DOI:
10.1039/C0LC00275E
(Paper)
Lab Chip, 2011,
11, 224-230
Stereomask lithography (SML): a universal multi-object micro-patterning technique for biological applications†
Received
4th August 2010
, Accepted 29th October 2010
First published on 26th November 2010
Abstract
The advent of biological micro-patterning techniques has given new impetus to many areas of biological research, including quantitative biochemical analysis, tissue engineering, biosensing, and regenerative medicine. Derived from photolithography or soft lithography, current bio-patterning approaches have yet to completely address the needs of out-of-cleanroom, universal applicability, high feature resolution, as well as multi-object placement, though many have shown great promise to precisely pattern one specific biomaterial. In this paper, we present a novel versatile biological lithography technique to achieve integrated multi-object patterning with high feature resolution and high adaptability to various biomaterials, referred to as stereomask lithography (SML). Successive patterning of multiple objects is enabled by using unique three-dimensional masks (i.e., the stereomasks), which lay out current micropatterns while protecting pre-existing biological features on the substrate. Furthermore, high-precision reversible alignment among multiple bio-objects is achieved by adopting a peg-in-hole design between the substrate and stereomasks. We demonstrate that the SML technique is capable of constructing a complex biological microenvironment with various bio-functional components at the single-cell resolution, which to the best of our knowledge has not been realized before.
Introduction
It is of increasing importance to engineer bio-functional platforms with precisely positioned biological substances (e.g., biomolecules, micro/nano particles, and living cells) for quantitative biochemical analysis, molecular sensing, tissue and stem cell engineering.1–7 For instance, micro/nano-patterned extracellular proteins have been widely employed in the investigation of biochemical control of cell growth, viability and responsiveness,8,9 in which manipulation of focal adhesion with high spatial accuracy directly affects cytoskeleton organization and cell polarization.10–14 In addition, micro-spotted combinatorial arrays have long been utilized in high-throughput multiplexed screening applications, such as immunological or cancerous marker detection, drug discovery, stem cell differentiation, genomic identification, etc.4,5,15–19 Moreover, generating a concentration gradient of signaling molecules (such as cell adhesive ligands, growth factors and chemokines) on an immobilized planar surface or under continuous flow enables quantitative study of cell adhesion, migration and stem cell differentiation in response to these stimuli.20–22 Furthermore, well aligned single- or multiple-cell arrays in an integrated culture medium permit on-chip screenings and diagnostics, quantitative biophysical and biochemical analyses, high-throughput bioreactors, and controlled cell interaction assays, all of which have extensive implications for fundamental and translational biomedical research. In particular, the micropatterned single cell arrays overcome the difficulty of traditional cell ensemble-based assay, that is, the key responses from an individual cell might be masked by the average behavior of a group of cells.23–26 Recently, the cell microarrays have been extensively used to create stem cell aggregates (such as an embryoid body and neurosphere) with defined size and shape in regenerative medicine, leading to better control of stem cell differentiation and proliferation.26–30
Over the last decade, a number of biological micropatterning techniques have been attempted to define micro/nanometre-resolution geometries of various substances for quantitative bio-investigations, most of which can be divided into two primary categories: photolithography-based31–34 and soft lithography-based35 approaches. Derived from the microelectronics manufacturing process, the photolithography-based techniques are well established to form sub-micrometre to millimetre patterns. However, unavoidable UV exposure, multiple baking steps, and solvent development could pose potential threats to biological substances by causing adverse optical, thermal, and/or chemical damage to the organic structures and biological functions. Moreover, high fabrication expense and limited cleanroom access further restrict them to less biologically oriented applications. Soft lithography, renovated from conventional elastomeric stamping, has offered a powerful alternative. Utilizing an elastomeric replica (of polydimethylsiloxane) from a photo-defined mold (of SU-8, a typical high-aspect-ratio photopolymer), soft lithography has successfully demonstrated its potential use to pattern proteins and other biomolecules with ultrahigh resolution (from microscale to nanoscale). Although it offers a more flexible and reliable scheme to micropattern biological substances, soft lithography still relies on cleanroom access and