Stimuli responsive gels based on low molecular weight gelators
Xingyuan
Yang
ab,
Guanxin
Zhang
a and
Deqing
Zhang
*a
aBeijing National Laboratory for Molecular Sciences, Organic Solids Laboratory, Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, China. E-mail: dqzhang@iccas.ac.cn; Tel: +86 6263 9355
bGraduate School of Chinese Academy of Sciences, Beijing, 100190, China
First published on 24th October 2011
Abstract
In this feature article we summarize the recent developments of stimuli responsive gels based on low molecular weight gelators (LMWGs) for which the gel-sol transitions can be triggered by other stimuli besides heating. These include gels responsive to redox reactions and light irradiations. Chemoresponsive gels are also introduced, including those for which the gel-sol transitions can be induced by reactions with neutral molecules and those for which the gel strength is improved following the addition of neutral molecules. Perspectives for such stimuli responsive gels are discussed with regard to their potential applications and remaining challenging issues.
 Xingyuan Yang Xingyuan Yang | (Right) Xingyuan Yang received his B.S. in chemistry from Beijing Normal University in 2005 and Ph.D. in organic chemistry from the Institute of Chemistry, Chinese Academy of Sciences in 2011 under the supervision of Prof. Deing Zhang and Prof. Daoben Zhu. Currently he is an engineer at the Research Institute of Petroleum Processing (RIPP). His research interests include design and synthesis oforganic functional molecules. |
(Left ) Guanxin Zhang was born in 1976 and received his M.Sc. in chemistry from Beijing Normal University in 2002 and Ph.D. in organic chemistry from ICCAS in 2005 under the supervision of Prof. Deing Zhang and Prof. Daoben Zhu. He is now an Associated Professor at the Institute. His research interests include design, synthesis and self-assembly of organic functional molecules, chemo/biosensors and nanomaterials. |
(Middle) Deqing Zhang received his Ph.D. from Ruprecht-Karls University Heidelberg in 1996 under the supervision of Prof. Dr H. A. Staab. He is currently a Research Professor at ICCAS. His research interests include development of external stimuli-responsive molecular systems for molecular switches, logic gates and chemo/biosensors. He is also interested in the design and synthesis of organic conjugated molecules toward functional assemblies and materials. He serves as an Editorial Advisory Board member of several scientific journals. |
1. Introduction
Gels as soft materials are continuous in structure and solid-like in rheological behavior.1–7 They can be categorized into chemical gels and physical gels.8–16 Usually, chemical gels are covalently cross-linked polymers and not able to be redissolved, whereas physical gels based on intermolecular interactions can be redissolved again showing cycled reversibility. Self-assembly of certain molecules, referred to as low-molecular weight gelators (LMWGs),1,2,9 through weak intermolecular interactions such as H-bonding, π–π stacking and van der Waals interactions, can lead to solvent gelation and formation of physical gels. In the gel phase, molecules of LMWGs are self-assembled into entangled three-dimensional networks, which entrap and immobilize solvent molecules by capillary forces. These physical gels are characterized by the following unique features: (1) they are generally composed of hierarchically fiber-like structures, which cross-link to form a three-dimensional (3D) network;6,8 (2) they are thermally responsive, namely, the gels are transformed into solutions after heating and they can be recovered after cooling, since molecules of LMWGs within the fibril structures are associated through weak non-covalent interactions such as H-bonding, π–π stacking and van der Waals;8,9,11 (3) they are viscoelastic owing to the fact that solvent molecules are macroscopically immobilized by surface tensions and capillary forces;1,2,17 (4) they possess the respective critical gelation temperatures (Tgel), at which the gel-sol transitions occur, and the respective critical gelator concentrations (CGC), above which gels form in certain solvents at room temperature.1,2,8,17Gels based on LMWGs have been investigated for tens of years. However a key question, “what are the structural requirements for a molecule to gel an organic liquid”, still remains. This is because the process of gelation is complicated and often in competition with other kinds of molecular aggregations such as crystallization. Development of LMWGs is usually regarded as serendipitous discovery. However, rational design of LMWGs is eagerly pursued and has played an important role in generation of new LMWGs. Chemical segments such as cholesterol and urea have been found favorable for gelation of solvents and thus they are widely employed for designing new LMWGs.
Recent studies have demonstrated that these physical gels may have potential applications in a number of areas including nanomaterials and delivery or modification agents for paints, inks, cleaning agents, drugs, etc.17–21 As a result, they have received increasing attention in recent years. Apart from studies on the gel formation and structures, extensive efforts have been made to investigate functional and stimuli responsive gels. Applications of these physical gels, for example as microreactors for chemo- and biotransformation, have also been intensively addressed. Several critical reviews have been published highlighting the advances of gel chemistry from different aspects.8,17,22–24 In this paper, we want to focus on recent developments of stimuli-responsive gels based on LMWGs.
Stimuli-responsive physical gels offer us promising opportunities for designing and constructing new functional materials, such as sensors, actuators, etc. Apart from thermal responsiveness, various physical gels also respond to other stimuli including light irradiation and redox reactions which can also trigger the gel-sol (solution) transitions. The molecular design rationale for these stimuli-responsive gels is to incorporate photoresponsive-/electroactive-/chemical reactive segments into the respective LMWGs derived from cholesterol, urea and other gelating moieties. For instance, organogels which respond to light irradiation have been achieved by incorporating photoresponsive moieties (e.g. azobenzene and stilbene)8,23 into the corresponding LMWGs. By designing LMWGs with electroactive moieties, organogels showing responsiveness to redox reactions have been described.23 In the following, we will introduce the representative examples of stimuli-responsive gels based on LMWGs with responsive groups to demonstrate the molecular design principle and perspectives for these fascinating physical gels.
