Heterologous production of thiostrepton A and biosynthetic engineering of thiostrepton analogs†‡
Chaoxuan
Li§
,
Feifei
Zhang§
and
Wendy L.
Kelly
*
School of Chemistry and Biochemistry and the Parker H. Petit Institute for Bioengineering and Bioscience, Georgia Institute of Technology, Atlanta, Georgia 30332, USA. E-mail: wendy.kelly@chemistry.gatech.edu; Fax: (001) 404-894-2295; Tel: (001) 404-783-4922
First published on 25th November 2010
Abstract
Thiostrepton A 1, produced by Streptomyces laurentii ATCC 31255 (S. laurentii), is one of the more well-recognized thiopeptide metabolites. Thiostrepton A 1 and other thiopeptides are of great interest due to their potent activities against emerging antibiotic-resistant Gram-positive pathogens. Although numerous lines of evidence have established that the thiopeptides arise from the post-translational modification of ribosomally-synthesized peptides, few details have been revealed concerning this elaborate process. Alteration to the primary amino acid sequence of the precursor peptide provides an avenue to probe the substrate specificity of the thiostrepton post-translational machinery. Due to the difficulties in the genetic manipulation of S. laurentii, the heterologous production of thiostrepton A 1 from an alternate streptomycete host was sought to facilitate the biosynthetic investigations of the peptide metabolite. The production of thiostrepton A 1 from the non-cognate hosts did not lend itself to be as robust as S. laurentii-based production, therefore an alternate strategy was pursued for the production of thiostrepton variants. The introduction of a fosmid used in the heterologous production of thiostrepton A 1, harboring the entire thiostrepton biosynthetic gene cluster, into the tsrA deletion mutant permitted restoration of thiostrepton A 1 production near to that of the wild-type level. The fosmid was then engineered to enable the replacement of wild-type tsrA. Introduction of expression fosmids encoding alternate TsrA sequences into the S. laurentii tsrA deletion mutant led to the production of thiostrepton variants retaining antibacterial activity, demonstrating the utility of this expression platform toward thiopeptide engineering.
Introduction
The thiopeptides are a family of highly modified peptide antibiotics, first discovered over half a century ago (Fig. 1).1 Dozens of thiopeptides have since been isolated from diverse genera of Gram-positive bacteria, including species of Bacillus, Micrococcus, Nocardia, Staphylococcus, and Streptomyces.2–7 From the viewpoint of human medicine, one of the most attractive characteristics of the thiopeptides is their highly potent activities against Gram-positive pathogens such as methicillin-resistant Staphylococcus aureus (MRSA) and penicillin-resistant Streptococcus pneumoniae (PRSP).8 Certain thiopeptides have also demonstrated anticancer and antimalarial properties.9–13 Clinical development of the thiopeptides, however, is currently limited in part due to the low water solubility and bioavailability of these agents. The complex structures of the thiopeptides place constraints on the ease in which analogs could be produced by total synthesis. Therefore, considerable interest lies in co-opting thiopeptide biosynthetic machinery toward the engineering of thiopeptide derivatives.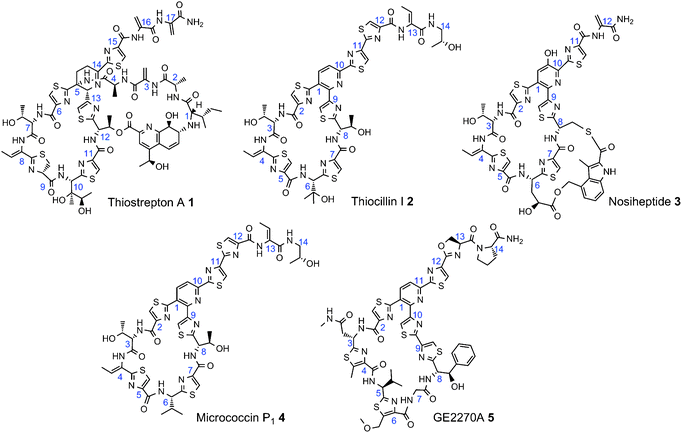 | ||
| Fig. 1 Examples of thiopeptide antibiotics. Numbers in blue are placed next to the α-carbon of the corresponding amino acid from the propeptide sequence. | ||
This latter goal is now feasible. Several thiopeptide biosynthetic gene clusters were recently reported, revealing that the thiopeptides arise from an array of post-translational modifications upon a ribosomally-synthesized precursor peptide.14–18 Thus, a site-directed mutant of the precursor peptide should enable the production of a corresponding thiopeptide derivative, provided that the amino acid substitution is tolerated by all (or nearly all) of the elements of the thiopeptide post-translational modification system. Indeed, thiocillin I 2 variants with comparable antibacterial activities to the parent thiopeptide have been generated by this approach.19,20
Thiostrepton A 1 (Fig. 1), a bacteriocin produced by Streptomyces laurentii ATCC 31255 (S. laurentii), is one of the more extensively studied members of the thiopeptide antibiotics.7 In addition to the distinctive core thiopeptide macrocycle, thiostrepton A 1 possesses the added structural complexity of a second macrocycle in which the quinaldic acid serves as a linker between a threonine side chain of the core macrocycle and the amino terminus of the structural peptide. The proposed thiostrepton biosynthetic gene (tsr) cluster is nearly 30 kb in length, containing 21 open reading frames.15,17 TsrA is the thiostrepton precursor peptide, proposed to be 58 amino acids in length and composed of a 41 amino acid leader peptide followed by a 17 amino acid propeptide (Fig. 2).15,17 Considerably more genetic and biochemical studies are required to uncover the details concerning the transformation of the preproantibiotic into thiostrepton A 1, a process requiring over one dozen chemical modifications. Although gene inactivation can be performed in the thiostrepton producer S. laurentii, this strain has been recalcitrant to further manipulation of the thiostrepton biosynthetic locus, including attempts to replace the gene encoding the precursor peptide with one encoding an alternate amino acid sequence.15,21 One strategy to circumvent this limitation of S. laurentii is to heterologously express the entire tsrcluster in a more genetically tractable host bacterium. Such an approach may permit the multiple genetic manipulations that would be necessary to engineer the production of thiostrepton analogs. Herein we first report the heterologous production of thiostrepton A 1 by two Streptomyceshost strains. We then adapt the platform developed for heterologous production toward a pliable system for the generation of thiostrepton analogs from S. laurentii itself.