microfabrication equipment to generate the photopatterned mold. In addition, the layer-to-layer alignment is not straightforward in the multi-object lithography processes. Furthermore, positioning living cells directly into arbitrary microscopic geometries cannot be accomplished by the process itself. Interestingly, Craighead's group has developed a shadow mask peel-off technology for high-resolution placement of biomolecules, including antibody, poly-L-lysine and lipid bilayers.36,37 To fabricate shadow masks, they deposit ultrathin parylene films on target substrates, and use photolithography and reactive ion etching to create through-hole patterns on the films. The substrates are subsequently incubated with biomolecules, and finally the shadow masks are peeled off, revealing the desired patterns of biomolecules. Other groups have extended this technique to cell patterning.38,39 However, since the shadow masks have to be formed in situ on patterning substrates, the parylene deposition followed by photolithography and dry etching processes might introduce unfavorable effects to the substrates. In addition, multi-object patternability is also not straightforward.40 To further investigate complex biological interactions in a highly controlled and integrated fashion (e.g., cellular responses to multiple biological stimuli in a high-throughput assay), a simple bio-oriented patterning technique is highly desired, which would ideally allow precise patterning of multiple bio-functional objects together with living cells into a hierarchically organized microenvironment.41
In the paper, we first present a universal biological lithography technique to achieve the aforementioned objectives, referred to as stereomask lithography (SML). Successive patterning of multiple objects is enabled by using a combination of the peel-off method and unique three-dimensional (3D) shadow masks (i.e., the stereomasks), which lay out current micropatterns while protecting pre-existing biological features on the substrate. Taking advantage of the previously reported direct projection on dry-film photoresist (DP2) process, such complex stereomasks can be completely fabricated by dry-film photoresist in an out-of-cleanroom setting.42 During the bio-patterning process, the stereomask is laminated onto the target substrate, followed by the perfusion of biological solution through microscale screen openings on the mask towards the substrate, from which the negative images of biological substances result. Importantly, the incorporated protective chambers keep the stereomask from making direct contact with the pre-existing biological features. In addition, an innovative peg-in-hole alignment strategy has been implemented between the substrate and stereomasks in multi-object processing. It is worth noting that the separated mask generation and biological patterning processes eliminate any potential exposure of sensitive biological elements to optical, thermal or chemical treatments usually involved in microfabrication. Combined with all the favorable features, this unique bio-patterning technique provides a powerful out-of-cleanroom bio-microfabrication solution to place various bio-functional substances with single-cell resolution (of 10 μm) and accurate alignment precision (of 10 μm).
Materials and methods
Materials
The PerMX dry film photoresist (with nominal thickness of 10 μm) was generously provided by DuPont. Bovine serum albumin conjugated with FITC (BSA-FITC, A9771, green fluorescence) and poly-D-lysine hydrobromide (P7886) were purchased from Sigma-Aldrich. Bovine serum albumin conjugated with Texas Red (BSA-TR, A23017, red fluorescence), streptavidin conjugated with Marina Blue (streptavidin-MB, S11221, blue fluorescence) and FluoSphere 200nm yellow-green fluorescent polystyrene beads with carboxylate surface groups (F8811) were purchased from Invitrogen. Protein solutions used for bio-patterning experiments were prepared by dissolving protein–fluorophore conjugates in phosphate buffer solution (BSA-FITC 400 μg ml−1, BSA-TR 200 μg ml−1 and streptavidin-MB 100 μg ml−1). Poly-D-lysine solution was prepared by dissolving poly-D-lysine solids in phosphate buffer solution at 400 μg ml−1. The fluorescent nano beads sample was used as received. Collagen I was purchased from BD Biosciences. The Sylgard 184 silicone elastomer (PDMS) was purchased from Dow Corning. The FNC coating mix (an aqueous solution of fibronectin and other cell adhesion proteins) was purchased from Athena Environmental Sciences. Transparency films (CG 3480) were from 3M.
Cell culture
The NIH 3T3 fibroblast cell line was purchased from American Type Culture Collection. Cells were maintained in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum and 1% Pen Strep, at 37 °C in a 95% air/5% CO2 humidified incubator. Cell cultures were passaged every two days at 90% confluence. Cells were harvested by trypsinization at 90% confluence prior to bio-patterning experiments. Dulbecco's modified eagle medium (DMEM), fetal bovine serum, 0.25% trypsin-EDTA and Pen Strep were purchased from Invitrogen.
