Mechanistic understanding of Pyrococcus horikoshii Dph2, a [4Fe–4S] enzyme required for diphthamide biosynthesis†
Received
29th June 2010
, Accepted 16th September 2010
First published on 8th October 2010
Abstract
Diphthamide, the target of diphtheria toxin, is a unique posttranslational modification on eukaryotic and archaeal translation elongation factor 2 (EF2). The proposed biosynthesis of diphthamide involves three steps and we have recently found that in Pyrococcus horikoshii (P. horikoshii), the first step uses an S-adenosyl-L-methionine (SAM)-dependent [4Fe–4S] enzyme, PhDph2, to catalyze the formation of a C–C bond. Crystal structure shows that PhDph2 is a homodimer and each monomer contains three conserved cysteine residues that can bind a [4Fe–4S] cluster. In the reduced state, the [4Fe–4S] cluster can provide one electron to reductively cleave the bound SAM molecule. However, different from classical radical SAM family of enzymes, biochemical evidence suggest that a 3-amino-3-carboxypropyl radical is generated in PhDph2. Here we present evidence supporting that the 3-amino-3-carboxypropyl radical does not undergo hydrogen abstraction reaction, which is observed for the deoxyadenosyl radical in classical radical SAM enzymes. Instead, the 3-amino-3-carboxypropyl radical is added to the imidazole ring in the pathway towards the formation of the product. Furthermore, our data suggest that the chemistry requires only one [4Fe–4S] cluster to be present in the PhDph2 dimer.
Introduction
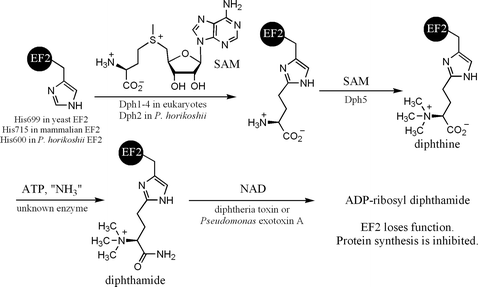
Diphthamide, found in both eukaryotes and archaea,1–3 is a unique posttranslational modification on a histidine residue of translational elongation factor 2 (EF2). EF2 is a GTPase required for ribosomal protein synthesis.4 Diphthamide is the target of diphtheria toxin,5 which ADP-ribosylates diphthamide and inhibits protein synthesis, leading to host cell death.6 Diphthamide was reported to help prevent -1 frame shift mutation during protein synthesis.7 The biosynthesis of the modification is still not completely understood,8 despite the fact that the modification has been identified for over three decades.1 Genetic studies have identified five genes, Dph1, Dph2, Dph3, Dph4, and Dph5, in eukaryotes required for the biosynthesis of diphthamide and a three-step biosynthesis pathway involving these genes has been proposed.9,10 The first step is the transfer of the 3-amino-3-carboxypropyl group from S-L-adenosylmethionine (SAM) to the C-2 position of the imidazole ring of the target histidine residue in EF2. Dph1-4 are known to be required for the first step.6,9,11–15 The second step, catalyzed by Dph5,16 is the trimethylation of the amino group to form an intermediate called diphthine. The last step is the ATP-dependent amidation of the carboxyl group of diphthine (Fig. 1). The enzyme that catalyzes the last step has not been identified. Diphthamide is also found in archaea. However, among the five eukaryotic genes required for diphthamide biosynthesis, only two orthologs can be found in archaea by BLAST search. One of them, Dph2, is homologous to eukaryotic Dph1 and Dph2 (Dph1 and Dph2 are homologous to each other), and the other one is the diphthine synthase, Dph5.