2. Redox-responsive gels based on LMWGs with electroactive groups
It is known that tetrathiafulvalene (TTF) and its derivatives can be reversibly transformed into the respective radical cations (TTF˙+) and dications (TTF2+) by either chemical or electrochemical redox reactions.25,26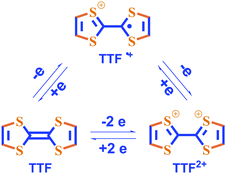
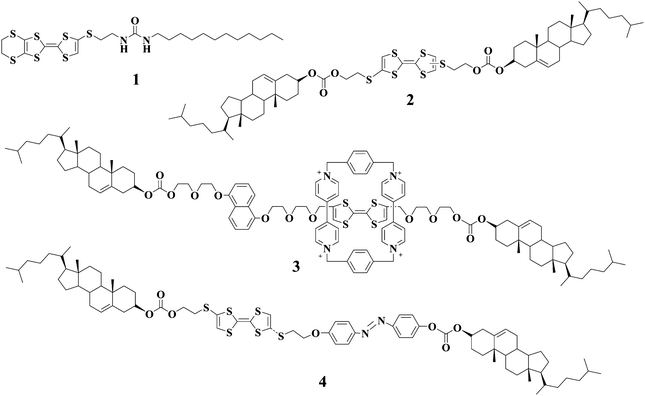
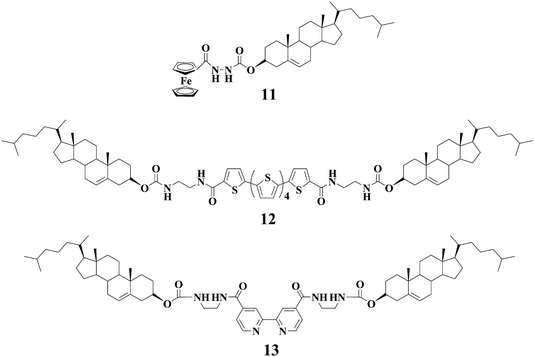
By taking advantage of this unique feature, we and other groups have reported the TTF-based gelators and the resulting gels that show responsiveness to redox reactions. LMWG 127 is one of such TTF-derived gelators. Apart from electroactive TTF segment it also contains a urea group that can form intermolecular extended H-bonging and accordingly is widely employed as one of gelating groups. LMWG 1 can gel several solvents such as cyclohexane and 1,2-dichloroethane. Either addition of Fe3+ or applying of an oxidation potential to the gel of 1 can lead to the gel-sol transition. Spectroscopic investigations show that TTF unit in 1 is oxidized into the corresponding TTF˙+ after inputs of either Fe3+ or an oxidation potential. Interestingly, the gel state can be restored by electrochemical reduction, followed by heating and cooling (see Fig. 1). Therefore, the gel-sol transition can be tuned as a function of the redox state of the TTF moiety in LMWG 1. Alternatively, the gel-phase of LMWG 1 can also be modulated by reaction with tetracyanoquinodimethane (TCNQ) which is a strong electron acceptor. Addition of TCNQ to the gel of 1 from 1,2-dichloroethane results in the destruction of the gel state, but the reaction of 1 with TCNQ in cyclohexane leads to a dark-green gel. This may be attributed to the fact that 1,2-dichloroethane is more polar than cyclohexane and as a result the TTF unit in 1 interacts more strongly with TCNQ in 1,2-dichloroethane. Apart from urea group cholesterol units were incorporated into TTF unit to generate the TTF-based gelator 2.28 The gel of 2 from hexane can be destructed upon addition of I2 which can oxidize the TTF unit in 2. This result provides another example of organogels that respond to redox reactions.
 | ||
| Fig. 1 Gel formation with 1 and tuning by the formation of charge-transfer complexes and chemical or electrochemical oxidation and reduction. | ||
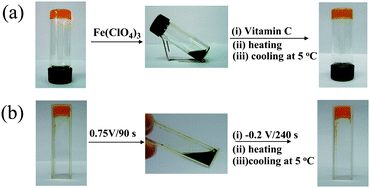
Stoddart and coworkers have built a fascinating chemistry of TTF-based rotaxanes.29–31 They have recently described a new gelator 3 with TTF-based bistable [2]rotaxane framework.32 The cholesterol units in 3 have dual functions, i.e. as stopper groups and gelating segments. LMWG 3 can gel a mixture of CH2Cl2/MeOH(3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, v/v), moreover, the gel can be transformed into the solution after oxidations with Fe(ClO4)3. This is likely due to the oxidation of the TTF unit and in turn the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring is switched from TTF station to 1,5-dioxynaphthalene (NDP) station as reported earlier.29–31 Accordingly, the intermolecular interactions may be altered, leading to the gel-sol transition. Although it has not been mentioned whether the gel phase can be restored by further reduction, it is expected that the gel-sol transition of LMWG 3 can be reversibly tuned by redox reactions of the TTF unit.
:2, v/v), moreover, the gel can be transformed into the solution after oxidations with Fe(ClO4)3. This is likely due to the oxidation of the TTF unit and in turn the cyclobis(paraquat-p-phenylene) (CBPQT4+) ring is switched from TTF station to 1,5-dioxynaphthalene (NDP) station as reported earlier.29–31 Accordingly, the intermolecular interactions may be altered, leading to the gel-sol transition. Although it has not been mentioned whether the gel phase can be restored by further reduction, it is expected that the gel-sol transition of LMWG 3 can be reversibly tuned by redox reactions of the TTF unit.
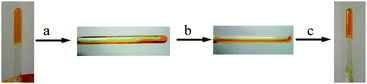
Some of us33 have just recently described a new gelator 4 featuring both electroactive TTF and photoresponsive azobenzene units with the end to generate multistimuli responsive gels. LMWG 4 can lead to gelation of several solvents and the gels become CD active because of the formation of chiral assemblies probably due to the cholesterol units in LMWG 4. The cis-trans photoisomerization can take place smoothly. As anticipated, the gel-sol transitions for gels of LMWG 4 can be reversibly tuned by either chemical or electrochemical oxidation/reduction reactions (Fig. 2). Additionally, the photoisomerization of the azobenzene group in 4 can also trigger the gel-sol transition (Fig. 3). Therefore, the gel-sol transition for gels of LMWG 4 induced by redox reactions and light irradiations can be operated separately. Physical gels of 4 respond not only to thermal stimuli but also to redox reactions and light irradiation.