Stereomask fabrication
As the core of SML, the stereomasks essentially consist of two layers of photo-defined dry film constructs. As shown in Fig. 1, both layers comprise identical through-pattern designs to deposit bio-substances, while additional recesses in the bottom layer are created to prevent stereomasks from making direct contact with pre-existing features on the substrate. Using the previously introduced DP2 process, the 3D stereomasks can be directly fabricated from multilayer dry film photoresists in an out-of-cleanroom setting, from which 10 μm resolution and 5 μm alignment precision have been reliably achieved.42 Briefly, the microfabrication process for stereomasks starts with thermal lamination of a PerMX dry film onto a 3M transparency film. After soft baking at 115 °C for 5 min, it is exposed through a direct projection lithography setup (365 nm, 220 mJ cm−2) as described previously42 to generate the cross-shaped through-patterns in the first layer, i.e., the top layer of the stereomask (Fig. 1a). Subsequently, another layer of dry film (which will finally become the bottom layer of the stereomask) is laminated on top of the first layer and the second exposure is conducted to define the identical through-patterns as well as the diamond-shaped protective chambers (Fig. 1b). In the following step, the dry film is post-baked at 95 °C for 2 min and developed in an ultrasonic bath with propylene glycol monomethyl ether acetate for 1 min, which reveals the through-patterns on both layers while protective recesses only on the second layer (Fig. 1c). Eventually, the two-layer stereomask can be peeled off from the transparency support, flipped over, and laminated onto the substrate with the protective features facing towards the substrate, ready for bio-patterning. It is worth noting that the height of the protective chambers can be adjusted by using a different thickness of the bottom dry-film layer to ensure that there is no direct contact with protein or cellular patterns pre-existing on the substrate.
 |
| | Fig. 1 Schematic illustration of stereomask fabrication: (a) the first layer of dry film is photopatterned with cross-shaped through-windows; (b) the second layer of dry film incorporates both the through-windows and the diamond-shaped protective chambers; and subsequently (c) the dry film stereomask is developed in PGMEA and a macroscopic picture of stereomask with an inset showing a microscopic view of through-patterns and protective chambers (d). Bar is 1 mm. | |
Reversible alignment
Importantly, high-precision alignment among multiple bio-objects is achieved by adopting a reversible peg-in-hole design between the substrate and stereomasks, analogous to the renowned mechanism that Lego® bricks interlock with each other. Micropillar structures made of dry film resists are applied onto the substrate prior to bio-patterning, while the complementary through-holes are microfabricated correspondingly on the stereomasks. Plugging the pillars into the holes, the stereomasks can be positioned easily at the desired location repeatedly, which ensures alignment precision during multi-object processing.
Bio-patterning
The single-object bio-patterning process begins with lamination of the stereomask onto the target substrate. To prevent air bubbles from being trapped in the micro through-patterns, the substrate is first wetted with ethanol, and then, rinsed with phosphate buffer solution. Biological solutions are deposited over the stereomask subsequently. Individual protocols have been applied to protein, nanobead and living cell micropatterning, respectively. For protein solutions, the samples are first incubated for 7–8 h at room temperature. Then, the substrate is rinsed with DI water 8 times and blow-dried. Finally, the stereomask is peeled off from the substrate. Prior to nanobeads patterning, the substrate is coated with a commercial FNC coat mix (an aqueous solution of fibronectin and other cell adhesion proteins) at five-times dilution for 2 h, which is proved to greatly increase the monolayer density of the nanobeads coating. Afterward, the nanobeads coating follows the same incubation and post-processing protocols as protein patterning. For cell patterning, 50 μg ml−1collagen in 0.2 M acetic acid (for glass or Petri dish substrates) or 400 μg ml−1 poly-D-lysine in PBS (for PDMS substrate) is first incubated for 1 h. The collagen/poly-D-lysine solution is then aspirated out, and the cell solution at 1 × 106cells per mL is incubated over the substrate in a humidified incubator at 37 °C for 1 h 10 min and then in a biosafety cabinet at room temperature for another 50 min. The suspended cells are aspirated from the substrate, and the stereomask is physically removed. Eventually, the cell patterns are immersed in fresh culture medium.