Recently, we have successfully reconstituted the first step of diphthamide biosynthesis using the Pyrococcus horikoshii Dph2 (PhDph2) and EF2 (PhEF2) (Fig. 1). PhDph2 forms a homodimer. Each monomer has three conserved cysteine residues (Cys59, Cys163 and Cys287) that can bind a [4Fe–4S] cluster. Similar to radical SAM enzymes, the [4Fe–4S] cluster in PhDph2 binds SAM and in the reduced state can provide one electron to reductively cleave SAM. However, unlike radical SAM enzymes, our evidence suggest that PhDph2 generates a 3-amino-3-carboxypropyl (ACP) radical, instead of a 5′-deoxyadenosyl radical.17
Although our previous study suggests that an ACP radical is responsible for the C–C bond formation reaction,17 details of the reaction mechanism still need to be determined. For example, how does the ACP radical react with the imidazole ring to form the C–C bond? Does the reaction require each of the monomers to contain a [4Fe–4S] cluster, or is only one [4Fe–4S] cluster per dimer sufficient? These two questions may be connected to each other, as indicated by the two possible mechanistic proposals shown in Fig. 2. Here we report our efforts in trying to differentiate these two mechanisms using mutagenesis to generate a “heterodimer” of PhDph2, with one wild type monomer that contains a [4Fe–4S] cluster and a mutant monomer that is incapable of binding a [4Fe–4S] cluster. Our evidence suggest that the chemistry only requires one [4Fe–4S] cluster to be present per PhDph2 dimer, consistent with the mechanism in Fig. 2a. In addition, in the presence of PhEF2, we showed that 2-aminobutyrate is not formed, further supporting the mechanism shown in Fig. 2a.
![Two possible mechanisms for PhDph2-catalyzed reaction. (a) One [4Fe–4S] cluster per PhDph2 dimer is sufficient for the reaction and the ACP radical adds to the imidazole ring; (b) Two [4Fe–4S] clusters per PhDph2 dimer are required for the reaction. The ACP radical abstracts a hydrogen atom from the imidazole ring. As a consequence, one reaction needs two SAM with one ACP transferred to PhEF2 and the other one released as 2-aminobutyric acid.](/image/article/2011/MB/c0mb00076k/c0mb00076k-f2.gif) |
| | Fig. 2 Two possible mechanisms for PhDph2-catalyzed reaction. (a) One [4Fe–4S] cluster per PhDph2 dimer is sufficient for the reaction and the ACP radical adds to the imidazole ring; (b) Two [4Fe–4S] clusters per PhDph2 dimer are required for the reaction. The ACP radical abstracts a hydrogen atom from the imidazole ring. As a consequence, one reaction needs two SAM with one ACP transferred to PhEF2 and the other one released as 2-aminobutyric acid. | |
Results
Single cysteine to alanine mutants of PhDph2 can still bind [4Fe–4S] clusters while double mutants cannot
Our previous work showed that PhDph2 forms a homodimer and each monomer contains three conserved cysteine residues (Cysteine59, Cysteine163, and Cysteine287) that can bind a [4Fe–4S] cluster. In order to make a PhDph2 heterodimer with only one subunit capable of binding a [4Fe–4S] cluster, we decided to mutate these conserved cysteine residues to alanine. All three single mutants (C59A, C163A and C287A) purified anaerobically have a brown color, similar to that of wild type PhDph2, indicative of the binding of a [4Fe–4S] cluster. Ultraviolet-visible (UV-Vis) spectroscopy and electron paramagnetic resonance (EPR) spectroscopy were obtained to further confirm that these single mutants can bind an Fe–S cluster. All three single mutants of PhDph2 have absorption at 400 nm, similar to the wild type PhDph2 (Fig. 3a). The absorption disappears upon reduction by 0.5 mM dithionite (Fig. 3b). The 400 nm absorption is consistent with several known radical SAM enzymes, including AtsB,18 HemN,19 and MoaA.20 Upon reduction with dithionite, the wild type PhDph2 shows a strong EPR absorption with an average g factor of ∼1.935 (Fig. 3c). The temperature dependence of the EPR spectrum, which is best resolved at ∼12 K and becomes unobservable above 30 K, is typical for rapidly relaxing [4Fe–4S]1+ clusters. EPR spectra of PhDph2 single mutants are in general very similar to wild type (Fig. 3c), suggesting that they do bind Fe–S cluster. The most remarkable difference in the EPR spectrum of the reduced form is observed for C163A, which exhibits pronounced rhombic main features with greatly increased g-value anisotropy. For both C59A and C287A, the g-tensors are more axial than wild type. The changes in EPR spectra in the mutants probably reflect the changes of the local environment introduced by the mutation.
 |
| | Fig. 3 Spectroscopic characterization of PhDph2 mutants. (a) UV-Vis spectra of purified PhDph2 wild type (black line), single mutants: C59A (blue line), C163A (red line), C287A (green line) and double mutant (pink line); (b) UV-Vis spectra of PhDph2 C59A with and without dithionite; (c) EPR spectra of wild type PhDph2 and its single mutants in the reduced dithionite reduced state at 12 K. The principal values of g-factor for the main spectral component (shown for wild type and C163A with arrows of corresponding color) are: WT: 2.03, 1.92, 1.86; C287A: 2.01, 1.94, 1.88. S59A: 2.03, 1.94, 1.90; C163A: 2.11, 1.91, 1.793. | |
We next constructed a double cysteine mutant of PhDph2, C59A/C287A. This double mutant, when anaerobically purified, has no color, indicating the lack of Fe–S cluster. UV-Vis spectrum (Fig. 3a, pink line) confirmed the lack of Fe–S cluster. Thus, the single cysteine mutant can bind Fe–S cluster whereas the double mutants cannot.