 | ||
| Fig. 2 Reversible tuning of the gel formation of 4 (8.0 mg mL−1) in CH2Cl2/CH3OH (3/1, v/v) (a) by chemical oxidation and reduction and (b) by sequential electrochemical oxidation and reduction. | ||
 | ||
| Fig. 3 Reversible tuning of the gel formation of 4 (8.0 mg mL−1) in CH2Cl2/CH3OH (3/1, v/v) by UV light irradiation and further visible light irradiation: (a) UV light irradiation for 30 min; (b) UV light irradiation for 1.0 h; (c) further visible light irradiation for 2.0 h and then being left in the dark at 5.0 °C for 18 h. | ||
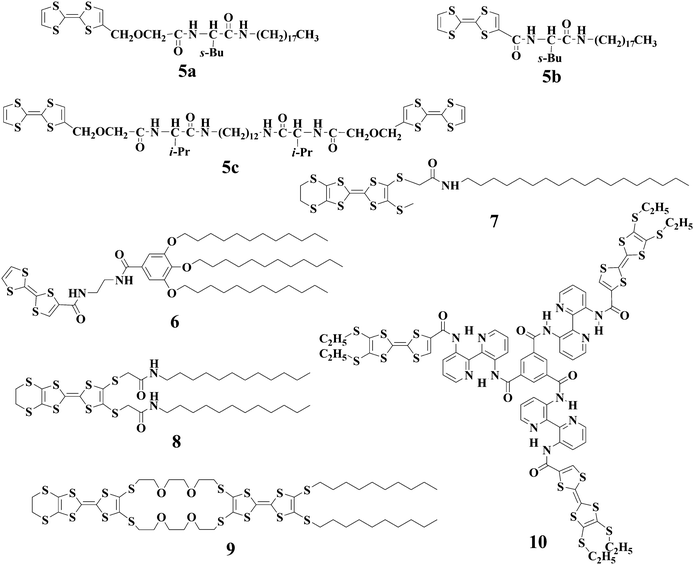
Other LMWGs with TTF units have also been reported in recent years. However, the motivation is to generate conducting nanostructures through gel formation. For instance, Kato and coworkers34 studied TTF derivatives 5a–c containing amino acid moieties. Gelation of 5a–c occurred for aromatic liquid crystals to form stable fibrous aggregates. Iodine-doping was successfully carried out for the solid-state self-assembled fibers, which showed conductivities in the range of 10−5–10−6 S cm−1 after doping. Meanwhile, Shinkai and coworkers35 reported another TTF-based gelator 6 bearing one 3,4,5-trialkoxybenzene group and two amide groups. Gelation of hydrocarbon solvents with LMWG 6 induced the formation of 1D nanostructures, which again could become conductive after exposure to I2 vapor. The TTF-drived gelator 7 containing one amide unit and one long alkyl chain can gel both hexane and decane.36,37 The resulting xerogel is composed of nanofibers in which a mixed-valence state due to the TTF unit in 7 is formed after doping with I2. Compound 8, the analogue of 7, can function as a gelator too.38 Direct casting of gel samples formed with 8 on the surface of HOPG led to the formation of fibrous materials. TTF-containing compounds 9 and 10 were also investigated as new gelators.39,40
Apart from TTF there are other electroactive units such as ferrocene and thiophene, which can also be reversibly tranformed into the respective cations by either chemical or electrochemical means; thus, the corresponding gels from LMWGs with these electroactive segments may also show responsiveness to redox reactions. For instance, Fang and coworkers41 studied LMWG 11 bearing a ferrocene segment. Their results manifest that the gel-sol transition of 11 can be reversibly tuned by chemical redox reactions with (NH4)2Ce(NO3)6 as oxidant and NH2·NH2 as reductant. In addition, the gel-sol transition is also affected by shear stress and sonication, and as a result the gels of 11 are multistimuli responsive.
Shinkai and his co-workers have also reported redox-stimuli responsive gels based on compounds 1242 and 13.43 Gelator 12 entails a redox active sexithiophene segment which enables gels of 12 to be responsive to redox reactions. For example, gel of 12 from 1,1,2,2-tetrachloroethane was changed to the corresponding solution by oxidation with FeCl3, and the gel state can be restored by further addition of ascorbic acid which can reduce the radical cation of sexithiophene segment to neutral. By taking advantage of the redox switching of Cu(I)/Cu(II) complexes, they reported another example of a heat-free sol–gel phase transition that can be induced by a redox stimulus. The Cu(I) coordination complex of 13 can gel solvents, and transformation of Cu(I) complex to the respective Cu(II) complex will induce structural distortion for the complex which in turn may destabilize the fibril structures leading to gel-sol transition.
3. Photoresponsive gels based on LMWGs
Azobenzene as a photoisomerization unit was incorporated into LMWGs to generate photoresponsive gels. Shinkai and coworkers44 initially designed LMWG 14 bearing an azobenzene segment and the gel-sol transitions for the resulting gels could be tuned by light irradiation. Later, Koumura and coworkers45 reported an azobenzene compound 15 with two urethane moieties linked to two cholesteryl ester units. They also found that the gel-sol transition for gels of 15 can take place upon photoirradiation as a result of trans-cis isomerization of the azobenzene unit. Further studies manifested that intermolecular H-bonding which was partly responsible for stabilizing the gels was broken or reformed during gel-sol transition induced by light irradiation.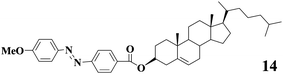
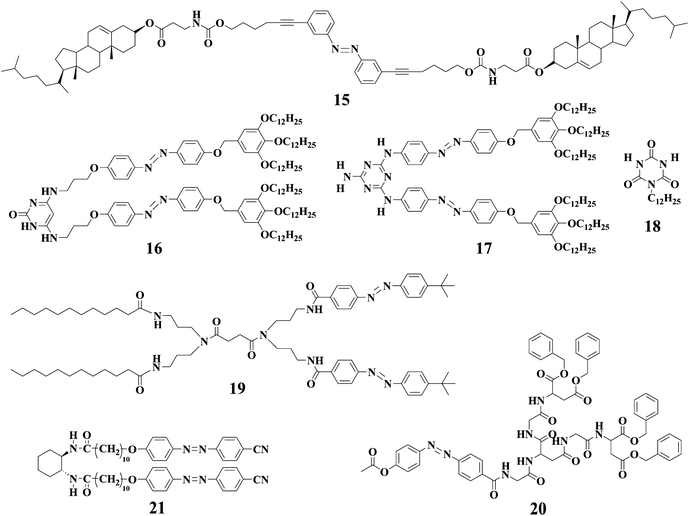
Yagai and coworkers46 described a gelator 16 derived from N,N′-disubstituted-4,6-diaminopyrimidin-2(1H)-one with two azobenzene units. 1H-NMR, absorption and dynamic light scattering studies clearly indicated that the self-assembly structures of 16 are strongly affected by the photoisomerization of the azobenzene units. This was ascribed to the variation of intermolecular H-bonding after trans-cis isomerization of azobenzene units. In fact, exposure of the gel of 16 from heptane resulted in the collapse of gel within a few hours. Further studies showed that the trans-cis isomerization of the azobenzene moieties induced the collapse of the lamellar structures into soluble supramolecular tapes. They also investigated the self-assembly structures from the mixture of 17 and 18.47 It was found that the concentrated cyclohexane solution of 17 and 18 gradually became viscous at room temperature and eventually a transparent gel was obtained after 24 h (Fig. 4). When the gel was irradiated by UV light for more than 15 min, the gel was transformed into the corresponding solution; the gel phase was regenerated after the photoinduced solution was kept in the dark for 48 h.