Multi-object bio-patterning offers accurate positioning of functional proteins, nanoparticles and living cells on the same substrate in sequence using SML, as illustrated in Fig. 2. With assistance of the peg-in-hole alignment, the stereomask with desired features of each object can be aligned and laminated onto the substrate. The corresponding biological sample is deposited using the single-object bio-patterning protocols as aforementioned. Following removal of the stereomask, the substrate is ready for the next patterning cycle. By simply repeating the three-step cycle of the stereomask placement, bio-substance deposition, and mask removal, a multi-object bio-patterning process can be established with high feature resolution and high alignment precision.
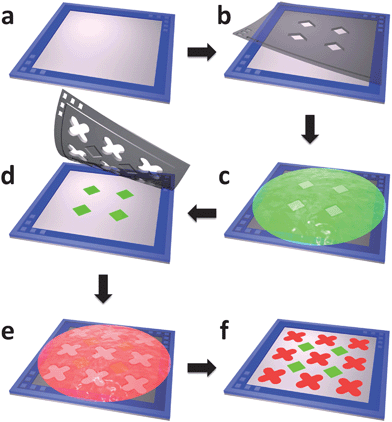 |
| | Fig. 2 The fabrication of two-protein micropatterns using stereomask lithography. (a) Aligning frame with micropillars is laminated onto target substrate. (b) First stereomask is aligned with aligning frame using peg-in-hole method and laminated onto substrate. (c) BSA-FITC is incubated. (d) Second stereomask is aligned using the same method and laminated onto substrate. (e) BSA-TR is incubated. (f) Second stereomask is peeled off and two-protein patterning is complete. | |
Results and discussion
Recent advances in both quantitative biology and biomedical research have illustrated the importance of high-throughput patterning of multiple biological objects with single-cell resolution.1–5 The reported stereomask lithography (SML) technique has addressed such a demand with process simplicity and high precision, as an outcome from the direct-projection lithography and the easy-processing dry film photoresist. The SML combines the desired features of the conventional shadow mask printing and peel-off processes, while the protective layer design as well as the peg-in-hole alignment enables the multi-object sequential patterning process.
Peg-in-hole alignment
The alignment process is crucial to the capacity of multi-object patterning. Precise placement of each stereomask to the desired location has been implemented utilizing the reversible peg-in-hole plug-in mechanism as shown in Fig. 2. In order to measure the misalignment, a testing alignment structure comprising micropillars of 250 μm in diameter is fabricated on the glass and a stereomask with micro holes of 300 μm in diameter is positioned with alignment pillars plugged in. In addition, vernier patterns with 10 μm measurement resolution and classic cross-bar designs are laid out to assist in the misalignment assessment. By calculating the relative displacement between the vernier patterns, the alignment precision within 10 μm has been demonstrated (as shown in Fig. S1, ESI†).
Adhesion of the stereomasks
Adhesion of the stereomasks to the commonly used biological substrates is of particular importance in the patterning process. The dry film stereomask exhibits adhesion after moderate lamination (under a pressure of 0.5 bar) to various substrates, e.g., Petri dish (polystyrene), glass (silicon dioxide), and PDMS (polydimethylsiloxane), which effectively prevents leakage of biological solution during patterning. The adhesion could potentially be attributed to the Van der Waals interaction between the epoxy network of the PerMX dry film and the substrate materials. The medium Young's modulus (3.2 GPa) of the photo-polymerized PerMX dry film also ensures intimate contact between the stereomasks and the substrates, which improves the adhesion strength. Assessed by a standard pull-out force measurement setup for three times on each configuration,43 the adhesion strengths of dry film stereomask are measured as 166 ± 72 kPa, 261 ± 19 kPa, and 37 ± 3 kPa on glass, Petri dish and PDMS substrate, respectively. After patterning, the laminated stereomask can be detached from the substrate by lifting with a razor blade. Based on microscopic inspection after each patterning step, there is no appreciable contamination of mask-related residues on the substrate.