 | ||
| Fig. 2 TsrA and thiostrepton A 1. | ||
Results and discussion
Heterologous production of thiostrepton A in Streptomyceshosts
An S. laurentii genomic fosmid library was previously constructed using the vector pCC1FOS (Epicentre Biotechnologies), and it was determined that fosmid JA3A10 possessed the entire proposed tsrcluster.15 Using λ Red-mediated recombination, the fosmid was retrofitted with the elements necessary to support the intergeneric transfer from E. coli into a Streptomyces species and the integration into a Streptomyceschromosome.22–25 The chloramphenicol resistance-imparting gene of the fosmid backbone was replaced with a cassette from pSET152 containing an integrase (int), an attP sequence, an origin of transfer (oriT), and an apramycin-resistance gene (aac(3)IV) to yield int-3A10.26 A control fosmid was also prepared in this fashion from pCC1FOS to provide int-pCC1FOS.The heterologous hosts chosen for this study were Streptomyces lividansTK24 (S. lividans) and Streptomyces actuosus ATCC 25421 (S. actuosus). Since the established resistance-imparting gene for thiostrepton A 1 is not co-localized with the tsrcluster, it was necessary to take additional measures in S. lividans to avoid any toxicity that would result from thiostrepton A 1 production.15,27 The vector pSE34, which harbors the rRNA methyltransferase imparting thiostrepton A 1 resistance, was introduced into S. lividans to yield S. lividans/pSE34. S. actuosus, on the other hand, produces another thiopeptide antibiotic, nosiheptide 3.28 This strain is inherently cross-resistant to thiostrepton A 1, negating the need to introduce a gene specifically conferring thiostrepton A resistance.29,30 The retrofitted fosmids were then introduced into S. lividans/pSE34 and S. actuosus by intergeneric conjugation to provide S. lividansFZ1 and S. actuosusFZ1 (strains containing int-3A10) and S. lividansFZ2 and S. actuosusFZ2 (strains containing int-pCC1FOS). Analysis of the culture extracts by HPLC and HPLC-MS revealed that thiostrepton A 1 was indeed produced in S. actuosusFZ1 and S. lividansFZ1 (Fig. 3). In contrast, none of the strains possessing int-pCC1FOS, which lacks the tsrcluster, produced thiostrepton A 1 (Fig. 3). Parallel with these studies, int-3A10 and int-pCC1FOS were introduced into Streptomyces coelicolor CH999 (S. coelicolor), but no thiostrepton A 1 was detected in the culture extract from the S. coelicolorhost strain (data not shown).
 | ||
| Fig. 3 HPLC analysis of extracts from the cultures of (A) wild-type S. laurentii, (B) S. lividansFZ2, (C) S.lividansFZ1, (D) S. actuosusFZ2, (E) S. actuosusFZ1. The asterisks indicate thiostrepton. Absorbance was monitored at 254 nm. | ||
The ability of int-3A10 to impart thiostrepton-production capabilities upon its heterologous host demonstrates that all essential genes for the biosynthesis of the antibiotic are contained within the fosmid int-3A10. Unfortunately, the production of thiostrepton A 1 in S. lividansFZ1 (0.2 mg L−1) and S. actuosus 3A10 (1.0 mg L−1) was significantly lower than the level of thiostrepton A 1 production in S. laurentii (44 mg L−1). A potential complication to the heterologous production of thiostrepton A 1 and other thiopeptides is the presence of the thiostrepton-inducible proteins in various streptomycetes, including strains of S. lividans and S. coelicolor.30–32 The TipAS and TipAL proteins from S. lividans 66 have been characterized to some detail. In response to complexation with thiostrepton A 1, TipAL activates its own promoter, ptipA, and induces the expression of two alternate translation products from the tipAgene: TipAL (itself) and TipAS.33 TipAS, the dominant thiostrepton-induced protein in S. lividans 66, forms an irreversible lanthionine adduct upon binding thiostrepton A 1, using a TipAS cysteine residue to attack a thiostrepton A 1dehydrolanine residue and sequester the thiopeptide.34 Although this may explain one limitation to achieving high-yield production of thiostrepton A 1 from non-cognate streptomycetes, it is unlikely to be the sole contributing factor. In addition to its interactions with TipAL and TipAS, thiostrepton A 1 has been implicated in modulating the expression of other genes in actinomycetes.31–33 Furthermore, it is not unusual to observe lower titres of a metabolite from a heterologous hostversus that of the native producer, and optimization of the fermentation conditions may overcome low production of the metabolite.35,36 For example, the production of epothilone D in Myxococcus xanthus was increased from less than 0.2 mg L−1 to 23 mg L−1 through a series of modifications to the culture conditions.37 In any event, the low titre of thiostrepton A production in S. lividansFZ1 and S. actuosusFZ1 restricts the utility of this integrative fosmid-based heterologous production platform to query the roles of the thiostrepton biosynthetic genes or to engineer thiostrepton derivatives. Genetic manipulations of the tsrcluster are apt to generate biosynthetic intermediates or analogs produced at lower levels relative to the wild-type biosynthetic system, and such metabolites may easily escape the limits of detection if using the heterologous hosts developed here. Therefore, rather than undergoing a complex and likely extensive effort in an attempt to improve thiostrepton production in either heterologous host, an alternative strategy for the generation of thiostrepton variants was pursued.