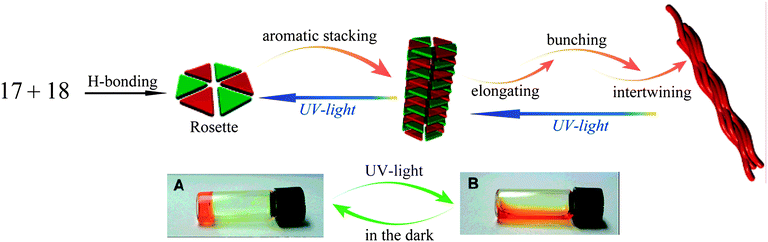 | ||
| Fig. 4 Illustration of the hierarchical self-assembly structures of 17 and 18, and the gel-sol transition under UV light irradiation. | ||
It usually takes a long period for the gel-sol transition of LMWGs with azobenzene moieties to complete by photoirradiation. However, rapid and reversible gel-sol transition was reported for gels derived from a bis-dendritic gelator 19 consisting of azobenzene dendron and aliphatic amide units; the collapse of the gel occurred in 2.0 min upon UV light irradiation, and gel regeneration was achieved by subsequent irradiation of visible light in just 5.0 s (Fig. 5).48 Another dendritic gelator 20 with one azobenzene unit was synthesized and the resulting gels were responsive to light irradiations as expected.49
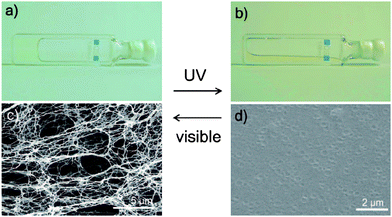 | ||
| Fig. 5 Photographs of the cyclohexane gel of 19 (0.05% w/v) (a) before and (b) after UV irradiation for 120 s, and FE-SEM images after drying (c) from the gel state and (d) from the sol state. | ||
Kato and coworkers50,51 designed a chiral gelator 21 with photochromic azobenzene moieties. Interestingly, they investigated the gelation of a liquid crystal, 4-cyano-4′-pentyl-biphenyl with 21. Nematic gel state was obtained by cooling the isotropic liquid state, and was transformed into the cholesteric liquid crystal phase after UV light irradiation. Cholesteric gel state was generated after keeping the cholesteric liquid state or visible light irradiation. Such photoresponsive soft materials may have potential applications in re-writable information recording.
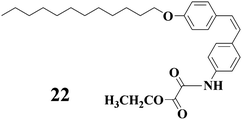
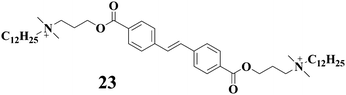
Gels that respond to light irradiation were also constructed with LMWGs containing stilbens moieties. As an example, compound 22 is composed of a cis-stilbenes group and one oxamide moiety, which can be dissolved in ethanol.52,53 However, the clear solution of 22 turned into an opaque gel after light irradiation with a high-pressure mercury lamp. Prolonged irradiation led to the crystallization of the gelator (the corresponding trans-form of 22) from the system with time. Heating a gel or a crystallized precipitate and further cooling resulted in the repeated gel formation. Compound 23 can gelate toluene-d8 with the aid of a small amount of N,N′-dimethyldodecylamine.54 The gel can be transformed into the solution after UV light irradiation. Interestingly, when the gel was irradiated through a mask, only within the illuminated areas did the gel-sol transition occur. 1H-NMR studies indicated that this gel-sol transition was due to the formation photodimer of 23 upon UV light irradiation.
Das and coworkers55 studied the self-assembly of the butadiene 24 and its analogues. The resulting gel of 24 from n-butanol was composed of globular aggregates rather than interconnected nanofibers. Rheological studies indicated the gels were stable and not thixotropic. Irradiation of the gel at wavelength longer than 385 nm led to gel-sol transition, as observed by the free-flowing nature of the irradiated sample. This is believed to be due to the trans-cis isomerization of one C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond in 24 as shown in Chart 1. Keeping the irradiated solution in the dark for 24 h resulted in the re-generation of the gel.
C bond in 24 as shown in Chart 1. Keeping the irradiated solution in the dark for 24 h resulted in the re-generation of the gel.
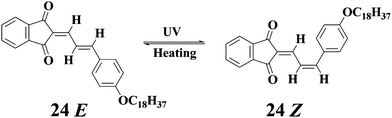 | ||
| Chart 1 Photo- and thermal isomerization of 24. | ||
Photodimerization may occur to anthracene-derived compounds when intermolecular arrangements are appropriate. Shinkai and coworkers56 described binary gelators 25a–c which were prepared from equimolar amounts of anthracene-9-carboxylic acid and the corresponding alkylamine. Their gelation abilities were found to be dependent on the lengths of the respective alkyl chains. The gels were converted to the solutions after UV light irradiation because of the generation of photodimers as shown in Chart 2. These photodimers can be dissociated by heating, but the gels cannot be regenerated directly. However, the gel phase can be recovered by further cooling the solution. Some of us reported gelator 2657 with an anthracene moiety. However, photodimerization did not happen for the gels of 26 and indeed the gel-sol transition did not take place for these gels upon UV light irradiation. Interestingly, gelator 26 became strongly emissive (green emission) after gelation (Fig. 6). This was attributed to the formation of emissive intermolecular anthracene-excimers in the gel phase, and as a result the photodimerization was inhibited.
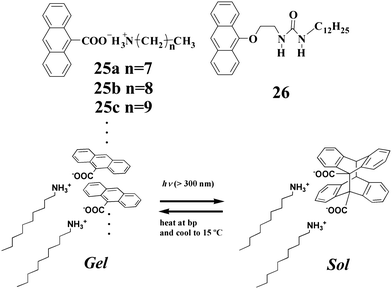 | ||
| Chart 2 Photodimerization and dissociation of 25c under UV light irradiation and heating. | ||
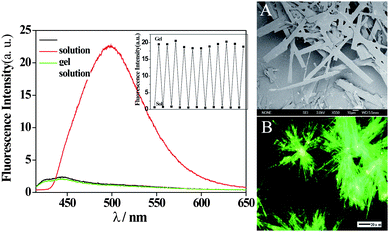 | ||
| Fig. 6 (Left) Fluorescence spectra of 26 (20 mg mL−1) in 1,2-dichloroethane (black), the gel phase (red), and the solution formed after heating the gel phase (green); (Right) SEM image of the xerogel based on 26 (A) and fluorescence microscopy image of the xerogel (B) from 1,2-dichloroethane. | ||
Compounds 27 and 2858 are photochromic because of the presence of 2H-chromene moieties which can be reversibly transformed between the closed and open forms as shown in Chart 3. The neutral carboxylic acid derivatives of 27 and 28 can be easily dissolved in DMF and DMSO. However, the corresponding sodium salts can gelate DMF and DMSO. Upon UV light irradiation the gels became rapidly colored and transformed into the solutions. The studies show that the transformation of the closed form into the open form of the 2H-chromene moieties in 27 and 28 affects strongly the intermolecular H-bonding interactions. The gel-phases can be restored by heating and further cooling the solutions. In addition, the gel-sol transitions for gels of 27 and 28 can also be triggered by lowering the pH values of the systems.