Single mutants of PhDph2 have activity and the double mutant does not
Wild type PhDph2 can catalyze the transfer of 3-amino-3-carboxypropyl group from SAM onto His600 of PhEF2, which can be detected by autoradiography if carboxy-14C-SAM is used. 5’-Deoxy-5’-methylthioadenosine (MTA), the small molecule product derived from SAM, can be monitored by HPLC.17 Using these two methods, we tested whether the single and double cysteine mutants of PhDph2 have activity or not (Fig. 4). All three single mutants (C59A, C163A, and C287A) were able to transfer the ACP group onto His600 of PhEF2 (Fig. 4a) and to produce MTA (Fig. 4b). However, the single mutants are not as active as the wild type PhDph2, as judged by the amount of MTA formed in the reaction (Fig. 4b). Unlike the single mutants, the double mutant of PhDph2 (C59A/C287A) has no activity (Fig. 7a, lanes 11 and 14), consistent with the finding that the double mutant cannot bind a [4Fe–4S] cluster.
 |
| | Fig. 4 Activity assay of PhDph2 single mutants monitored by autoradiography and HPLC. (a) 14C-labeling of PhEF2 by PhDph2 wild type and three single mutants. The reactions contain PhEF2, PhDph2 (WT or mutants), SAM, and dithionite. Left panel shows the Coomassie Blue-stained gel and right panel shows the autoradiography. (b) Reaction product methylthioadenosine (MTA) formation was detected by HPLC. SAM was eluted at 2 min and MTA was eluted at 21.5 min. | |
Expression and purification of PhDph2 heterodimer
To obtain a PhDph2 heterodimer with only one subunit capable of binding a [4Fe–4S] cluster to investigate the reaction mechanism, we co-expressed PhDph2 wild type with a His6 tag (WT-His6) and the C59A/C287A double mutant with a GST tag (DM-GST). The proteins were first purified by a Ni affinity purification to give a mixture of PhDph2 WT-His6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :WT-His6 homodimer and WT-His6:DM-GST heterodimer. The mixture was then subjected to a glutathione affinity purification to give the DM-GST:WT-His6 heterodimer (Fig. 5). The homodimers WT-His6:WT-His6 and DM-GST:DM-GST can be obtained by further purifying the flow through from the two affinity purification steps (Fig. 5a). The homodimer and heterodimer after such purifications were checked by SDS-PAGE as shown in Fig. 5b and c, which demonstrates that our tandem purification strategy works as outlined in Fig. 5a.
:WT-His6 homodimer and WT-His6:DM-GST heterodimer. The mixture was then subjected to a glutathione affinity purification to give the DM-GST:WT-His6 heterodimer (Fig. 5). The homodimers WT-His6:WT-His6 and DM-GST:DM-GST can be obtained by further purifying the flow through from the two affinity purification steps (Fig. 5a). The homodimer and heterodimer after such purifications were checked by SDS-PAGE as shown in Fig. 5b and c, which demonstrates that our tandem purification strategy works as outlined in Fig. 5a.