Deletion of tsrA in S. laurentii
Given the concentration-dependent limitations of thiostrepton production encountered in the heterologous hosts, the approach used for the genetic inactivation of tsrA was re-evaluated. Previously, an apramycin resistance cassette was inserted into the tsrA sequence, leading to the loss of thiostrepton production.15In trans complementation of tsrA in this mutant strain, however, never met with success, raising the possibility of a secondary effect within the insertional mutant.21 In an effort to overcome this limitation of complementation and, ultimately, to spur the production of thiostrepton variants, a markerless, in-frame deletion mutant of tsrA was constructed to provide S. laurentiiNDS1. As observed in the tsrA insertional mutant, deletion of tsrA in S. laurentii also abolished thiostrepton production (Fig. 4). Unfortunately, the re-introduction of tsrA alone into S. laurentiiNDS1 was still insufficient to revive thiostrepton production (data not shown). Introduction of the integrative fosmid int-3A10 into S. laurentiiNDS1 to yield S. laurentiiNDS1/int-3A10, however, did permit the robust production of thiostrepton A 1 at 113 ± 35 mg L−1 (Fig. 4). | ||
| Fig. 4 HPLC analysis of extracts from the cultures of (A) wild-type S. laurentii, (B) S. laurentiiNDS1, (C) S. laurentiiNDS1/int-3A10 (encoding wild-type TsrA), (D) S. laurentiiNDS1/int-3A101 (encoding TsrA Ala2Gly), (E) S. laurentiiNDS1/int-3A102 (encoding TsrA Ala4Gly), (F) S. laurentiiNDS1/int-3A103 (encoding TsrA Thr7Gly). The asterisks indicate either thiostrepton or a thiostrepton analog. Absorbance was monitored at 254 nm. | ||
Production of thiostrepton analogs in S. laurentii
With a functional platform in hand for complementation of S. laurentiiNDS1, it is now feasible to interrogate the tolerance of the thiostrepton biosynthetic machinery toward alternate precursor peptide substrates. To facilitate the replacement of the precursor peptide-encoding gene, wild-type tsrA in int-3A10 was replaced with a dual-selection cassette imparting resistance to chloramphenicol (chlR) and a widely-used counterselection marker, the Bacillus subtilis sacBgene.38 When E. coli and other Gram-negative bacteria are grown in media containing sucrose, the presence of sacB inflicts a lethal effect upon its host.38,39 Thus, the loss of sacB permits growth of E. coli on sucrose-containing medium and provides a useful screen for its allelic replacement. An advantage to this fosmid-based system for tsrA replacement is that all genetic manipulations involving tsrA can now be conducted in E. coli in a comparatively facile manner by PCR-targeted gene replacement.22 The engineered fosmid is introduced into S. laurentiiNDS1 only following the successful replacement of wild-type tsrA with a variant tsrA to assess analog production.An examination of the TsrA precursor peptide sequence and the structure of thiostrepton A 1 revealed that only three amino acid residues out of seventeen remain untouched by the enzymes of the thiostrepton post-translational machinery: Ala2, Ala4, and Thr7 (Fig. 5). These three residues were chosen for the initial round of mutagenesis, since none are expected to be directly involved in a potentially critical transformation in precursor peptide processing, such as dehydration or cyclization. Conservative alterations at these positions may therefore elicit a minimal disturbance upon the maturation of a thiopeptide variant. Three integrative fosmids, each encoding a separate mutant of TsrA, were constructed and designated as follows: int-3A101 (encoding TsrA Ala2Gly), int-3A102 (encoding TsrA Ala4Gly), and int-3A103 (encoding TsrA Thr7Gly (Fig. 5). The fosmids int-3A101 to int-3A103 were individually introduced into S. laurentiiNDS1 by intergeneric conjugation to provide S. laurentii/int-3A101 to S. laurentii/int-3A103, respectively. Two new metabolites with similar UV-visible absorption spectra to thiostrepton A 1 were observed in the HPLC analysis of the culture extracts of S. laurentiiNDS1/int-3A101 and S. laurentiiNDS1/int-3A102 (Fig. 4). Further analysis of the extracts by HPLC-MS confirmed that the masses of the two metabolites were consistent with those expected for the engineered thiostreptons Ala2Gly 6 and Ala4Gly 7, ESI.‡ The thiostrepton Thr7Gly 8 analog, on the other hand, was not detected either by HPLC or HPLC-MS.