Microfeatures with 10 μm minimum resolution have been fabricated using the SML. Fig. 3 shows linear protein features of 10 μm in width and square dot patterns of 20 μm in side length, which are printed on a PDMS substrate. Incubation procedures of 400 μg ml−1 BSA-FITC in PBS and 200 μg ml−1 BSA-TR in PBS are described in the Materials and Methods section. As can been seen, no appreciable leakage has been observed after 8 h incubation, which verifies reliable adhesion between the stereomask and the substrate.
 |
| | Fig. 3
Protein line array (a, d) of 10 μm in width and dot array (b, c) of 20 μm in side length. Bars are 20 μm. Green patterns are BSA-FITC and red patterns are BSA-TR. | |
 |
| | Fig. 4 20 μm nanobead dot array with inset showing an enlarged view. Bars are 40 μm. | |
 |
| | Fig. 5 Single cell (a) and arbitrary cell patterns (b–d). Bars are 120 μm. | |
Building a complex multi-object micropattern platform
In this section, we demonstrate the applicability of the SML platform to multi-object biological patterning. Multiplexed protein micropatterns and the cell–nanobead–protein complex have been constructed, respectively. Fig. 6 shows multi-protein microarrays fabricated by the SML, which can be potentially used for high-throughput sensing of cellular metabolites. The BSA-FITC micropatterns are first placed on the substrate and subsequently, BSA-TR micropatterns are created next to the BSA-FITC patterns. For three-protein microarrays, one more step is needed to position streptavidin-MB micropatterns onto the substrate. The misalignment between different protein patterns has been measured to be less than 10 μm.
 |
| | Fig. 6 Two-protein (a) and three-protein (b, c) micropatterns. Bars are 100 μm. Green patterns are BSA-FITC, red patterns are BSA-TR and blue patterns are streptavidin-MB. | |
In building the cell–nanobead–protein complex, the protein–fluorophore conjugates of BSA-TR and yellow-green fluorescent nanobeads are first micropatterned using two stereomasks in sequence. The fibroblast cells are subsequently patterned through the third stereomask to ensure the cell viability. As shown in Fig. 7, BSA-TR protein patterns (100 μm red circles in Fig. 7a and 80 μm red squares in Fig. 7b) and yellow-green nanobead patterns (100 μm circles in Fig. 7a and 50 × 100 μm ovals in Fig. 7b) are placed next to living cell islands (100 μm in Fig. 7a and 70 μm in Fig. 7b). The ability to engineer multiplexed patterns of living cells, signaling molecules and functional nanoparticles with micrometre precision allows implementation of complex microenvironment for quantitative biological assessment, which has indicated its potential use in the emerging directions of stem cell differentiation and cancer development.41,48
 |
| | Fig. 7
Cell–nanobead–protein micropattern complex, bars are 100 μm. | |
The dry film material (PerMX) used for the SML process is mechanically robust and flexible while presenting outstanding chemical resistance to organic and inorganic solvents once photo-polymerized, which allows the stereomasks to be used repetitively. In this case, thorough cleaning with detergent (e.g., 2% soap solution) and then DI water is necessary to remove organic residues on the mask surface, which would adversely affect adhesion between the stereomask and substrate and potentially cross-contaminate the following biological patterns.
Conclusions
We have developed a versatile biological lithography technique, stereomask lithography, which enables universal biologically friendly micropatterning of multiple bio-objects with high resolution and exceptional flexibility. The 3D dry-film masks employed by the SML technique include unique features of (a) the high-resolution patterning for any aqueous dissolvable bio-substances, (b) the protective recess designs to prevent the masks from making direct contact with the pre-existing substances on substrates, (c) the peg-in-hole structures for facile alignment, and (d) the adhesion to various biological substrates, from which single-cell resolution (of 10 μm) and high-accuracy alignment (less than 10 μm) of sequential multi-bio-object processing have been achieved. Its applicability has been demonstrated by constructing arbitrary multiplexed protein patterns and cell–nanoparticle–protein complex. The SML technique offers a simple, low-cost, yet powerful solution for out-of-cleanroom biological microfabrication, which can lead to potential applications in quantitative biochemical analysis, tissue engineering, biosensing, and regenerative medicine.
