (dd, 1H, J = 11.8, 2.1 Hz), 4.20 (dd, 1H, J = 11.8, 6.0 Hz), 3.88 (dd, 1H, J = 10.7, 3.6 Hz), 3.77 (ddd, 1H, J = 9.9, 6.0, 2.0 Hz), 3.7–3.6 (m, 2H), 3.5–3.3 (m, 2H, H-4), 2.2–2.0 (m, 7H), 1.98 (s, 3H), 1.7–1.5 (m, 2H), 1.4–1.3 (m, 22H), 0.90 (t, 3H, J = 6.8 Hz) ppm; 13C NMR (100 MHz, CD3OD): δ 173.6, 172.7, 130.9, 130.9, 98.6, 72.7, 72.5, 71.3, 69.2, 65.0, 55.5, 33.7, 33.2, 31.0, 30.9, 30.9, 30.8, 30.7, 30.6, 30.6, 30.6, 30.5, 30.5, 30.3, 28.3, 28.2, 27.4, 23.8, 22.7, 20.9, 14.6 ppm; HRMS (ESI) m/z Anal. calcd. for C28H52NO7: 514.3743, found:: 514.3745 (M + H)+.
3.1.5
Oleyl 2-acetamido-6-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-2-deoxy-α-D-glucopyranoside (5).
4 (622 mg, 1.21 mmol) was dissolved in anhydrous CH2Cl2 (36 ml) and penta-O-acetyl-β-D-galactopyranose (946 mg, 2.42 mmol) was added. The mixture was cooled at 0 °C and then treated with BF3·OEt2 (1.26 ml, 10.9 mmol) and stirred at that temperature for 6 h. After this time, the reaction mixture was treated with Et3N (1.5 ml), concentrated in vacuo and the residue was purified by silica gel column chromatography (Hexane-EtOAc, 1:1 → 0:1) to give 5 (319 mg, 31%) as a white solid. mp: 53–55 °C; [α]D = +60.9° (C 3.0, MeOH); 1H NMR (400 MHz, CD3OD): δ: 5.40 (dd, 1H, J = 6.1, 1.6 Hz), 5.4–5.3 (m, 2H), 5.2–5.1 (m, 2H), 4.81 (d, 1H, J = 7.7 Hz), 4.77 (d, 1H, J = 3.6 Hz), 4.34 (dd, 1H, J = 11.8, 1.9 Hz), 4.3–4.2 (m, 1H), 4.2–4.1 (m, 2H), 4.09 (dd, 1H, J = 11.8, 5.9 Hz), 3.92 (dd, 1H, J = 10.7, 3.6 Hz), 3.9–3.7 (m, 2H), 3.66 (dt, 1H, J = 9.9, 6.6 Hz), 3.6–3.5 (m, 1H), 3.41 (dt, 1H, J = 9.9, 6.3 Hz), 2.21 (s, 3H), 2.1–1.9 (m, 19 H), 1.7–1.5 (m, 2H), 1.4–1.3 (m, 22H), 0.90 (t, 3H, J = 6.9 Hz) ppm; 13C NMR (100 MHz, CD3OD): δ 173.4, 172.5, 172.2, 171.8, 171.3, 171.3, 130.8, 130.8, 102.5, 98.1, 83.4, 72.4, 72.4, 70.8, 70.5, 69.3, 69.1, 68.8, 64.3, 62.8, 54.9, 33.6, 33.1, 30.9, 30.8, 30.8, 30.7, 30.6, 30.5, 30.5, 30.4, 30.4, 30.2, 28.2, 28.1, 27.3, 23.7, 22.5, 20.9, 20.7, 20.6, 20.5, 20.4, 14.5 ppm; HRMS (ESI) m/z Anal. calcd. for C42H70NO16: 844.4689, found: 844.4699 (M + H)+.