 | ||
| Fig. 5 Thiostrepton A 1 and the expected analogs to be generated by site-directed mutagenesis of TsrA. (A) Comparison of wild-type TsrA with the TsrA mutants encoded in the four fosmids (int-3A101 to 103). The mutated amino acid residues are indicated in red, blue and green. (B) Structures of the expected thiostrepton analogs. | ||
An initial assessment of the antibacterial activity of the new thiostrepton variants was conducted by disc-diffusion of the S. laurentii culture extracts against bacteria-infused solid media revealed that the thiostrepton variants Ala2Gly 6 and Ala4Gly 7 both retained antibacterial properties (Fig. 6). Thiostrepton Ala2Gly 6 and thiostrepton Ala4Gly 7 were purified and their structures were verified by one-dimensional and two-dimensional NMR analysis, ESI.‡ The minimum inhibitory concentrations (MICs) of the thiostrepton analogs were determined against methicillin-resistant Staphylococcus aureus ATCC 10537, vancomycin-resistant Enterococcus faecium ATCC 12952, Bacillus sp. ATCC 27859, and Escherichia coli ATCC 27856 (E. coli 27856) (Table 1). None of the thiostreptons demonstrated antibacterial activity against E. coli 27856 (Data not shown). Both engineered thiopeptides still retained antibacterial activity against the tested Gram-positive strains, although thiostrepton Ala4Gly 7 did reveal a reduction in efficacy (Table 1). Both Ala2 and Ala4 reside within thiostrepton's second macrocycle, the quinaldic acid loop. Much of this region, excepting quinaldic acid, is likely exposed to solvent upon complexation to the ribosome and is not likely to participate in any direct interactions with either the ribosomal protein L11 or the 23S rRNA.40Retention of antibacterial activity in the Ala4Gly and Ala2Gly variants is consistent with this expectation. It is anticipated that the position of the core thiopeptide macrocycle corresponding to Thr7 of thiostrepton A 1 will be critical for antibacterial activity. This unmodified amino acid residue of the ribosome-binding thiopeptides is highly conserved and substitutions at the corresponding position of thiocillin I 2 resulted in a loss of antibacterial activity.19,20 The precise orientation of the thiopeptide macrocycle, when bound to the ribosome, appears to dictate the specific contacts assigned to this conserved threonine residue. For micrococcin P14 and thiocillin I 2, it is presumed that the threonine-containing edge of the macrocyclic loop forms several contacts with ribosomal protein L11.20,40 In contrast, this conserved edge in the thiostrepton macrocycle forms several contacts with the 23S rRNA.40,41 Furthermore, the Thr7 side chain comes within 4–5 Å of A1067 and A1095, two RNA nucleotides known to be critical to the activities of thiostrepton A 1 and nosiheptide 3.40,41 An analysis of the requirement for Thr7 in thiostrepton awaits the successful production of variants containing altered side chains at this position.
 | ||
| Fig. 6 Disc diffusion antimicrobial assays of culture extracts from S. laurentii strains producing thiostrepton variants. (A) Enterococcus faecium ATCC 12952, (B) Staphylococcus aureus ATCC 10537 and (C) Bacillus sp. ATCC 27859. For region 1 through region 4, culture extracts of the following strains were added, respectively: wild-type S. laurentii, S. laurentiiNDS1, S. laurentiiNDS1/int-3A101 (encoding TsrA Ala2Gly), and S. laurentiiNDS1/int-3A102 (encoding TsrA Ala4Gly); region 5, chloramphenicol (in A and C), vancomycin (in B); region 6, dimethyl sulfoxide. | ||
| Compound | MICa (μg mL−1) | ||
|---|---|---|---|
| MRSAb | VRE c | Bacillus d | |
| a Minimum inhibitory concentration. b Staphylococcus aureus ATCC 10537. c Enterococcus faecium ATCC 12952. d Bacillus sp. ATCC 27859. e ND - Not determined. | |||
| Thiostrepton | 0.012 | 0.012 | 0.025 |
| Thiostrepton Ala2Gly | 0.023 | 0.019 | 0.19 |
| Thiostrepton Ala4Gly | 0.12 | 0.12 | 0.46 |
| Vancomycin | 0.39 | NDe | ND |
| Chloramphenicol | ND | 3.9 | 0.98 |
The substitutions introduced thus far into the thiostrepton precursor peptide are relatively modest. Nevertheless, this initial study lays the groundwork for more radical substitutions at each of the three unmodified residues in thiostrepton A 1. The thiostrepton post-translational machinery may demonstrate flexibility toward the second and fourth positions of the propeptide region, given that the Ala2Gly and Ala4Gly variants did accumulate to appreciable levels in S. laurentii extracts, at about 19 and 11 mg L−1, respectively. In contrast, the Thr7Gly variant of a mature thiostrepton scaffold was not detected in S. laurentii culture. This latter observation may suggest a greater stringency within the thiostrepton biosynthetic system for the side chain at the seventh residue. With the ability to generate alternate precursor peptides in a relatively efficient fashion now established, we can thoroughly probe the substrate requirements of the collective post-translational machinery responsible for thiostrepton generation.