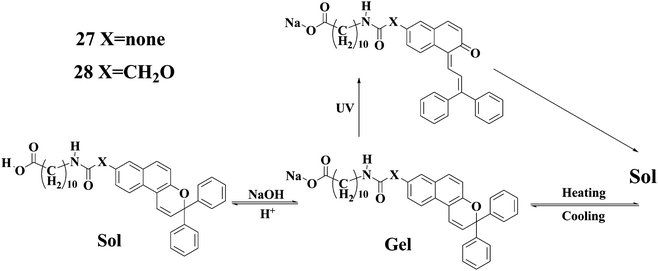 | ||
| Chart 3 Illustration of interconversion of compounds 27 and 28. | ||
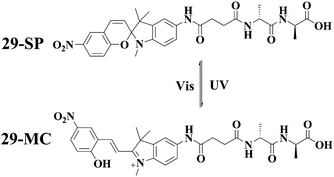
Spiropyran (SP) is another type of photochromic molecules. The respective closed form and open form (merocyanine form) can be reversibly interconverted by alternating UV and visible light irradiations. By taking advantage of the photochromic behavior of spiropyran, Zhang and her coworkers developed a photoresponsive hydrogelator 2959 in which the spiropyran unit was converted to the corresponding merocyanine form (MC). The gel-sol transition occurred upon exposure of the gel to visible light irradiation, and the gel phase could be regenerated by further UV light irradiation (Fig. 7). The light responsiveness of the hydrogel based on 29 is due to the fact that merocyanine moiety has a strong tendency to form π–π stacking compared to the closed form (spiropyran).
 | ||
| Fig. 7 Reversible transformation of the SP and MC forms of 29, and an illustration of the gel-sol transition after photoirradiation. | ||
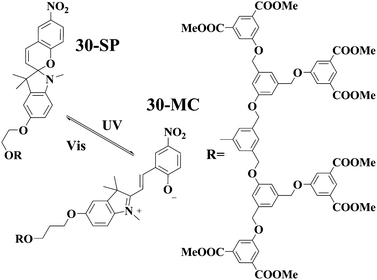
Some of us reported a spiropyan-functionalized dendron 3060 which can gel toluene and benzene. Multivalent π–π interactions due to the dendron segment in 30 may be the driving force for the gelation. Absorption spectral studies showed that the photochromic reaction of the spiropyran unit in 30 could take place both in solution and gel phase. However, the gel phase was not destroyed after UV light irradiation, and the yellow gel was transformed into the purple-blue gel as illustrated in Fig. 8. Further visible light irradiation of the purple-blue gel led to the yellow gel again. The results also manifest that the merocyanine form in the gel phase becomes more stable compared to that in solution. Of particular interest the purple-blue gel exhibits relatively strong red fluorescence and the red fluorescence of the gel can be switched off by visible light irradiation.
 | ||
| Fig. 8 Illustration of the gel-to-gel transformation after heating, cooling, UV and visible light irradiations. | ||
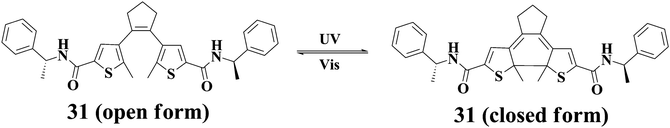
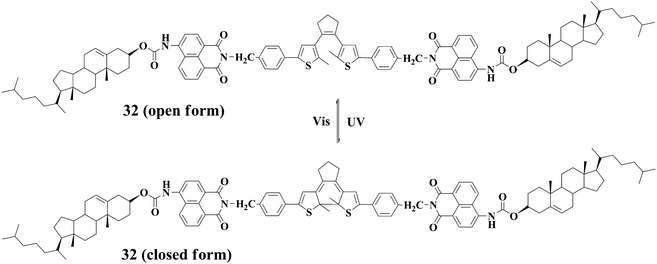
Dithienylethene derivatives as photochromic molecules have been extensively investigated. Gelators containing photochromic dithienylethene units are available. For instance, Feringa and coworkers reported gelator 3161 featuring one dithienylethene moiety. Photochromic transformations can easily take place for 31 both in the solution and gel states accompanying the reversible spectral changes. However, the gel-sol transition cannot be triggered by light irradiation. Interestingly, the chirality of the gel phase is preserved during the photochromic reactions; moreover, the chirality is lost after gel-sol transition, and gels with opposite chirality are generated after further cooling the respective solutions as illuatrated in Chart 4. Tian and his co-workers62 described another dithienylethene-derived gelator 32. Similarly, photochromic transformations can occur for 32 both in the solution and gel states. Again, the gel-sol transition cannot be induced by light irradiations. However, the gel can be converted to the solution after introducing F−, and the gel phase can be restored by addition of acid (Fig. 9).
 | ||
| Chart 4 Reversible transformation of the open and closed forms of 31, and the gel-sol transitions with chirality variation. | ||
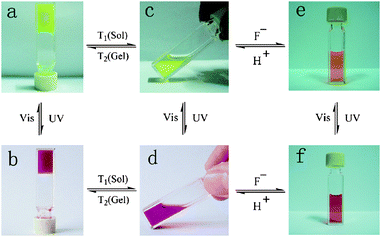 | ||
| Fig. 9 Reversible transformation of the open and closed forms of 32, and the gel-sol transitions under the cooperative effects of light, thermal, fluoride anions, protons: (a) Gel(open); (b) Gel(closed); (c) Sol(open); (d) Sol(closed); (e) Sol(open) + F−; (f) Sol(closed) + F−. | ||
4. Chemoresponsive gels based on LMWGs
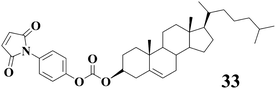
Chemoresponsive gels based on LMWGs are herein referred to as gels that exhibit responsiveness to chemical species (neutral molecules or metal ions or anions). Clarke, Steed and their coworkers17 have just recently published a detailed review on the progresses of metal- and anion-binding supramolecular gels, and they have already introduced various gels based on LMWGs which respond to metal- and anion-binding. Xu and coworkers63–69 have reported series of hydrogels in the presence of enzymes. In this section we will just focus on gels that are responsive to neutral molecules through either chemical reactions or intermolecular interactions with LMWGs.Some of us70 designed the cholesterol-based gelator 33 with maleimide unit which can easily react with thiols under basic conditions to form the corresponding Michael adducts. Gelator 33 can gel several solvents including cyclohexane leading to transparent gels. It is interesting to note that the gel of 33 from cycohexane can be gradually transformed into the solution after addition of n-hexylthiol (ca. 2.1 eq.) and triethylamine (ca. 2.1 eq.) onto the top layer of the gel as demonstrated in Fig. 10. Such gel-sol transition is attributed to the Michael reaction between the maleimide unit in 33 and thiol, which is facilitated by the presence of triethylamine; accordingly the intermolecular interactions of 33 will be altered. However, if n-hexylthiol is added alone, the gel-sol transition can not occur even after several days under the same conditions. This is probably because the Michael reaction between maleimide and thiol is slow in the absence of bases such as triethylamine. When only triethylamine is added the gel phase can not be destroyed either.
 | ||
| Fig. 10 Chemical structure of 33 and gel-sol phase transition triggered by the addition of n-hexylthiol and triethylamine after 2 h for the organogel of 33 (5.0 mg mL−1) in cyclohexane. | ||
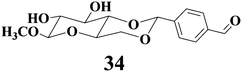
Compound 34 was a saccharide-derived hydrogelator containing an aldehyde group which can react with cysteine to form thiazolidine derivative.71Fig. 11 displays the variation of hydrogels of 34 after the careful addition of the corresponding aqueous solution of amino acids on the top of the hydrogels and further incubation at 37 °C for 4 h. The hydrogel in the vial containing cysteine was completely transformed into solution, and the hydrogels in other vials were unaffected as demonstrated in Fig. 11. Such selective responsiveness of the hydrogel toward cysteine should be related to the specific reaction of aldehyde with cysteine to form the thiazolidine derivative. The aldehyde group cannot react with other amino acids including the acidic and basic ones under the same conditions. In addition, the hydrogel of 34 is sensitive to acidic solutions. The hydrogel of 34 can be converted to the solution after mixing the aqueous solution with pH = 1.18 at 37 °C for 60 h.