3.1.6 Oleyl 2-acetamido-2-deoxy-4-O-(β-D-galactopyranosyl-α-D-glucopyranoside (6).
5 (115 mg, 0.14 mmol) was dissolved in 20 mL MeOH and treated with a 1 M solution of NaOMe (2 ml). The reaction mixture was stirred at room temperature for 30 min. After this time, the mixture was neutralized with Amberlite IR-120 (hydrogen form). The Amberlite was filtered, the solvent was removed in vacuo and the residue was purified by silica gel column chromatography (EtOAc–MeOH 5:1 → 2:1) to give 6 (83.9 mg, 97%) as a white solid. mp: 162–169 °C; [α]D = +67.5° (C 1, MeOH); 1H NMR (400 MHz, CD3OD, COSY): δ: 5.4–5.3 (m, 2H), 4.79 (d, 1H, J = 3.5 Hz), 4.37 (d, 1H, J = 7.4 Hz), 3.91 (dd, 1H, J = 10.8, 3.6 Hz), 3.9–3.8 (m, 3H), 3.8–3.5 (m, 8H), 3.49 (dd, 1H, J = 9.7, 3.2 Hz), 3.4–3.3 (m, 1H), 2.1–2.0 (m, 4H), 1.98 (s, 3H), 1.6–1.5 (m, 2H), 1.5–1.25 (m, 22H), 0.90 (t, 3H, J = 6.9 Hz) ppm; 13C NMR (100 MHz, CD3OD): δ 172.3, 129.7, 129.6, 103.9, 97.0, 80.1, 75.9, 73.6, 71.4, 71.0, 69.8, 69.1, 67.9, 61.3, 60.7, 53.9, 31.9, 29.70, 29.6, 29.6, 29.5, 29.4, 29.4, 29.3, 29.2, 29.1, 27.0, 26.9, 26.1, 22.6, 21.4, 13.3 ppm; HRMS (ESI) m/z Anal. calcd. for C32H60NO11: 634.4166, found: 634.4166 (M + H)+.
3.1.7
Oleyl 2-acetamido-3,6-di-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl)-2-deoxy-α-D-glucopyranoside (7).
5 (19 mg, 0.02 mmol) was dissolved in pyridine (0.5 ml) and acetic anhydride (0.5 ml) was added. The reaction mixture was stirred at room temperature for 4 h. After this time, the solvent was evaporated in vacuo and the crude was purified by silica gel column chromatography (EtOAc–hexane 2:1) to give 7 (19.5 mg, 98%) as a white solid. 1H NMR (500 MHz, CD3OD, COSY): δ: 5.4–5.3 (m, 3H), 5.18 (dd, 1H, J = 10.9, 8.9 Hz), 5.11 (dd, 1H, J = 10.4, 3.5 Hz), 5.02 (dd, 1H, J = 10.4, 7.9 Hz), 4.74 (d, 1H, J = 3.7 Hz), 4.69 (d, 1H, J = 7.9 Hz), 4.47 (dd, 1H, J = 11.8, 2.0 Hz), 4.2–4.1 (m, 5H), 3.9–3.8 (m, 1H), 3.79 (dd, 1H, J = 10.0, 9.0 Hz), 3.68 (dt, 1H, J = 9.8, 6.9 Hz), 3.5–3.4 (m, 1H), 2.2–2.1 (m, 6H), 2.1–2.0 (s, 3H), 2.0–1.9 (m, 16H), 1.7–1.6 (m, 2H), 1.5–1.2 (m, 22H), 0.93 (t, 3H, J = 6.9 Hz) ppm, 13C NMR (126 MHz, CD3OD): δ 173.5, 172.3, 172.2, 172.0, 171.9, 171.4, 171.1, 130.9, 130.8, 102.1, 98.3, 78.1, 72.7, 72.5, 71.7, 70.8, 70.0, 69.5, 68.6, 63.9, 62.3, 53.2, 33.0, 30.9, 30.8, 30.8, 30.7, 30.6, 30.6, 30.5, 30.4, 30.4, 30.3, 30.3, 28.1, 28.1, 27.3, 24.2, 23.7, 22.4, 21.2, 20.7, 20.7, 20.6, 20.5, 14.4 ppm.