Experimental
General
Unless specified, common chemicals, solvents, restriction enzymes, DNA ligase and other materials were purchased from standard commercial sources and used as provided. The QIAprep Spin Miniprep Kit (Qiagen, Valencia, CA) was used for the isolation of plasmids and fosmids from E. coli strains. Streptomycesgenomic DNA was isolated using the Wizard®Genomic DNA Purification Kit (Promega, Madison, WI) according to the manufacturer's recommendations. High performance liquid chromatography (HPLC) analysis was performed on a Beckman Coulter System Gold instrument. HPLC-MS was performed at the Georgia Institute of Technology Bioanalytical Mass Spectrometry Facility with a Phenomenex Syngeri RP column (250 mm × 2 mm, 4 μm) (Torrance, CA) and developed with 20% Buffer B in Buffer A for 8 min followed by a gradient from 20–100% Buffer B over 35 min at 0.25 mL min−1 (Buffer A: 5% acetonitrile and 0.1% formic acid; Buffer B: 95% acetonitrile and 0.1% formic acid). High-resolution mass spectrometry was performed at the Emory University Mass Spectrometry Center. Proton and carbon NMR spectra were recorded on a Bruker 500 MHz spectrometer. All NMR experiments were performed according to standard pulse sequences supplied with each instrument.Bacterial strains plasmids and growth medium
Streptomyces laurentii ATCC 31255 (S. laurentii) and Streptomyces actuosus ATCC 25421 (S. actuosus) were obtained from American Type Culture Collection (ATCC). All strains and plasmids are listed in Table S3, and primers are listed in Table S4, ESI.‡ All Escherichia coli (E. coli) strains were grown in Luria-Bertani liquid or solid medium with the appropriate antibiotic(s). For the selective growth of E. coli or Streptomyces, the following antibiotics and concentrations were used: kanamycin (50 μg mL−1), apramycin (50 μg mL−1), ampicillin (100 μg mL−1), nalidixic acid (25 μg mL−1), chloramphenicol (30 μg mL−1), thiostrepton (20 μg mL−1), spectinomycin (50 μg mL−1 for E. coli, 100 μg mL−1 for Streptomyces) and streptomycin (50 μg mL−1 for E. coli, 10 μg mL−1for Streptomyces). R2YE solid medium was used for the growth and sporulation of S. lividans and S. actuosus, and for the protoplast transformation of S. lividans.42MS agar was used for the intergeneric conjugation of all Streptomyces strains.23Fosmid engineering
Fosmids pCC1FOS and JA3A10 were retrofitted for integration into the chromosomes of various Streptomyceshosts by employing λ Red-mediated recombination.22,23 First, a 4.4 kb fragment containing aac(3)IV, int, attP and oriT was amplified from pSET152 by polymerase chain reaction (PCR) using the primers CTSR1-F and CTSR1-R. Next, the resulting PCR product was used to replace the chloramphenicol resistance gene (chlR) located on the fosmid backbone, generating the fosmids int-3A10 and int-pCC1FOS. The allelic replacements in the resulting fosmids were confirmed by PCR and by sequence analysis of the amplified products.Generation of Streptomyces strains for heterologous thiostrepton A production
As an initial step only for S. lividans, pSE34 was introduced by protoplast transformation following an established protocol.42 Each fosmid (int-3A10 or int-pCC1FOS) was introduced by intergeneric conjugation into S. lividans and S. actuosus, providing the strains S. lividansFZ1, S. lividansFZ2, S. actuosusFZ1 and S. actuosusFZ2. The presence of int-3A10 in FZ1 strains was confirmed by PCR amplification of tsrK, tsrN, and tsrV, and by sequence analysis of the amplified products.Deletion of tsrA in S. laurentii
Deletion of tsrA followed the method for PCR-targeted gene replacement, ESI.‡22 The upstream and downstream regions flanking tsrA were both amplified by PCR using primer pairs DTSR1-F/DTSR1-R for the upstream segment, and DTSR2-F/DTSR2-R for the downstream segment. The resulting two PCR products were cloned into pSC-B-amp/kan to yield pLeft and pRight, respectively, and confirmed by sequence analysis. The plasmid pRight was digested with NdeI and SbfI, and the resulting 1 kb fragment ligated into pLeft to generate pLR. The 2 kb fragment following HindIII digestion of pLR was ligated into pGM160HKss, yielding pNDS1. Following transformation into E. coliET12567/pUZ8002, the plasmid pNDS1 was introduced into S. laurentii by intergeneric conjugation.23 The resulting strain, S. laurentii/pNDS1, was grown at 37 °C in tryptic soy broth containing kanamycin to force the integration of pNDS1 into the chromosome of S. laurentii. Next, the S. laurentii mutant was grown at 28 °C in tryptic soy broth lacking any antibiotic to generate a kanamycin-sensitive double crossover mutant of tsrA. The tsrA deletion mutant, S. laurentiiNDS1, was confirmed by PCR amplification of the affected region and sequence analysis of the amplified product (Fig. S6, ESI‡).Site-directed mutagenesis of tsrA
Three pairs of PCR primers were employed for the mutagenesis of the thiostrepton prepeptide: A2G-F and A2G-R; A4G-F and A4G-R; T7G-F and T7G-R (Table S4, ESI‡). Using the appropriate primers, mutants were amplified from pJP11 by adapting the QuikChange site-directed mutagenesis strategy (Agilent Technologies, La Jolla, CA). DNA sequence analysis confirmed successful mutagenesis of tsrA in each case, including the absence of any undesired mutations. The three plasmids carrying the tsrA mutants were designated as pCL61 for thiostrepton Ala2Gly, pCL62 for thiostrepton Ala4Gly, and pCL63 for thiostrepton Thr7Gly (Table S3, ESI‡).The chlR and levansucrase-encoding sacBgenes were amplified using primers Chl-F and Chl-R; SacB-F and SacB-R from pCC1FOS and pEX100T, respectively. The resulting PCR products were cloned into pSC-B-amp/kan, to provide pDC1 and pDC2. The plasmid pDC2 was digested with NdeI and SbfI, and the resulting 2 kb fragment ligated into pDC1, yielding a dual-marker disruption cassette in pDC3. The dual-marker disruption cassette was then amplified from pDC3 by PCR using primers SD1-F and SD1-R. The resulting PCR product was used to replace tsrA in fosmid int-3A10 by PCR-targeted gene replacement, generating int-3A100.22 The three tsrA mutants were each amplified by PCR from plasmids pCL61 to pCL63 with primers SD2-F and SD2-R. The resulting PCR products were then used to replace the dual marker cassette in int-3A100, yielding three sucrose-tolerant fosmids: int-3A101 for TsrA Ala2Gly, int-3A102 for TsrA Ala4Gly, and int-3A103 for TsrA Thr7Gly. The allelic replacement of wild-type tsrA by the site-directed mutants of tsrA was confirmed in each fosmid by sequence analysis. Fosmids int-3A10 (for wild-type tsrA) and int-3A101 to int-3A103 were first transformed into E. coliET12567/pUZ8002, and then introduced into S. laurentiiNDS1 through intergeneric conjugation. Colonies resistant to apramycin were selected for fermentation analysis.