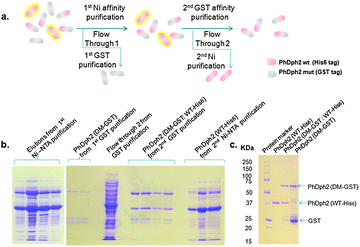 |
| | Fig. 5 Tandem purification to get different PhDph2 dimers. (a) Diagram showing the tandem purification strategy to get different PhDph2 dimers; (b) Tandem purification of PhDph2 dimers: left, elutions from Ni-NTA purification by using different concentrations of imidazole solutions to get PhDph2 (WT-His6) and PhDph2 (DM-GST:WT-His6) mixture; Right, purification of PhDph2 (DM-GST), PhDph2 (DM-GST:WT-His6), and PhDph2 (WT-His6). PhDph2 (WT-His6). (c) Purified PhDph2 dimers. The PhDph2 (WT-His6) shown here was further purified by heating at 95 °C. | |
PhDph2 heterodimer is stable
In order to use the PhDph2 heterodimer to study the reaction mechanism, we have to make sure that the heterodimer is stable. If it is not stable and subunits can dissociate and reassociate, then homodimers of WT and double mutant will form in the reaction and complicate the data analysis. The homodimers WT-His6:WT-His6 and DM-GST:DM-GST were first mixed to allow subunits exchange, if there was any. The mixture was then incubated with Ni-NTA resin. If subunits exchange occurs, the heterodimer DM-GST:WT-His6 will form, which will bind to the Ni-resin, leading to the retaining of DM-GST on the resin. If subunits exchange did not occur, only the WT-His6 will be retained on the Ni-resin. Proteins retained on Ni-resin can then be eluted with imidazole and analyzed by SDS-PAGE. The detection of the GST-tagged protein in the imidazole elutions on SDS-PAGE would indicate that subunits exchange has occurred. Based on this analysis, PhDph2 WT-His6 (∼34 kDa per monomer, Fig. 5C, lane 2) and DM-GST (about 60 kDa per monomer, Fig. 5C, lane 4) can indeed undergo subunit exchange when mixed together, because DM-GST was detected on SDS-PAGE in the imidazole elutions (Fig. 6b, lane 2 and 3). By the same analysis, if subunit exchange occurs to the PhDph2 heterodimer (WT-His6:DM-GST), then the homodimer with GST-tagged double mutant will form which cannot be retained on the Ni-resin. When we carried out the experiment with the heterodimer, no DM-GST was detected in the flow through on SDS-PAGE (Fig. 6b, lane 5) and both WT-His6 and DM-GST were found in the elutions from Ni-resin (Fig. 6b, lane 6). As a control, when we used the homodimer of DM-GST, we can readily detect it in the flow through (Fig. 6a, lane 1). Taken together, we found that the homodimers are not stable, but the heterodimer is stable.
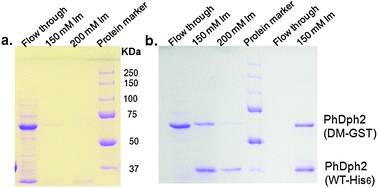 |
| | Fig. 6 Stability of PhDph2 homodimer and PhDph2 (DM-GST:WT-His6) heterodimer. (a) PhDph2 (DM-GST) cannot bind to Ni resin, it was found in the flow through of Ni-affinty purification but not in the elutions. (b) Stability test. Left of the protein marker: PhDph2 (WT-His6) and (DM-GST) were mixed, incubated for 90 min, and then purified with Ni-resin. PhDph2 (DM-GST) was found in flow through; PhDph2 (DM-GST:WT-His6) and PhDph2 (WT-His6) were eluted from Ni resin by 150 mM and 200 mM imidazole; Right of the protein marker: PhDph2 (DM-GST:WT-His6) heterodimer from tandem purification was incubated with Ni-resin then eluted with 150 mM imidazole. No homodimer was found in the flow through. | |
The PhDph2 heterodimer is active
To test whether the first step of diphthamide biosynthesis requires the PhDph2 dimer to have two [4Fe–4S] clusters or only one is sufficient, we set up reactions either with the PhDph2 heterodimer (WT-His6:DM-GST) or with the WT homodimer. The heterodimer, like wild type PhDph2, can catalyze the transfer of 3-amino-3-carboxylpropyl from SAM to EF2, as indicated by the 14C-labeling experiment (Fig. 7a). Similar results were obtained when we monitored the formation of MTA, by HPLC (Fig. 7c blue and brown line). In contrast, the DM-GST homodimer has no activity (Fig. 7c lane4, pink line). In order to make sure that the reaction rates catalyzed by heterodimer and WT homodimer was the same, we monitored the reaction time. The results shown in Fig. 7a and b indicate that the rate catalyzed by heterodimer was essentially the same as that catalyzed by wild type. These results suggest that only one [4Fe–4S] cluster per PhDph2 dimer is required for the reaction, which is consistent with the mechanism shown in Fig. 2a.