3.2 Analytical experiments
3.2.2
Liquid chromatography-mass spectrometry analysis of sphingolipids.
A549
cells (2.5 × 105cells mL−1) were seeded in 1 mL of medium with 10% FBS in a 6-well plates. Twenty-four hours later, media were replaced with fresh medium containing test compound or vehicle solvent and cultured for 48 h at 37 °C/5% CO2. Then, cells were washed in PBS, collected by brief trypsinization and transferred to glass vials. An aliquot of the cells was taken for cell counting, using a Countess automatic cell counter. Sphingolipid extracts, spiked with internal standards, were prepared as described26 and analysed by liquid chromatography–mass spectrometry using a Waters Aquity UPLC system connected to a Waters LCT Premier orthogonal accelerated time of flight mass spectrometer (Waters, Millford, MA), operated in positive electrospray ionisation mode in the conditions previously reported.27
3.2.3 C6 and A375 ganglioside analysis.
C6 and A375 cells (2 × 105cells cm−2) were seeded in 30 mL of DMEM medium with 10% FBS in plastic flasks and allowed to attach for 6 h. After the cells were attached to the substrate, the medium was changed to DMEM without serum. Twenty-four hours later, media were replaced with fresh medium containing compound 1 (10 μM) and cultured for 48 h (A375) or 96 h (C6) at 37 °C/5% CO2. Then, cells were washed twice with 15 mM PBS (140 mM NaCl, pH = 7.4), collected by brief trypsinization and pelleted by centrifugation at 2000 g for 5 min at 4 °C. Then, cells were lyophilized, finely powdered and extracted twice with chloroform–methanol (1:1, 2.5 mL) to obtain the total lipid extract. The combined extracts were cooled at 4 °C overnight, and centrifuged to remove insoluble material. Total lipid extract was dried completely under reduced pressure and gangliosides were isolated by extracting with diisopropyl ether-1-butanol (6:4) and 0.1% aqueous NaCl (1 mL) and subsequent filtration on Sephadex G-50 gel.13Gangliosides were lyophilized and analyzed by HPLC. HPLC analysis. All analyses were performed using a Jasco Pu-2089 Plus pump equipped with an ultraviolet Jasco UV-2075 Plus detector and a 20 μL Rheodyne injector. Data acquisition and processing were accomplished with the Jasco ChromPass Chromatography Data System 1.8.6.1 software. A normal phase Kromasil-NH2 (5 μm, 4.6 × 250 mm) column was used as stationary phase. Ganglioside separation was carried out with a mobile phase made with the following gradient mixture: solvent A, acetonitrile–5 mM sodium phosphate buffer, pH 5.6 (83:17); solvent B, acetonitrile-20 mM sodium phosphate buffer, pH 5.6 (50:50). The gradient elution program was the same as described earlier.28 The flow rate was 1.0 mL min−1 and the elution profile was monitored by recording the UV absorbance at 215 nm. Retention times of individual gangliosides were compared with the retention times of commercially available pure gangliosides. The isolated gangliosides were collected into fractions for MALDI-TOF mass spectrometric analysis.
3.3 Enzymatic activity
3.3.1
Cells and cultures.
Human alveolar epithelial A549, rat glioma C6 cells and human melanoma A375 cells were obtained from the American Type Culture Collection (ATCC) and grown in medium HAM F12 (A549) and DMEM (C6 and A375) with glutamine medium supplemented with 10% foetal bovine serum (FBS). Cell proliferation assays were performed as described.27 The number of viable and dead cells was determined by Trypan Blue staining and using a Countess® Cell Counter (Invitrogen), following the manufacturer's recommendations.
3.3.2
Glucosylceramide and sphingomyelin synthase activity (A549).