Evaluation of thiostrepton production in Streptomyces
Growth of S. actuosus and S. laurentii strains were performed as described previously.15,43 The fermentation of S. lividans was carried out in a three-step process. First, 50 mL of tryptic soy broth in a 250 mL Erlenmeyer flask was inoculated with an S. lividans strain and grown at 28 °C and 220 rpm for 48 h. Next, 0.5 mL of the S. lividans preculture was used to inoculate 50 mL of seed medium (50 g L−1glucose, 15 g L−1 soybean flour, 15 g L−1 soluble starch, pH 7.2) in a 250 mL Erlenmeyer flask.44 After 7 days at 28 °C and 220 rpm, 1 mL of this seed culture was then used to inoculate 100 mL of fermentation medium in a 500 mL Erlenmeyer flask.43 The resulting culture was incubated at 28 °C and 220 rpm for 7 days. To harvest thiopeptide metabolites, the whole culture was extracted twice with an equal volume of chloroform. The chloroform layers were pooled together and the solvent removed in vacuo. The solid residue was dissolved in 2 mL of chloroform. Samples were analyzed by HPLC with a Phenomenex Luna C18(2) column (250 × 4.6 mm, 5 μm). The column was developed using a gradient of 0–100% acetonitrile in water over 30 min at a flow rate of 1 mL min−1. Absorbance was monitored at 254 nm. Under these conditions, thiostrepton A 1 elutes with a tR of about 22 min. Under the conditions used for HPLC-MS, thiostrepton A 1 elutes at a tR of about 28 min providing ions at m/z 1664.4 [M + H]+ and m/z 832.9 [M + 2H]2+. The predominant ion for thiostrepton A 1 was [M + 2H]2+, whereas [M + H]+ was only a minor species.Purification and structural determination of thiostrepton analogues
Crude extracts from S. laurentiiNDS1 int-3A101 (5 L) and S. laurentiiNDS1 int-3A102 (4 L) fermentation culture were purified by HPLC with a Phenomenex Luna C18(2) RP-Aqueous column (250 × 10 mm, 5 μm) while monitoring absorbance at 254 nm. For the purification of thiostrepton Ala2Gly, the column was developed using solvents A (water) and B (acetonitrile) at a flow rate of 4.7 mL min−1 as follows: the mobile phase was increased from 10% to 40% solvent B over 5 min, then held constant at 40% solvent B for 5 min, then increased from 40% to 50% solvent B over 5 min, then held constant at 50% solvent B for 5 min, then increased from 50% to 100% solvent B over 5 min, and finally held constant at 100% solvent B for 5 min. For the purification of thiostrepton Ala4Gly, the column was also equilibrated with 10% solvent B and developed as follows: the mobile phase was increased to 40% solvent B over 5 min, then held constant at 40% solvent B for 5 min, then increased to 45% solvent B over 5 min, then held constant at 45% solvent B for 5 min, then increased to 100% solvent B over 2 min, and finally held at 100% solvent B for 5 min. Purified samples were analyzed by HPLC-MS, HR-ESI-MS and NMR and were stored under argon at −20 °C. NMR data is included in the ESI.‡HR-ESI-MS of thiostrepton Ala2Gly: C71H83O18N19Na1S5, m/z 1672.4689 [M+Na]+ (calculated 1672.4670). HR-ESI-MS of thiostrepton Ala4Gly: C71H83O18N19Na1S5, m/z 1672.4664 [M+Na]+ (calculated 1672.4670).Antibacterial activity of thiostrepton analogs
Antimicrobial activities of S. laurentii culture extracts were qualitatively assessed by solid agar disc-diffusion assays. 3 mL of Luria-Bertani (LB) liquid medium was inoculated with 3 μL of a cell stock of either Bacillus sp. ATCC 27859 (Bacillus sp.), or Escherichia coli ATCC 27856 (E. coli 27856). Brain heart infusion broth (3 mL) was inoculated with 3 μL of a cell stock of vancomycin-resistant Enterococcus faecium ATCC 12952 (VRE) or methicillin-resistant Staphylococcus aureus ATCC 10537 (MRSA). All strains were incubated at 37 °C and 220 rpm for 18 h. Infused solid medium was prepared by adding 200 μL of overnight bacterial culture to 20 mL of respective molten medium agar cooled to 42 °C. Eight 7 mm filter paper disks were spaced evenly on the surface of the solid medium. To each disk, 10 μL of S. laurentii strain extract in DMSO was deposited and the plates were then incubated at 37 °C for 18 h. The positive antibiotic control used for Bacillus sp., E. coli 27856, and VRE was 10 μL of chloramphenicol (1 mg mL−1) and the positive antibiotic control used for MRSA was 10 μL of vancomycin (0.5 mg mL−1). The negative control used for all bacterial strains was 10 μL of DMSO. Antibacterial activity was qualitatively determined by the presence or absence of a growth inhibition zone.Minimum inhibitory concentrations (MICs) of thiostrepton analogs against MRSA, VRE and Bacillus sp. were further determined using liquid microdilution method. Briefly, MRSA, VRE, and Bacillus sp. were grown overnight at 37 °C in nutrient broth, brain heart infusion broth and LB liquid medium, respectively. Overnight cultures of the individual strains were each diluted 1000-fold with the respective medium and transferred to a microtiter plate. Thiostrepton A 1 and thiostrepton analogs were prepared in DMSO and quantified by UV spectroscopy using an extinction coefficient at 280 nm of 0.027 cm−1 μM−1.45 Serial dilutions of test samples and controls were performed to assess growth inhibition. The positive antibiotic control used for Bacillus sp., and VRE was chloramphenicol, whereas the positive control for MRSA was vancomycin. DMSO was used as the negative control in all assays. Cell growth was monitored by comparing the optical density at 600 nm at the time of treatment and after 18 h incubation at 37 °C. A difference in OD600 was considered growth and the lowest concentration causing complete suppression of visible bacterial growth defined MIC.