No aminobutyric acid is formed when excess PhEF2 is present
Our previous results showed that in the absence of PhEF2, PhDph2 can catalyze the cleavage of SAM to form 2-aminobutyric acid (ABA).17 However, whether ABA is formed when excess PhEF2 is present will depend on the detailed reaction mechanism. In the mechanism shown in Fig. 2a, no ABA will form in the presence of excess PhEF2. In contrast, in the mechanism shown in Fig. 2b, ABA will form in the presence of excess PhEF2. To further differentiate the two mechanisms shown in Fig. 2, we detected the formation of ABA by LCMS after dansylation. In the absence of PhEF2, ABA was formed, consistent with our previous results.17 With excess PhEF2, no ABA was detected (the level is the same as the negative control, Fig. 8). The data provide further support for the mechanism shown in Fig. 2a.
 |
| | Fig. 7 Activity assay of PhDph2 heterodimer. (a) Time course of reactions catalyzed by PhDph2 wild type (WT-His6) homodimer, PhDph2 double mutant and wild type heterodimer (DM-GST:WT-His6) and PhDph2 double mutant (DM-GST) homodimer by using 14C-labeled SAM. The upper panel shows the Coomassie blue-stained gel and the lower panel shows the autoradiography. (b) Plot of the intensity of labelled PhEF2 (shown on the autoradiography in (a)) versus time. (c) The formation of MTA catalyzed by different PhDph2 dimers was detected by HPLC. | |
 |
| | Fig. 8 Detection of dansylated 2-aminobutyric acid (ABA) by LCMS. Left panel shows the MS traces and right panel shows the LC traces. Components used in different reactions were labeled in the figure. ABA was only formed in the reaction with PhDph2 but without PhEF2. | |
Discussion
Our previous work showed that PhDph2 forms a homodimer.17 Each subunit consists of three domains. Three cysteine residues (Cysteine59, Cysteine163, and Cysteine287), each from a different domain, are clustered in the center of each PhDph2 subunit to bind a [4Fe–4S] cluster. The three cysteine residues are conserved in eukaryotic Dph1 proteins but only two of the three cysteine residues are conserved in eukaryotic Dph2 proteins. It is intriguing whether all three cysteine residues in archaeal PhDph2 are required for enzyme activity and whether the PhDph2 dimer requires one or both subunits to contain a [4Fe–4S] cluster for activity.
To test whether all the three conserved cysteine residues are required for the activity of PhDph2, we performed site directed mutagenesis to change the three cysteine residues to alanine. We then studied how such mutations affect the enzyme activity by monitoring the reaction products (the modified PhEF2 and MTA). The results showed that mutating one of the three cysteine residues did not affect the activity much (Fig. 4). Thus, two conserved cysteine residues are sufficient for the activity of PhDph2. The spectroscopic data also suggest that the single cysteine mutants can still bind a [4Fe–4S] cluster (Fig. 3). However, after changing two of the cystein residues to alanine (C59A/C287A), PhDph2 cannot bind to the Fe–S cluster and is inactive.
To gain further insights into the detailed reaction mechanism of PhDph2, we examined the activity of a heterodimer of PhDph2 consisting of one wild type subunit and one C59A/C287A double mutant. In this heterodimer, the wild type subunit can bind a [4Fe–4S] cluster while the double mutant cannot. Our stability measurement suggests that the heterodimer is more stable than the homodimers. The reason for this stability is not clear. It is possible that the PhDph2 dimer with one [4Fe–4S] cluster is thermodynamically more stable. Alternatively, the GST tag on the double mutant may somehow disfavor the formation of the double mutant homodimer. Under the experimental conditions tested, the heterodimer is as active as the wild type PhDph2 homodimer (Fig. 7). The results support the mechanism shown in Fig. 2a and indicates that only one [4Fe–4S] cluster per PhDph2 dimer is sufficient for the reaction. This conclusion is consistent with our previous structure of PhDph2, in which only one [4Fe–4S] is found to be present in the dimer.17 Furthermore, we demonstrated that when excess PhEF2 was present, no ABA was formed in the reaction (Fig. 8). This result indicates that the ACP radical generated does not undergo hydrogen abstraction in the presence of PhEF2 and thus the addition to the imidazole ring is more likely, which further supports the mechanism shown in Fig. 2a. It is interesting to note that in classical Radical SAM enzymes, the deoxyadenosyl radical generated always does hydrogen abstraction to carry out the enzymatic reaction.21,22 In contrast, in PhDph2, all our evidences suggest that the 3-amino-3-carboxypropyl radical should undergo an addition reaction instead of hydrogen abstraction. Thus, the non-classical Radical SAM enzyme PhDph2 differ not only in the radical species derived from SAM,17,21 but also in the downstream chemistry that the radical undergoes.