A549
cells cultured as described above were washed with sodium phosphate (PBS) (10 mM, 137 mM NaCl, pH = 7.4) and collected by brief trypsinization. The cells were then washed twice with PBS and resuspended in 50 mM TRIS-HCl, pH = 7.4, 10 mM MgCl2 and lysed. The cell lysate (100 μL) was incubated with inhibitors for 10 min at 37 °C and NAD (25 μL, 16 mM in 50 mM TRIS-HCl buffer, pH = 7.4, 10 mM MgCl2), BSA/Cer-C6-NBD complex (52 μL, 1:1, 20 μM in 50 mM TRIS-HCl buffer, pH = 7.4, 10 mM MgCl2) and UDP-Glucose (25 μL, 2 mM in 50 mM TRIS-HCl buffer, pH = 7.4, 10 mM MgCl2) were added and incubations were prolonged for 35 min. The reaction was stopped by adding methanol (800 μL) to each reaction mixture and centrifuging at 13000 g for 3 min. HPLC coupled to a fluorescence detector was used to measure the glucosylceramide synthase and sphingomyelin synthase activities by injecting 25 μL of the supernatant. HPLC analysis: 15 cm reverse-phase C18 column, flow rate 1 mL min−1, column temperature 25 °C, mobile phase 25% H2O-75% acetonitrile, both with a 0.1% trifluoroacetic acid. Fluorescence detector, excitation at 470 nm and emission at 530 nm. Calibration curves showed linear response in the concentration ranges of the analyses.
3.4
Luciferase reporter assays in C6 glioma cells
To evaluate the activation of IRE1-XBP1 and ATF6 pathways by compound 1 treatment, we used a UPRE luciferase reporter (5xUPRE-pGL3) and ERSE-pGL3 reporter, respectively. C6 glioma cells (2 × 104cells per well) were cotransfected with 5 × UPRE-pGL3 or ERSE-pGL3 constructs, and a β-galactosidase construct, as a control for transfection efficiency. As a transfection reagent, we used Fugene HD (Roche, Mannheim, Germany) according to the manufacturer's protocol. After 48 h of treatment, the cells were washed with PBS, passive lysis buffer (Promega) was added to each well and the samples were stored at −80 °C until use. Luciferase activity was measured in a Sirius luminometer (Berthold detection systems, Pforzheim, Germany). We used a Multiskan Ascent (Thermo Electron Corporation, Shanghai, China) to detect β-galactosidase activity in the samples.
Acknowledgements
The financial support provided by the Servicio de Salud de Castilla La Mancha Community (SESCAM), FISCAM (PI-2008/18 and PI-2008/19), and the Ministry of Ciencia e Innovación (CTQ2007-67403/BQU and SAF2008-00706) is greatly appreciated. We thank Dr Gemma Fabriàs for her encouragement and insightful suggestions and Mrs Eva Dalmau for excellent technical assistance.
Notes and references
- P. Boyle and J. Ferlay, Ann. Oncol., 2005, 16, 481–488 CrossRef CAS.
- J. Ferlay, P. Autier, M. Boniol, M. Heanue, M. Colombet and P. Boyle, Ann. Oncol., 2007, 18, 581–592 CAS.
- A. Jemal, R. Siegel, E. Ward, T. Murray, J. Xu and M. J. Thun, Ca–Cancer J. Clin., 2007, 57, 43–66 Search PubMed.
- D. E. Maziak, B. R. Markman, J. A. MacKay and W. E. Evans, Ann. Thorac. Surg., 2004, 77, 1484–91 CrossRef.
- A. Delgado, J. Casas, A. Llebaria, J. L. Abad and G. Fabriàs, ChemMedChem, 2007, 2, 580–606 CrossRef CAS.
- S. Hakomori, Cancer Res., 1996, 56, 5309–5318 CAS.
- S. Birklé, G. Zeng, L. Gao, R. K. Yu and J. Aubry, Biochimie, 2003, 85, 455–463 CrossRef CAS.