Conclusions
Thiostrepton A 1 and other thiopeptides are highly complex peptide metabolites arising from the extensive post-translational modification of a precursor peptide. The thiopeptides possess potent antibacterial activities against Gram-positive pathogens, including MRSA and VRE. Nonetheless, the poor water solubility and the architectural complexity of these metabolites have hindered the development of a sophisticated thiopeptide structure–activity relationship and the development of a clinically useful antibiotic. We have been able to develop a methodology for the heterologous production of thiostrepton A 1 in a Streptomyceshost and have adapted that same methodology toward the engineering of thiostrepton variants in the native thiostrepton-producing bacterium, S. laurentii. In the course of this work, two different thiostrepton analogs were generated. These variants were structurally characterized and the MICs against indicator bacterial strains were determined to be comparable to that of thiostrepton A 1. We can discern that the thiostrepton biosynthetic machinery does possess some tolerance toward substitution of at least two residues of the TsrA precursor peptide in the production of the thiostrepton scaffold. The platform developed here now permits an extensive analysis of the thiostrepton post-translational modification system's ability to elaborate an alternate substrate precursor peptide into a mature thiopeptide. The thiostrepton analogs generated by such efforts can be applied to probe the structural features of the piperidine-containing thiopeptides required for ribosome inhibition and antimicrobial activity.Acknowledgements
This work was supported in part by the Camille and Henry Dreyfus Foundation and the Defense Advanced Research Projects Agency Grant (N6601-09-1-2086) to W. L. K. Ms. Kathryn E. Rommel is thanked for her assistance in acquiring the NMR spectra.References
- T. L. Su, Br. J.Exp. Pathol., 1948, 29, 473–481 Search PubMed.
- M. C. Bagley, J. W. Dale, E. A. Merritt and X. Xiong, Chem. Rev., 2005, 105, 685–714 CrossRef CAS.
- J. E. Biskupiak, E. Meyers, A. M. Gillum, L. Dean, W. H. Trejo and D. R. Kirsch, J. Antibiot., 1988, 41, 684–687 CAS.
- M. C. Carnio, A. Höltzel, M. Rudolf, T. Henle, G. Jung and S. Scherer, Appl. Environ. Microbiol., 2000, 66, 2378–2384 CrossRef CAS.
- A. Mukai, T. Fukai, Y. Hoshino, K. Yazawa, K. Harada and Y. Mikami, J. Antibiot., 2009, 62, 613–619 CrossRef CAS.
- J. Shoji, H. Hinoo, Y. Wakisaka, K. Koizumi, M. Mayama, S. Matsura and K. Matsumoto, J. Antibiot., 1976, 29, 366–374 CAS.
- W. H. Trejo, L. D. Dean, J. Pluscec, E. Meyers and W. E. Brown, J. Antibiot., 1977, 30, 639–643 CAS.
- W. Li, J. E. Leet, H. A. Ax, D. R. Gustavson, D. M. Brown, L. Turner, K. Brown, J. Clark, H. Yang, J. Fung-Tomc and K. S. Lam, J. Antibiot., 2003, 56, 226–231 CAS.
- B. Clough, M. Strath, P. Preiser, P. Denny and I. R. Wilson, FEBS Lett., 1997, 406, 123–125 CrossRef CAS.
- J. M.-M. Kwok, S. S. Myatt, C. M. Marson, R. C. Coombes, D. Constantinidou and E. W.-F. Lam, Mol. Cancer Ther., 2008, 7, 2022–2032 CrossRef CAS.
- G. A. McConkey, M. J. Rogers and T. F. McCutchan, J. Biol. Chem., 1997, 272, 2046–2049 CrossRef CAS.
- S. K. Radhakrishnan, U. G. Bhat, D. E. Hughes, I.-C. Wang, R. H. Costa and A. L. Gartel, Cancer Res., 2006, 66, 9731–9735 CrossRef CAS.