Experimental procedures
Cloning, expression, and purification of PhDph2, PhDph2 mutants and PhEF2
Cloning, protein expression and purification of PhDph2 and PhEF2 were reported.17 PhDph2 site directed mutants (C59A, C163A, C287A and C59A/C287A) were carried out by overlap-extension polymerase chain reaction (PCR)23 with AccuPrime Pfx DNA polymerase (Invitrogen). The constructed mutant genes were inserted into pENTR™/TEV/D-TOPO® entry vector (Invitrogen). The amplified plasmid was purified using QIAprep® Spin Miniprep Kit (QIAGEN) and its sequence was confirmed by DNA sequencing (performed by Cornell University Life Sciences Core Laboratories Center). PhDph2 C59A and C287A single mutants were recombined with destination vector pDESTF1 to create expression clones with an N-terminal His6 tag. The double mutant was recombined with pDEST15 with an N-terminal GST tag. A different cloning method was used for PhDph2 C163A. The PCR product was digested by EcoRI and SalI and the mutant gene was ligated into pET28a vector by T4 DNA ligase.
Construction and tandem purification of PhDph2 heterodimer
The plasmids containing PhDph2 wild type with His6 tag (WT-His6) in pCDFduet vector and PhDph2 C59A/C287A double mutant with GST tag (DM-GST) in pDEST15 vector were co-transformed into the E. coli expression strain BL21(DE3) with pRARE2. The cells were grown in LB media with 100 μg/ml ampicillin, 25 μg/ml chloramphenicol, and 50 μg/ml streptomycin at 37 °C and induced at OD600 of 0.8 with 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG). The induced cells were incubated in a shaker (New Brunswick Scientific Excella E25) at 37 °C and 200 rpm for 3 h. Cells were harvested by centrifugation at 6371 g (Beckman Coulter Avanti J-E) and 4 °C for 10 min. Cell pellets were transferred into an anaerobic chamber (Coy Laboratory Products) and pellets from 2L of LB culture were re-suspended in 30 ml lysis buffer (500 mM NaCl, 10 mM MgCl2, 5 mM imidazole, and 20 mM Tris-HCl at pH 8.0) with 0.01% lysozyme, which were stored and de-oxygenated in the anaerobic chamber. The resuspended cells were sealed in a polypropylene tube and taken out for freezing with liquid nitrogen and then thawed at 26 °C in the anaerobic chamber to lyse the cells. The sealed tube containing the cell lysis were taken out of the anaerobic chamber and centrigued at 48400 g (Beckman Coulter Avanti J-E) for 30 min and then taken back into the anaerobic chamber for further processing. The supernatant was incubated for 1 h with 1.2 ml Ni-NTA resin (Qiagen) pre-washed with water and equilibrated with the lysis buffer. The resin was then loaded onto a polypropylene column and washed with 20 ml lysis buffer. PhDph2 heterodimer (WT-His6:DM-GST) and wild type homodimer (WT-His6:WT-His6) were eluted from the column with elution buffers (100 mM, 150 mM and 200 mM imidazole in the lysis buffer, 1.5 ml each). The protein was buffer-exchanged to GST bind/wash buffer (43 mM Na2HPO4, 14.7 mM KH2PO4, 1.37 M NaCl and 27 mM KCl) using a Bio-Rad 10–DG desalting column. The heterodimer was further purified by incubating with GST binding resin (Novagen) for 2 h at 26 °C in the anaerobic chamber. The resin was then loaded onto a polypropylene column and washed with 20 ml GST bind/wash buffer. PhDph2 heterodimer was eluted from the column with 4 ml GST elution buffer (10 mM glutathione in 50 mM Tris-HCl buffer, pH 8.0). DM-GST in the flow-through from the Ni resin purification was purified with GST binding resin. WT-His6 from the flow through of the GST purification was incubated with Ni-Resin to concentrated and then further purified by heating at 90 °C for 5 min.
PhDph2 dimer stability test
PhDph2 homodimers (DM-GST:DM-GST and WT-His6:WT-His6) were mixed in 1:1 ratio and incubated at 26 °C for 1.5 h in an anaerobic chamber (Coy Laboratory Products). The mixture was then incubated with Ni-NTA resin for 1 h at 26 °C and loaded onto a polypropylene column, washed with lysis buffer (20 mM Tris-HCl with 500 mM NaCl, 10 mM MgCl2 and 5 mM imidazole, pH 8.0), and eluted with 100 mM, 150 mM and 200 mM imidazole in the lysis buffer. Both flow through and elution fractions were checked by SDS-PAGE. PhDph2 (DM-GST:WT-His6) from the tandem purification was incubated at both 26 and 37 °C for 1.5 h before incubating with Ni-resin and elution from Ni-resin.