- S. Lahiri and A. H. Futerman, Cell. Mol. Life Sci., 2007, 64, 2270–2284 CrossRef CAS.
- E. V. Dyatlovitskaya and A. G. Dandyyba, Biochemistry (Moscow), 2006, 71, 347–343 CrossRef CAS.
- S. Ruggieri, G. Mungai, A. Mannini, L. Calorini, A. Fallani, E. Barietta, G. Mannori and O. Cecconi, Clin. Exp. Metastasis, 1999, 17, 271–276 CrossRef CAS.
- S. Laferte, M. N. Fukuda, M. Fukuda, A. Dell and J. W. Dennis, Cancer Res., 1987, 47, 150–159 CAS.
- S. Saha and D. C. Mohanty, J. Exp. Clin. Cancer Res., 2003, 22, 125–134 Search PubMed.
- M. Guerrera and S. Ladisch, Cancer Lett., 2003, 201, 31–40 CrossRef CAS.
- I. García-Álvarez, G. Corrales, E. Doncel-Pérez, A. Muñoz, M. Nieto-Sampedro and A. Fernández-Mayoralas, J. Med. Chem., 2007, 50, 364–373 CrossRef CAS.
- M. L. López Donaire, J. Parra-Cáceres, B. Vázquez-Lasa, I. García-Álvarez, A. Fernández-Mayoralas A. López-Bravo and J. San Román, Biomaterials, 2009, 30, 1613–1626 CrossRef CAS.
- M. L. López-Donaire, M. Fernández-Gutiérrez, J. Parra-Cáceres, B. Vázquez-Lasa, I. García-Álvarez, A. Fernández-Mayoralas and J. San Román, Acta Biomater., 2010, 6, 1360–1369 CrossRef CAS.
- I. García-Álvarez, L. Garrido, E. Doncel-Pérez, M. Nieto-Sampedro and A. Fernández-Mayoralas, J. Med. Chem., 2009, 52, 1263–1267 CrossRef CAS.
- F. Natali, L. Siculella, S. Salvati and G. V. Gnoni, J. Lipid Res., 2007, 48, 1966–1975 CAS.
- J. A. Menendez and R. Lupu, Nat. Rev., 2007, 7, 763–777 CAS.
- F. P. Kuhajda, Cancer Res., 2006, 66, 5977–5980 CrossRef CAS.
- Y. Gu, J. Zhang, W. Mi, J. Yang, F. Han, X. Lu and W. Yu, Breast Cancer Res., 2008, 10, R1 CrossRef.
- A. Fernández-Mayoralas, N. De la Figuera, M. Zurita, J. Vaquero, G. A. Abraham, J. San Román and M. Nieto-Sampedro, J. Med. Chem., 2003, 46, 1286–1288 CrossRef CAS.
- A.-H. Lee, E. F. Scapa, D. E. Cohen and L. H. Glimcher, Science, 2008, 320, 1492–1496 CrossRef CAS.
- M. Goldfinger, E. L. Laviad, R. Hadar, M. Shmuel, A. Dagan, H. Park, A. H. Merrill, Jr, I. Ringel, A. H. Futerman and B. Tirosh, J. Immunol., 2009, 182, 7038–7047 CrossRef CAS.
- C. Bello and P. Vogel, PCT Int. Appl. Search PubMed
WO 2009/118712 A2, 2009.
- R. L. Shaner, J. C. Allegood, H. Park, E. Wang, S. Kelly, C. A. Haynes, M. C. Sullards and A. H. Merril, Jr., J. Lipid Res., 2009, 50, 1692–1707 CAS.
- D. Canals, D. Mormeneo, G. Fabriàs, A. Llebaria, J. Casas and A. Delgado, Bioorg. Med. Chem., 2009, 17, 235–41 CrossRef CAS.
- G. Gazzotti, S. Sonnino and R. Ghidoni, J. Chromatogr., A, 1985, 348, 371–378 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here. 