- M. J. Rogers, Y. V. Bukhman, T. F. McCutchan and D. E. Draper, RNA, 1997, 3, 815–820 CAS.
- L. C. W. Brown, M. G. Acker, J. Clardy, C. T. Walsh and M. A. Fischbach, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2549–2553 CrossRef.
- W. L. Kelly, L. Pan and C. Li, J. Am. Chem. Soc., 2009, 131, 4327–4334 CrossRef CAS.
- C. Li and W. L. Kelly, Nat. Prod. Rep., 2010, 27, 153–164 RSC.
- R. Liao, L. Duan, C. Lei, H. Pan, Y. Ding, Q. Zhang, D. Chen, B. Shen, Y. Yu and W. Liu, Chem. Biol., 2009, 16, 141–147 CrossRef CAS.
- R. P. Morris, J. A. Leeds, H. U. Naegeli, L. Oberer, K. Memmert, E. Weber, M. J. LaMarche, C. N. Parker, N. Burrer, S. Esterow, A. E. Hein, E. K. Schmitt and P. Krastel, J. Am. Chem. Soc., 2009, 131, 5946–5955 CrossRef CAS.
- M. G. Acker, A. A. Bowers and C. T. Walsh, J. Am. Chem. Soc., 2009, 131, 17563–17565 CrossRef CAS.
- A. A. Bowers, M. G. Acker, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2010, 132, 7519–7527 CrossRef CAS.
- C. Li and W. L. Kelly, unpublished work.
- K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef CAS.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS.
- Y. Zhang, F. Buchholz, J. P. P. Muyrers and A. F. Stewart, Nat. Genet., 1998, 20, 123–128 CrossRef CAS.
- Y. Zhang, J. P. P. Muyrers, G. Testa and A. F. Stewart, Nat. Biotechnol., 2000, 18, 1314–1317 CrossRef CAS.
- M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. N. Rao and B. E. Schoner, Gene, 1992, 116, 43–49 CrossRef CAS.
- T. M. Smith, Y. F. Jiang, P. Shipley and H. G. Floss, Gene, 1995, 164, 137–142 CrossRef CAS.
- F. Benazet, M. Cartier, J. Florent, C. Godard, G. Jung, J. Lunel, D. Mancy, C. Pascal, J. Renaut, P. Tarridec, J. Theilleux, R. Tissier, M. Dubost and L. Ninet, Experientia, 1980, 36, 414–416 CrossRef CAS.
- E. Cundliffe and J. Thompson, J. Gen. Microbiol., 1981, 126, 185–192 CAS.
- B. S. Yun, T. Hidaka, T. Kuzuyama and H. Seto, J. Antibiot., 2001, 54, 375–378 CAS.
- C. V. Kumar and J. F. Martin, FEMS Microbiol. Lett., 1994, 118, 107–112 CrossRef CAS.
- T. Murakami, T. G. Holt and C. J. Thompson, J. Bacteriol., 1989, 171, 1459–1466 CAS.
- D. J. Holmes, J. L. Caso and C. J. Thompson, EMBO J., 1993, 12, 3183–3191 CAS.
- M. L. Chiu, M. Folcher, P. Griffin, T. Holt, T. Klatt and C. J. Thompson, Biochemistry, 1996, 35, 2332–2341 CrossRef CAS.
- B. Julien and S. Shah, Antimicrob. Agents Chemother., 2002, 46, 2772–2778 CrossRef CAS.
- Z. Xu, K. Jakobi, K. Welzel and C. Hertweck, Chem. Biol., 2005, 12, 579–588 CrossRef CAS.
- J. Lau, S. Frykman, R. Regentin, S. Ou, H. Tsuruta and P. Licari, Biotechnol. Bioeng., 2002, 78, 280–288 CrossRef CAS.
- H. P. Schweizer and T. T. Hoang, Gene, 1995, 158, 15–22 CrossRef CAS.
- P. Gay, D. Le Coq, M. Steinmetz, T. Berkelman and C. I. Kado, J. Bacteriol., 1985, 164, 918–921 CAS.
- J. M. Harms, D. N. Wilson, F. Schluenzen, S. R. Connell, T. Stachelhaus, Z. Zaborowska, C. M. T. Spahn and P. Fucini, Mol. Cell, 2008, 30, 26–38 CrossRef CAS.
- G. Lentzen, R. Klinck, N. Matassova, F. Aboul-ela and A. I. H. Murchie, Chem. Biol., 2003, 10, 769–778 CrossRef CAS.
- T. Kieser, M. J. Bibb, M. J. Buttner, K. F. Chater and D. A. Hopwood, Practical Streptomyces Genetics, John Innes Foundation, Norwich, UK, 2000 Search PubMed.
- D. R. Houck, L.-C. Chen, P. J. Keller, J. M. Beale and H. G. Floss, J. Am. Chem. Soc., 1988, 110, 5800–5806 CrossRef CAS.
- N. D. Priestley, T. M. Smith, P. R. Shipley and H. G. Floss, Bioorg. Med. Chem., 1996, 4, 1135–1147 CrossRef CAS.
- P. C. Ryan, M. Lu and D. E. Draper, J. Mol. Biol., 1991, 221, 1257–1268 CAS.
Footnotes |
| † This article is part of a themed issue of Molecular BioSystems on post-translational modifications. |
| ‡ Electronic supplementary information (ESI) available: Fig. S1–S23 and Tables S1–S4. See DOI: 10.1039/c0mb00129e |
| § These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2011 |