Acknowledgements
This work is supported in part by National Science Foundation CAREER ECCS-0846502 and EFRI BSBA- 0937997. The authors would like to thank Dr Julie Sutcliffe and Dr Laura Marcu for use of the microscopes, and Dr Jiyoun Lee and Ms Jun Yan for technical assistance with cell culture and fluorescent microscopy. In addition, we would acknowledge DuPont Company for generously providing PerMX dry film samples.
Notes and references
- M. P. Lutolf and H. M. Blau, Adv. Mater., 2009, 21, 3255–3268 CrossRef CAS.
- R. A. Marklein and J. A. Burdick, Adv. Mater., 2010, 22, 175–189 CrossRef CAS.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS.
- S. Choudhuri, J. Biochem. Mol. Toxicol., 2004, 18, 171–179 CrossRef CAS.
- M. J. Heller, Annu. Rev. Biomed. Eng., 2002, 4, 129–153 CrossRef CAS.
- T. M. Blicharz, W. L. Siqueira, E. J. Helmerhorst, F. G. Oppenheim, P. J. Wexler, F. F. Little and D. R. Walt, Anal. Chem., 2009, 81, 2106–2114 CrossRef CAS.
- K. L. Gunderson, Methods Mol Biol, 2009, 529, 197–213 CAS.
- C. S. Chen, M. Mrksich, S. Huang, G. M. Whitesides and D. E. Ingber, Science, 1997, 276, 1425–1428 CrossRef CAS.
- J. C. Chang, G. J. Brewer and B. C. Wheeler, Biosens. Bioelectron., 2001, 16, 527–533 CrossRef CAS.
- M. Thery, V. Racine, A. Pepin, M. Piel, Y. Chen, J. B. Sibarita and M. Bornens, Nat. Cell Biol., 2005, 7, 947–953 CrossRef CAS.
- M. Thery, A. Jimenez-Dalmaroni, V. Racine, M. Bornens and F. Julicher, Nature, 2007, 447, 493–496 CrossRef CAS.
- R. McBeath, D. M. Pirone, C. M. Nelson, K. Bhadriraju and C. S. Chen, Dev. Cell, 2004, 6, 483–495 CrossRef CAS.
- S. A. Ruiz and C. S. Chen, Stem Cells, 2008, 26, 2921–2927 Search PubMed.
- C. M. Nelson, R. P. Jean, J. L. Tan, W. F. Liu, N. J. Sniadecki, A. A. Spector and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11594–11599 CrossRef CAS.
- H. Zhu, G. Stybayeva, M. Macal, E. Ramanculov, M. D. George, S. Dandekar and A. Revzin, Lab Chip, 2008, 8, 2197–2205 RSC.
- Y. Chen, C. Miller, R. Mosher, X. Zhao, J. Deeds, M. Morrissey, B. Bryant, D. Yang, R. Meyer, F. Cronin, B. S. Gostout, K. Smith-McCune and R. Schlegel, Cancer Res., 2003, 63, 1927–1935 CAS.
- H. C. Ma and K. Y. Horiuchi, Drug Discovery Today, 2006, 11, 661–668 CrossRef CAS.
- C. J. Flaim, S. Chien and S. N. Bhatia, Nat. Methods, 2005, 2, 119–125 CrossRef CAS.
- Y. Soen, A. Mori, T. D. Palmer and P. O. Brown, Mol. Syst. Biol., 2006, 2, 37.
- J. A. Burdick, A. Khademhosseini and R. Langer, Langmuir, 2004, 20, 5153–5156 CrossRef.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5, 401–406 RSC.
- N. Li Jeon, H. Baskaran, S. K. Dertinger, G. M. Whitesides, L. Van de Water and M. Toner, Nat. Biotechnol., 2002, 20, 826–830 CAS.
- D. Di Carlo, L. Y. Wu and L. P. Lee, Lab Chip, 2006, 6, 1445–1449 RSC.
- I. Inoue, Y. Wakamoto, H. Moriguchi, K. Okano and K. Yasuda, Lab Chip, 2001, 1, 50–55 RSC.
- B. Dykstra, J. Ramunas, D. Kent, L. McCaffrey, E. Szumsky, L. Kelly, K. Farn, A. Blaylock, C. Eaves and E. Jervis, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8185–8190 CrossRef CAS.