 | ||
| Fig. 11 Hydrogels with 34 covered with the aqueous solutions of different amino acids; the sequence of amino acids added to the hydrogels from left to right is as follows: aspartic acid, lysine, alanine, serine, cysteine, histidine, arginine, proline, and phenylalanine. | ||
Alkylamines are generally nongelators or inefficient ones. However, Weiss and coworkers72,73 found that the amines can react with carbon dioxide to afford ammonium carbamates (see Chart 5) which are more efficient LMWGs than the respective amines. In fact, these ammonium carbamates from the reaction between the respective amines and CO2 can gel several solvents. Interestingly, the ammonium carbamates can be transformed into the corresponding amines again by slight heating. In this manner, reversible formation of organogels is realized by employing ‘latent’ gelators (amines) and CO2.
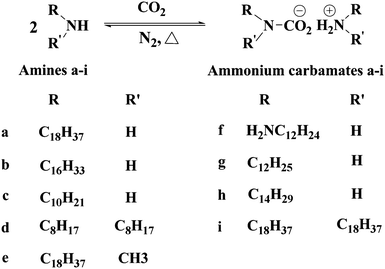 | ||
| Chart 5 The reaction of amines and carbon dioxide generating ammonium carbamates. | ||
Several reports indicate that it is possible to improve the gel stabilities by introducing certain molecules to the systems. For instance, Shinkai and coworkers74 reported porphyrin-derived gelator 35 with amide groups. Compound 35 can gel benzene and toluene, but not p-xylene and anisole. However, all four of the solvents noted above can be gelled by 35 in the presence of C60. Moreover, the gel-to-sol transition temperature for the gel of 35 from benzene was enhanced from 79 to 120 °C after addition of C60. A striking morphological change was induced for the self-assembly structures of 35 after introducing C60. They attributed these changes to the strong porphyrin-C60 interactions.
Compounds 36a–d are series of Zn(II) porphyrin-based gelators with cholesterol segments reported by Shinkai and coworkers.75 Interestingly, the influence of C60 on the gelation abilities of 36a–d are dependent on the spacer groups in 36a–d. The gelation abilities of 36a and 36c in which –(CH2)2– and –(CH2)4– are the space groups, respectively, are enhanced in the presence of C60. For 36b and 36d in which the space groups are –(CH2)3– and –(CH2)5–, respectively, however, their gelation abilities are not obviously affected by C60. The influence of C60 on the gelation abilities of 36a and 36c is ascribed to the binding of Zn(II) porphyrin units with C60, forming a 2:1 Zn(II) porphyrin/C60 sandwich complex.
9,10-bis(1,3-Dithiol-2-ylidene)-9,10-dihydroanthracene, which is usually referred to as ex-TTF, is unique in terms of its structure and electrochemical behavior. Martín et al.76–81 have presented series of reports with regard to the binding of ex-TTF with fullerene and assembly of ex-TTF-fullerene derivatives into nanostructures comprising supramolecular dendrimers. We have very recently reported an ex-TTF derived gelator 37 with L-glutamid-derived lipid segment which can facilitate the gelation through hydrophobic and H-bonding interactions.82 Compound 37 can gel several organic solvents such as toluene, DMSO and ethanol. Notably, the gelation ability of 37 can be enhanced after addition of C60. The gel melting temperature increases gradually by introducing C60 reaching the maximum after addition of 0.5 eq. of C60.
Compound 38 is a new C60-derived gelator with L-glutamid-derived lipid unit reported by us just recently.83 The results manifest that the gelation ability of 38 can be improved by addition of an exTTF compound 39, for which the intermolecular interaction between the C60 unit in 38 and exTTF unit in 39 is responsible based on the absorption and 1H-NMR spectral studies.
Ajayaghosh and his collaborators84 have intensively investigated the self-assembly and gel formation of oligo(p-phenylene vinylene)s (OPVs). For instance, compound 4084d with two –OH groups can gel several nonpolar solvents, but the gels of 40 from relatively polar solvents are thixotropic and mechanically unstable. They have recently shown that the self-assembly of OPV molecules is accelerated in the presence of carbon nanotubes (CNTs) through the interactions of OPV molecules and CNTs. In this way, CNTs which are dispersed and aligned within the gel can on one hand reinforce the self-assembly structures of OPV molecules, and may on the other hand act as physical cross-links between these self-assembly structures, thus enhancing the gel stability.
Shinkai and coworkers85 have described a versatile gelator 41 with an electron accepting naphthalenediimide moiety. It is interesting to note that gels of 41 can form charge-transfer complexes with dihydroxynaphthalenes, but not with alkoxy- and hydroxynaphthalenes. Moreover, different color gels are generated with different isomers of dihydroxynaphthalenes. Thus, naked-eye differentiation of positional isomers of dihydroxynaphthalenes becomes possible with the gels of 41. The authors also mentioned that the gel superstructure was extremely sensitive to the amount of dihydroxynaphthalene compounds added to the gel system. For example, when more than 1.2 equiv of dihydroxynaphthalene were added, the gel-sol transition occurred easily.
5. Conclusion and perspectives
In summary, we have introduced recent developments of stimuli-responsive gels based on LMWGs for which the gel-sol transitions can be triggered by other stimuli except heating. These stimuli-responsive gels are prepared from LMWGs with electroactive or photoresponsive or reactive groups. However, molecular design of LMWGs leading to stimuli-responsive gels is still not straightforward since incorporation of functional groups into gelator-frameworks may perturb the self-assembly processes in a destructive way. Additional studies will be required, especially those dealing with the kinetics and mechanisms of gel formation. These studies may offer guiding principles for rationally designing functional LMWGs and responsive gels.Clearly, stimuli-responsive gels based on LMWGs deserve more exploitations since their potential applications are enormous. For instances, such gels may find uses in areas where on-demand flow is desirable. They may also be useful as reversibly formed membranes or media for the controlled release of drugs. When functional solvents such as cholesteric or ferroelectric liquid crystals are gelled with these functional gelators, more advanced applications can be imagined.
Further studies in this attractive area need the endeavours of scientists of different disciplines including organic chemistry, physical chemistry, nanoscience and materials science. Stability and mechanical strength are among the issues that must be well addressed for realizing the practical applications of such gels. Design and synthesis of functional gelators that will lead to gels responsive to more than two external stimuli still remain challenging.
References
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3159 CrossRef CAS.
- D. J. Abdallah and R. G. Weiss, Adv. Mater., 2000, 12, 1237 CrossRef CAS.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- J. H. van Esch and B. L. Feringa, Angew. Chem., Int. Ed., 2000, 39, 2263–2266 CrossRef CAS.