Samples of PhDph2 and mutants (50 μM), with and without dithionite, were prepared anaerobically in 150 mM NaCl solution and 200 mM Tris-HCl at pH 7.4. The sample treated with dithionite was allowed to incubate for 30 min after adding the reducing reagent at a final concentration of 0.5 mM. The samples were sealed in a quartz cell (100 μl each) before taking out from the anaerobic chamber. UV-Vis spectra were obtained on a Cary 50 Bio UV-Visible spectrophotometer (Varian), scanning from 200 nm to 800 nm. The baseline was corrected with the buffer used to prepare the samples.
ESR spectra were recorded on a Bruker EMX (BRUKER, Billerica, MA) spectrometer at a frequency of 9.24 GHz under standard conditions in 4 mm ID quartz tubes. A liquid helium cryostat, ESR-10 (Oxford Instruments Ltd, England) was used to stabilize the temperature in the range of 4–50 K. Spectra shown in Fig. 2d are recorded with a modulation amplitude of 8G and microwave power 0.63 mW.
Activity assay of PhDph2 wild type and mutants by autoradiography and HPLC
The reaction components, 12 μM PhEF2, 40 μM PhDph2 mutants (C59A and C287A), and 10 mM dithionite, were mixed in 150 mM NaCl, 1 mM DTT, and 200 mM Tris-HCl at pH 7.4 to a final volume of 15 μl in the anaerobic chamber under strictly anaerobic conditions. The reaction vials were sealed before taking out of the anaerobic chamber. Carboxy-14C-SAM (2 μL, final concentration of 27 μM) was injected into each reaction vial to initiate the reaction. The reaction mixtures were vortexed briefly to mix and incubated at 65 °C for 40 min. The reaction was stopped by adding loading buffer to the reaction mixture and subsequent heating at 100 °C for 10 min, which was resolved by 12% SDS-PAGE. The dried gel was exposed to a PhosphorImaging screen (GE Healthcare, Piscataway, NJ) and the radioactivity was detected using a STORM860 phosphorimager (GE Healthcare, Piscataway, NJ).
The reactions of PhEF2 and PhDph2 single mutants/double mutant/heterodimer analyzed by HPLC are similar to those of analyzed by radio labeling except that they were scaled up to a final volume of 60 μl and a normal SAM instead of a 14C SAM was used. The reaction mixture was quenched by 5% TFA and centrifuged to separate the precipitated proteins and the supernatant. The supernatant was analyzed by HPLC (Shimadzu) on a C18 column (HαSprite) monitored at 260 nm absorbance, using a linear gradient from 0 to 40% buffer B in 20 min at a flow rate of 0.3 ml min−1 (buffer A: 50 mM ammonium acetate, pH 5.4; buffer B, 50% (v/v) methanol/water).
Time course reaction of PhDph2 wild type and heterodimer
PhDph2 wild type (10 μM)/heterodimer (20 μM)/double mutants (10 μM) was incubated with PhEF2 (25 μM), carboxy-14C-SAM (20 μM) and dithionite (5 mM) in a vial sealed with a rubber cap at 37 °C. The amount of iron sulfur cluster contained in wild type and heterodimer are the same (based on the absorbance at 400 nm). Eight microliter reaction mixture was taken out at 2 min, 4 min, 6 min, 12 min, 20 min, and 50 min and the reaction was stopped by freezing at −20 °C. Reaction with PhDph2 double mutants was monitored at 20 min and 50 min. Reaction mixtures at different time slots were mixed with gel loading dye and subsequent heated at 100 °C for 10 min, then resolved by 12% SDS-PAGE. The dried gel was exposed to a PhosphorImaging screen (GE Healthcare, Piscataway, NJ) and the radioactivity was detected using a STORM860 phosphorimager (GE Healthcare, Piscataway, NJ). The signal was quantified by Image Quant TL (Amersham Biosciences). The intensity of 14C-labeled PhEF2 was plotted vs. time.