- M. Cordey, M. Limacher, S. Kobel, V. Taylor and M. P. Lutolf, Stem Cells, 2008, 26, 2586–2594 Search PubMed.
- J. C. Mohr, J. J. de Pablo and S. P. Palecek, Biomaterials, 2006, 27, 6032–6042 CrossRef CAS.
- J. M. Karp, J. Yeh, G. Eng, J. Fukuda, J. Blumling, K. Y. Suh, J. Cheng, A. Mahdavi, J. Borenstein, R. Langer and A. Khademhosseini, Lab Chip, 2007, 7, 786–794 RSC.
- H. C. Moeller, M. K. Mian, S. Shrivastava, B. G. Chung and A. Khademhosseini, Biomaterials, 2008, 29, 752–763 CrossRef CAS.
- A. Khademhosseini, L. Ferreira, J. Blumling, 3rd, J. Yeh, J. M. Karp, J. Fukuda and R. Langer, Biomaterials, 2006, 27, 5968–5977 CrossRef CAS.
- R. Derda, L. Li, B. P. Orner, R. L. Lewis, J. A. Thomson and L. L. Kiessling, ACS Chem. Biol., 2007, 2, 347–355 CrossRef CAS.
- S. S. Shah, M. C. Howland, L. J. Chen, J. Silangcruz, S. V. Verkhoturov, E. A. Schweikert, A. N. Parikh and A. Revzin, ACS Appl. Mater. Interfaces, 2009, 1, 2592–2601 CrossRef CAS.
- J. Y. Lee, S. S. Shah, J. Yan, M. C. Howland, A. N. Parikh, T. Pan and A. Revzin, Langmuir, 2009, 25, 3880–3886 CrossRef CAS.
- J. Y. Lee, S. S. Shah, C. C. Zimmer, G. Y. Liu and A. Revzin, Langmuir, 2008, 24, 2232–2239 CrossRef CAS.
- Y. N. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 551–575 CrossRef CAS.
- B. Ilic and H. G. Craighead, Biomed. Microdevices, 2000, 2, 6.
- R. N. Orth, J. Kameoka, W. R. Zipfel, B. Ilic, W. W. Webb, T. G. Clark and H. G. Craighead, Biophys. J., 2003, 85, 3066–3073 CrossRef CAS.
- S. Jinno, H. C. Moeller, C. L. Chen, B. Rajalingam, B. G. Chung, M. R. Dokmeci and A. Khademhosseini, J. Biomed. Mater. Res. A, 2008, 86, 278–288 CrossRef.
- C. P. Tan, B. R. Seo, D. J. Brooks, E. M. Chandler, H. G. Craighead and C. Fischbach, Integr. Biol., 2009, 1, 587–594 RSC.
- C. P. Tan, B. R. Cipriany, D. M. Lin and H. G. Craighead, Nano Lett., 2010, 10, 719–725 CrossRef CAS.
- M. P. Lutolf, R. Doyonnas, K. Havenstrite, K. Koleckar and H. M. Blau, Integr. Biol., 2009, 1, 59–69 RSC.
- S. W. Zhao, H. L. Cong and T. R. Pan, Lab Chip, 2009, 9, 1128–1132 RSC.
- T. Pan, A. Baldi and B. Ziaie, J. Microelectromech. Syst., 2006, 15, 267–272 CrossRef.
- S. Naffar-Abu-Amara, T. Shay, M. Galun, N. Cohen, S. J. Isakoff, Z. Kam and B. Geiger, PLoS One, 2008, 3, 1–9.
- P. Vulto, N. Glade, L. Altomare, J. Bablet, L. Del Tin, G. Medoro, I. Chartier, N. Manaresi, M. Tartagni and R. Guerrieri, Lab Chip, 2005, 5, 158–162 RSC.
- J. R. Rettig and A. Folch, Anal. Chem., 2005, 77, 5628–5634 CrossRef CAS.
- A. Revzin, R. G. Tompkins and M. Toner, Langmuir, 2003, 19, 9855–9862 CrossRef CAS.
- D. M. Pirone and C. S. Chen, J. Mammary Gland Biol. Neoplasia, 2004, 9, 405–417 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: A microscopic picture showing the alignment precision of the peg-in-hole method. See DOI: 10.1039/c0lc00275e |
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.