- O. Gronwald, E. Snip and S. Shinkai, Curr. Opin. Colloid Interface Sci., 2002, 7, 148–156 CrossRef CAS.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS.
- C. Sanchez, B. Julian, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS.
- J. D. Mackenzie and E. P. Bescher, Acc. Chem. Res., 2007, 40, 810–818 CrossRef CAS.
- W. Cai, G. T. Wang, P. Du, R. X. Wang, X. K. Jiang and Z. T. Li, J. Am. Chem. Soc., 2008, 130, 13450–13459 CrossRef CAS.
- M. Suzuki, M. Yumoto, H. Shirai and K. Hanabusa, Chem.–Eur. J., 2008, 14, 2133–2144 CrossRef CAS.
- L. Yu and J. D. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2009, 38, 967–975 RSC.
- P. C. Xue, R. Lu, X. C. Yang, L. Zhao, D. F. Xu, Y. Liu, H. Zhang, H. Nomoto, M. Takafuji and H. Ihara, Chem.–Eur. J., 2009, 15, 9824–9835 CrossRef CAS.
- M. O. M. Piepenbrock, G. O. Lloyd, N. Clarke and J. W. Steed, Chem. Rev., 2010, 110, 1960–2004 CrossRef CAS.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 455–463 RSC.
- M. Suzuki and K. Hanabusa, Chem. Soc. Rev., 2010, 39, 5067–5067 RSC.
- Q. Chen, D. Q. Zhang, G. X. Zhang, X. Y. Yang, Y. Feng, Q. H. Fan and D. B. Zhu, Adv. Funct. Mater., 2010, 20, 3244–3251 CrossRef CAS.
- D. J. Adams, Macromol. Biosci., 2011, 11, 160–173 CrossRef CAS.
- A. Dawn, T. Shiraki, S. Haraguchi, S. Tamaru and S. Shinkai, Chem.–Asian J., 2011, 6, 266–282 CrossRef CAS.
- D. D. Diaz, D. Kuhbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- (a) M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC; (b) J. L. Segura and N. Martín, Angew. Chem., Int. Ed., 2001, 40, 1372–1409 CrossRef CAS; (c) A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433–444 CrossRef CAS; (d) J. Jeppesen, M. Nielsen and J. Becher, Chem. Rev., 2004, 104, 5115–5132 CrossRef CAS; (e) N. Martín, L. Sánchez, M. A. Herranz, B. Illescas and D. Guldi, Acc. Chem. Res., 2007, 40, 1015–1024 CrossRef; (f) W. Dichtel, O. Miljanić, W. Zhang, J. Spruell, K. Patel, I. Aprahamian, J. Health and J. Stoddart, Acc. Chem. Res., 2008, 41, 1750–1761 CrossRef CAS . and reference therein.
- (a) D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC; (b) X. Y. Yang, D. Q. Zhang, G. X. Zhang and D. B. Zhu, Sci. China Chem., 2011, 54, 596–602 CrossRef CAS.
- C. Wang, D. Q. Zhang and D. B. Zhu, J. Am. Chem. Soc., 2005, 127, 16372–16373 CrossRef CAS.
- C. Wang, F. Sun, D. Q. Zhang, G. X. Zhang and D. B. Zhu, Chin. J. Chem., 2010, 28, 622–626 CrossRef CAS.
- J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Angew. Chem., Int. Ed., 2001, 40, 1216 CrossRef CAS.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924–1936 CrossRef CAS.
- R. E. Gillard, F. M. Raymo and J. F. Stoddart, Chem.–Eur. J., 1997, 3, 1933–1940 CrossRef CAS.
- Y. L. Zhao, I. Aprahamian, A. Trabolsi, N. Erina and J. F. Stoddart, J. Am. Chem. Soc., 2008, 130, 6348 CrossRef CAS.
- C. Wang, Q. Chen, F. Sun, D. Q. Zhang, G. X. Zhang, Y. Y. Huang, R. Zhao and D. B. Zhu, J. Am. Chem. Soc., 2010, 132, 3092–3096 CrossRef CAS.
- T. Kitamura, S. Nakaso, N. Mizoshita, Y. Tochigi, T. Shimomura, M. Moriyama, K. Ito and T. Kato, J. Am. Chem. Soc., 2005, 127, 14769–14775 CrossRef CAS.
- T. Kitahara, M. Shirakawa, S. Kawano, U. Beginn, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2005, 127, 14980–14981 CrossRef CAS.
- J. Puigmarti-Luis, E. E. Laukhina, V. N. Laukhin, A. P. del Pino, N. Mestres, J. Vidal-Gancedo, C. Rovira and D. B. Amabilino, Adv. Funct. Mater., 2009, 19, 934–941 CrossRef CAS.
- J. Puigmarti-Luis, V. Laukhin, A. P. del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238–241 CrossRef CAS.
- J. Puigmarti-Luis, A. P. del Pino, V. Laukhin, L. N. Feldborg, C. Rovira, E. Laukhina and D. B. Amabilino, J. Mater. Chem., 2010, 20, 466–474 RSC.
- T. Akutagawa, K. Kakiuchi, T. Hasegawa, S. Noro, T. Nakamura, H. Hasegawa, S. Mashiko and J. Becher, Angew. Chem., Int. Ed., 2005, 44, 7283–7287 CrossRef CAS.
- I. Danila, F. Riobe, J. Puigmarti-Luis, A. P. del Pino, J. D. Wallis, D. B. Amabilino and N. Avarvari, J. Mater. Chem., 2009, 19, 4495–4504 RSC.
- J. Liu, P. L. He, J. L. Yan, X. H. Fang, J. X. Peng, K. Q. Liu and Y. Fang, Adv. Mater., 2008, 20, 2508 CrossRef CAS.
- S. Kawano, N. Fujita and S. Shinkai, Chem.–Eur. J., 2005, 11, 4735–4742 CrossRef CAS.
- S. Kawano, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2004, 126, 8592–8593 CrossRef CAS.
- K. Murata, M. Aoki, T. Suzuki, T. Harada, H. Kawabata, T. Komori, F. Ohseto, K. Ueda and S. Shinkai, J. Am. Chem. Soc., 1994, 116, 6664–6676 CrossRef CAS.
- N. Koumura, M. Kudo and N. Tamaoki, Langmuir, 2004, 20, 9897–9900 CrossRef CAS.
- S. Yagai, T. Iwashima, K. Kishikawa, S. Nakahara, T. Karatsu and A. Kitamura, Chem.–Eur. J., 2006, 12, 3984–3994 CrossRef CAS.
- S. Yagai, T. Nakajima, K. Kishikawa, S. Kohmoto, T. Karatsu and A. Kitamura, J. Am. Chem. Soc., 2005, 127, 11134–11139 CrossRef CAS.
- J. H. Kim, M. Seo, Y. J. Kim and S. Y. Kim, Langmuir, 2009, 25, 1761–1766 CrossRef CAS.