Dansylation of the reaction products and detection by LC-MS
To differentiate the two possible mechanisms, reactions catalyzed by PhDph2 with excess of PhEF2 or without PhEF2 were set up anaerobically. The reaction products were characterized by dansylation reaction which was monitored by LC-MS as previously described.17
Abbreviations
Acknowledgements
This work is partly supported by the Camille and Henry Dreyfus New Faculty Award Program and NIH (R01GM088276) to H.L. EPR measurement was supported by NIH/NCRR P41-RR016292 for the ACERT Center Grant to J.H.F.
References
- E. A. Robinson, O. Henriksen and E. S. Maxwell, J. Biol. Chem., 1974, 249, 5088–5093 CAS.
- B. G. Van Ness, J. B. Howard and J. W. Bodley, J. Biol. Chem., 1980, 255, 10710–10716.
- B. G. Van Ness, J. B. Howard and J. W. Bodley, J. Biol. Chem., 1980, 255, 10717–10720.
- M. G. Gomez-Lorenzo, C. M. T. Spahn, R. K. Agrawal, R. A. Grassucci, P. Penczek, K. Chakraburtty, J. P. G. Ballesta, J. L. Lavandera, J. F. Garcia-Bustos and J. Frank, EMBO J., 2000, 19, 2710–2718 CrossRef CAS.
- R. J. Collier, Toxicon, 2001, 39, 1793–1803 CrossRef CAS.
- S. Liu, G. T. Milne, J. G. Kuremsky, G. R. Fink and S. H. Leppla, Mol. Cell. Biol., 2004, 24, 9487–9497 CrossRef CAS.
- P. A. Ortiz, R. Ulloque, G. K. Kihara, H. Zheng and T. G. Kinzy, J. Biol. Chem., 2006, 281, 32639–32648 CrossRef CAS.
-
C. T. Walsh, Posttranslational Modifications of Proteins: Expanding Nature's Inventory, Roberts and Company Publishers, Englewood, Colorado, 2006 Search PubMed.
- T. J. Moehring, D. E. Danley and J. M. Moehring, Mol. Cell. Biol., 1984, 4, 642–650 CAS.
- P. C. Dunlop and J. W. Bodley, J. Biol. Chem., 1983, 258, 4754–4758 CAS.
- J. M. Moehring, T. J. Moehring and D. E. Danley, Proc. Natl. Acad. Sci. U. S. A., 1980, 77, 1010–1014 CrossRef CAS.
- J. Y. Chen, J. W. Bodley and D. M. Livingston, Mol. Cell. Biol., 1985, 5, 3357–3360 CAS.
- L. C. Mattheakis, F. Sor and R. J. Collier, Gene, 1993, 132, 149 CrossRef CAS.
- N. J. Phillips, M. R. Ziegler and L. L. Deaven, Cancer Lett., 1996, 102, 85 CrossRef CAS.
- D. C. Schultz, B. R. Balasara, J. R. Testa and A. K. Godwin, Genomics, 1998, 52, 186 CrossRef CAS.
- L. C. Mattheakis, W. H. Shen and R. J. Collier, Mol. Cell Biol., 1992, 12, 4026–4037 CAS.
- Y. Zhang, X. Zhu, A. T. Torelli, M. Lee, B. Dzikovski, R. M. Koralewski, E. Wang, J. Freed, C. Krebs, S. E. Ealick and H. Lin, Nature, 2010, 465, 891–896 CrossRef CAS.
- T. L. Grove, K.-H. Lee, J. St. Clair, C. Krebs and S. J. Booker, Biochemistry, 2008, 47, 7523–7538 CrossRef CAS.
- G. Layer, K. Verfurth, E. Mahlitz and D. Jahn, J. Biol. Chem., 2002, 277, 34136–34142 CrossRef CAS.
- P. Hanzelmann, H. L. Hernandez, C. Menzel, R. Garcia-Serres, B. H. Huynh, M. K. Johnson, R. R. Mendel and H. Schindelin, J. Biol. Chem., 2004, 279, 34721–34732 CrossRef.
- K. S. Duschene, S. E. Veneziano, S. C. Silver and J. B. Broderick, Curr. Opin. Chem. Biol., 2009, 13, 74–83 CrossRef CAS.
- P. A. Frey, A. D. Hegeman and F. J. Ruzicka, Crit. Rev. Biochem. Mol. Biol., 2008, 43, 63–88 CrossRef CAS.
- M. M. Ling and B. H. Robinson, Anal. Biochem., 1997, 254, 157–178 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.