- Y. Ji, G. C. Kuang, X. R. Jia, E. Q. Chen, B. B. Wang, W. S. Li, Y. Wei and J. Lei, Chem. Commun., 2007, 4233–4235 RSC.
- M. Moriyama, N. Mizoshita, T. Yokota, K. Kishimoto and T. Kato, Adv. Mater., 2003, 15, 1335–1338 CrossRef CAS.
- M. Moriyama, N. Mizoshita and T. Kato, Bull. Chem. Soc. Jpn., 2006, 79, 962–964 CrossRef CAS.
- S. Miljanić, L. Frkanec, Z. Meić and M. Žinić, Langmuir, 2005, 21, 2754–2760 CrossRef.
- S. Miljanić, L. Frkanec, Z. Meić and M. Žinić, Eur. J. Org. Chem., 2006, 1323–1334 CrossRef.
- J. Eastoe, M. Sanchez-Dominguez, P. Wyatt and R. K. Heenan, Chem. Commun., 2004, 2608–2609 RSC.
- N. S. S. Kumar, S. Varghese, G. Narayan and S. Das, Angew. Chem., Int. Ed., 2006, 45, 6317–6321 CrossRef CAS.
- M. Ayabe, T. Kishida, N. Fujita, K. Sada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 2744–2747 CAS.
- C. Wang, D. Q. Zhang, J. F. Xiang and D. B. Zhu, Langmuir, 2007, 23, 9195–9200 CrossRef CAS.
- S. A. Ahmed, X. Sallenave, F. Fages, G. Mieden-Gundert, W. M. Müller, U. Müller, F. Vögtle and J. L. Pozzo, Langmuir, 2002, 18, 7096–7101 CrossRef CAS.
- Z. J. Qiu, H. T. Yu, J. B. Li, Y. Wang and Y. Zhang, Chem. Commun., 2009, 3342–3344 RSC.
- Q. Chen, Y. Feng, D. Q. Zhang, G. X. Zhang, Q. H. Fan, S. N. Sun and D. B. Zhu, Adv. Funct. Mater., 2010, 20, 36–42 CrossRef CAS.
- J. J. D. de Jong, L. N. Lucas, R. M. Kellogg, J. H. van Esch and B. L. Feringa, Science, 2004, 304, 278–281 CrossRef CAS.
- S. Wang, W. Shen, Y. L. Feng and H. Tian, Chem. Commun., 2006, 1497–1499 RSC.
- M. L. Ma, Y. Kuang, Y. Gao, Y. Zhang, P. Gao and B. Xu, J. Am. Chem. Soc., 2010, 132, 2719–2728 CrossRef CAS.
- Y. Gao, Y. Kuang, Z. F. Guo, Z. H. Guo, I. J. Krauss and B. Xu, J. Am. Chem. Soc., 2009, 131, 13576 CrossRef CAS.
- Z. M. Yang, K. M. Xu, Z. F. Guo, Z. H. Guo and B. Xu, Adv. Mater., 2007, 19, 3152 CrossRef CAS.
- Y. Zhang, Z. M. Yang, F. Yuan, H. W. Gu, P. Gao and B. Xu, J. Am. Chem. Soc., 2004, 126, 15028–15029 CrossRef CAS.
- Y. Zhang, H. W. Gu, Z. M. Yang and B. Xu, J. Am. Chem. Soc., 2003, 125, 13680–13681 CrossRef CAS.
- Z. M. Yang, G. L. Liang, L. Wang and B. Xu, J. Am. Chem. Soc., 2006, 128, 3038–3043 CrossRef CAS.
- Z. M. Yang, P. L. Ho, G. L. Liang, K. H. Chow, Q. G. Wang, Y. Cao, Z. H. Guo and B. Xu, J. Am. Chem. Soc., 2007, 129, 266–267 CrossRef CAS.
- Q. Chen, D. Q. Zhang, G. X. Zhang and D. B. Zhu, Langmuir, 2009, 25, 11436–11441 CrossRef CAS.
- Q. Chen, Y. X. Lv, D. Q. Zhang, G. X. Zhang, C. Y. Liu and D. B. Zhu, Langmuir, 2010, 26, 3165–3168 CrossRef CAS.
- M. George and R. G. Weiss, Langmuir, 2002, 18, 7124–7135 CrossRef CAS.
- M. George and R. G. Weiss, J. Am. Chem. Soc., 2001, 123, 10393–10394 CrossRef CAS.
- M. Shirakawa, N. Fujita and S. Shinkai, J. Am. Chem. Soc., 2003, 125, 9902–9903 CrossRef CAS.
- T. Ishi-i, R. Iguchi, E. Snip, M. Ikeda and S. Shinkai, Langmuir, 2001, 17, 5825–5833 CrossRef CAS.
- H. Isla, M. Gallego, E. M. Perez, R. Viruela, E. Orti and N. Martin, J. Am. Chem. Soc., 2010, 132, 1772 CrossRef CAS.
- S. S. Gayathri, M. Wielopolski, E. M. Perez, G. Fernandez, L. Sanchez, R. Viruela, E. Orti, D. M. Guldi and N. Martin, Angew. Chem., Int. Ed., 2009, 48, 815–819 CrossRef CAS.
- G. Fernandez, E. M. Perez, L. Sanchez and N. Martin, J. Am. Chem. Soc., 2008, 130, 2410–2411 CrossRef CAS.
- E. M. Perez, L. Sanchez, G. Fernandez and N. Martin, J. Am. Chem. Soc., 2006, 128, 7172–7173 CrossRef CAS.
- G. Fernandez, L. Sanchez, E. M. Perez and N. Martin, J. Am. Chem. Soc., 2008, 130, 10674–10683 CrossRef CAS.
- G. Fernandez, E. M. Perez, L. Sanchez and N. Martin, Angew. Chem., Int. Ed., 2008, 47, 1094–1097 CrossRef CAS.
- X. Y. Yang, G. X. Zhang, D. Q. Zhang and D. B. Zhu, Langmuir, 2010, 26, 11720–11725 CrossRef CAS.
- X. Y. Yang, G. X. Zhang, D. Q. Zhang, J. F. Xiang, G. Yang and D. B. Zhu, Soft Matter, 2011, 7, 3592–3598 RSC.
- (a) A. Ajayaghosh, V. K. Praveen and C. Vijayakumar, Chem. Soc. Rev., 2008, 37, 109–122 RSC; (b) A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS; (c) S. Srinivasan, P. A. Babu, S. Mahesh and A. Ajayaghosh, J. Am. Chem. Soc., 2009, 131, 15122–15123 CrossRef CAS; (d) S. Srinivasan, S. S. Babu, V. K. Praveen and A. Ajayaghosh, Angew. Chem., Int. Ed., 2008, 47, 5746–5749 CrossRef CAS.
- P. Mukhopadhyay, Y. Iwashita, M. Shirakawa, S. Kawano, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2006, 45, 1592–1595 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |