Reconstruction of the carbon sp2 network in graphene oxide by low-temperature reaction with CO†
Angeles
Pulido
a,
Patricia
Concepción
a,
Mercedes
Boronat
a,
Cristina
Botas
b,
Patricia
Alvarez
b,
Rosa
Menendez
b and
Avelino
Corma
*a
aInstituto de Tecnología Química, Universidad Politécnica de Valencia-Consejo Superior de Investigaciones Científicas, Av. de los Naranjos s/n, E-46022, Valencia, Spain. E-mail: acorma@itq.upv.es
bInstituto Nacional del Carbón, CSIC, Apartado 73, E-33080, Oviedo, Spain
First published on 7th November 2011
Abstract
Low-temperature (−176 °C) CO adsorption on graphene oxide and partially reduced graphene oxide sheets was investigated in a combined IR spectroscopic and DFT study. The reactivity of the carbon vacancies in the network was observed to be extremely high, causing the CO molecules to dissociate in a barrier-less process that leads to the reconstruction of the sp2graphene network. After the adsorption of CO on the graphene oxide materials, the intensity of the FTIR bands is lower in the case of partially reduced graphene oxide sheets, indicating that there are fewer active sites available for CO interaction.
The unique electronic properties of graphene, a zero band gap semiconductor, have generated enormous interest due to its multiple potential applications in the fields of photonics and electronics.1–4 However, the industrial implementation of graphene (a two-dimensional honeycomb network of sp2-hybridized carbon atoms) has been restricted as there is a lack of a large-scale production of single layer graphene sheets with a high quality 2D crystal sp2 network. Although single-layer graphene sheets were first isolated via mechanical exfoliation from graphite by Geim and co-workers in 2004,5 this route is not practical for large-scale production. To overcome this limitation, alternative routes to mechanical exfoliation such as chemical exfoliation, the use of organic precursors or in situ catalyzed growth on a substrate have been proposed.6–9
Single-layer graphene oxide sheets can be obtained from graphite in a cost-efficient process with a large scalability.10 The properties of graphite oxide are highly dependent on the synthetic route (process and conditions) and on the characteristics of the parent graphite, the resultant structure still being under debate.11–19 It is commonly accepted that the basal plane of the carbon honeycomb is mainly decorated with hydroxyl and epoxy groups,12 although the presence of ketones and phenols has also been reported.15,16 Carboxyls, ketones, lactones, lactols, phenols, pyrones and anhydrates have been found at the sheet edges.16Graphene oxide contains sp2- and sp3-hybridized C atoms and it is an insulator; however electrical conductivity can be increased by reducing the oxygen content and restoring the C sp2graphene network.20Oxygen can be removed from the graphene oxide sheet via thermal or chemical reduction. Reduced graphene oxides still contain a residual amount of oxygen (∼5–10 atomic%), where the structural reconstruction process, the functional groups and their spatial distribution remain unclear.20–22
Thus, large scale production of single-layer graphene sheet derived from graphene oxide materials is indeed a very promising route; but implementation of graphene for industrial applications requires an elevated control of its properties (such as size of the sheet or quality of the carbon sp2 network) which is still a challenging task due to the complexity of the overall production and characterization processes. In the present work, a combined experimental and theoretical investigation of low-temperature (−176 °C) CO adsorption on sheets of partially reduced graphene oxide and graphene oxide is presented that we considered would help to get a better understanding of the structure of graphene oxide materials and its reactivity. From a joint analysis of the experimental spectroscopic and periodic DFT results, we report here on the extremely high reactivity of carbon vacancies in the graphene sheets and the reconstruction of the graphene sp2 network by CO.
The parent graphite (99.9 wt% C, 0.01 wt% ash) was prepared under argon flow at 2800 °C from a coal-based pitch coke.23 The size, morphology and orientation of the crystalline structures (optical texture)24 were determined by polarized light microscopy. The optical texture observed in the blocks of graphite confirms a homogeneous crystallinity of small size, identified as mosaics, 1.5–5 μm. These results are supported by the values of the sizes along the c (Lc) and a axis (La) obtained by X-ray diffraction (XRD), 190 Å and 435 Å, respectively; see the ESI† for additional details. The graphene oxide (GO) was obtained from synthetic graphite by a modified version of Hummer's method. This method makes use of Hummer's reagents25 with additional amounts of NaNO3 and KMnO4. Concentrated H2SO4 (360 mL) was added to a mixture of graphite (7.5 g) and NaNO3 (7.5 g), and the mixture was cooled down to 0 °C using an ice bath. KMnO4 (45 g) was added slowly in small doses to keep the reaction temperature below 20 °C. The solution was then heated to 35 °C and stirred for 3 h. Next, 1.5 L of 3% H2O2 was slowly added, giving rise to a pronounced exothermal effect up to 98 °C. The reaction mixture was then stirred for 30 min and, finally, the mixture was centrifuged (3700 rpm for 30 min), the supernatant being decanted away. The remaining solid material was then washed with 600 mL of water and centrifuged again, this process being repeated until the pH was neutral. Finally, a colloidal suspension of individual graphene oxide sheets in purified water (1 mg mL−1) was prepared in 1 L batches, and kept under ultrasound for 10 h. Finally the suspension was centrifuged (3700 rpm for 30 min), the supernatant was filtered over cellulose, and the solid was discarded. The partially reduced graphene oxide (RGO) sheets were prepared by reducing the GO with hydrazine monohydrate in an aqueous solution26 in the presence of a base.27 To this end, 500 mL of an aqueous solution of GO (1 mg mL−1) was loaded in a 250 mL round-bottom flask and hydrazine hydrate (5 mL, ratio 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), ammonia (200 μL) and toluene (10 mL) were then added. Finally, the solution was heated in an oil bath at 100 °C under a water-cooled condenser for 24 h. The base was added to the reaction solution to increase the pH to around 10, the electrostatic repulsions between the GO sheets being maximal at this pH. Toluene was added to the reaction mixture to avoid the agglomeration of graphene sheets upon the evaporation of water.28
:1), ammonia (200 μL) and toluene (10 mL) were then added. Finally, the solution was heated in an oil bath at 100 °C under a water-cooled condenser for 24 h. The base was added to the reaction solution to increase the pH to around 10, the electrostatic repulsions between the GO sheets being maximal at this pH. Toluene was added to the reaction mixture to avoid the agglomeration of graphene sheets upon the evaporation of water.28
The size and height of the GO and RGO samples were measured by means of atomic force microscopy (AFM) imaging and profiling. The dominant presence of “monolayers” was identified in GO, with the expected average height of 0.90 nm for graphene oxide.17,29–31 However, close examination of the profiles of some areas of the AFM images of RGO reveals that there are areas that look like sheets of thick layers. This is confirmed by analyzing the histogram of heights, which shows peaks for sheets of between 2–3 layers (3.39 nm) and 3–4 layers (4.28 nm). Additional AFM details are available as the ESI†. A more pronounced tendency to agglomeration is observed in RGO by AFM which may be related to the lower content of oxygen functional groups and a smaller number of defects in the reduced graphene layer. First insights into the structure of the GO and RGO samples were obtained from Raman spectroscopy analysis, available as the ESI†. The most prominent bands in the GO and RGO Raman spectra are the so-called G band (characteristic of sp2carbon networks) and the D band (induced by local defects and disorder in sp2 phases) at 1599 cm−1 and 1346 cm−1, respectively.32–34 An overtone (2D) at ∼2680 cm−1 and combination bands (D + G) at ∼2890 cm−1 were also observed. Bands due to sp3 and sp networks at 1333 cm−1 and in the 1850–2100 cm−1 region were not observed, discriminating any amorphization of the material or the formation of linear carbon chains. To evaluate the functional groups of the graphene oxide and partially reduced graphene oxide samples, X-ray photoelectron spectroscopy (XPS) measurements were carried out. All of the spectra were energy calibrated by assigning 284.5 eV to the C1s binding energy of the ‘graphitic’ peak, details available as the ESI†. The binding energy of the C–C (sp2) is assigned to 284.5 eV and chemical shifts of +0.5, + 1.5, +2.5 and +4.0 eV are typically assigned to the C (sp3) hybridisation, C–OH, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, and COOH functional groups, respectively.35 Most of the structural models of graphene oxide also include an epoxy group (C–O–C), the most probable hybridisation at the basal planes, which can be expected to have a C1s binding energy similar to that of C–OH,36 but with the possibility of exhibiting a larger chemical shift than what might be expected of the emission of C–O–C, that is, inside the range of the CO emission.37 In the case of the sample reduced with hydrazine the increment in nitrogen content calculated by XPS (to 5.7%) indicates the presence of C–N bonds which exhibit bonding energies mainly in the range of the C–OH bonds and/or contribute to the binding energies of the Csp3 bonds.38,39 Functional groups (%) of GO and RGO obtained from the curve fitting of C1s XPS spectra are shown in Table 1. The oxygen functional groups on the GO sample mainly consist of a high proportion of hydroxyl (20.3%), carboxyl (19.5%) and epoxy groups (13.8%) as observed by XPS analysis. The RGO sample still contains residual oxygen functional groups: mainly hydroxyl (11.5%) and epoxy groups (8%), whereas only 4% of carboxylic groups still remain in the sample.
O, and COOH functional groups, respectively.35 Most of the structural models of graphene oxide also include an epoxy group (C–O–C), the most probable hybridisation at the basal planes, which can be expected to have a C1s binding energy similar to that of C–OH,36 but with the possibility of exhibiting a larger chemical shift than what might be expected of the emission of C–O–C, that is, inside the range of the CO emission.37 In the case of the sample reduced with hydrazine the increment in nitrogen content calculated by XPS (to 5.7%) indicates the presence of C–N bonds which exhibit bonding energies mainly in the range of the C–OH bonds and/or contribute to the binding energies of the Csp3 bonds.38,39 Functional groups (%) of GO and RGO obtained from the curve fitting of C1s XPS spectra are shown in Table 1. The oxygen functional groups on the GO sample mainly consist of a high proportion of hydroxyl (20.3%), carboxyl (19.5%) and epoxy groups (13.8%) as observed by XPS analysis. The RGO sample still contains residual oxygen functional groups: mainly hydroxyl (11.5%) and epoxy groups (8%), whereas only 4% of carboxylic groups still remain in the sample.
| Csp2 | Csp3 | C–OH | C–O–C | COOH | C/O | |
|---|---|---|---|---|---|---|
| GO | 36.2 | 11.3 | 20.3 | 13.8 | 19.5 | 1.8 |
| RGO | 46.2 | 30.8 | 11.5 | 8.0 | 4.4 | 5.1 |
Functionalization of GO and RGO materials before and after CO adsorption was followed by Fourier-Transform infrared (FTIR) spectroscopy. An FTIR cell allowing in situ treatments in controlled atmospheres and temperatures from −176 °C to 500 °C was connected to a vacuum system with a gas dosing facility. A drop of a highly diluted water solution of the graphenic samples was deposited onto a Germanium disc and a slow air drying process was then applied. The samples were pre-treated in a vacuum (10−5 mbar) at 120 °C for 1 h. After activation, the samples were cooled down to −176 °C under dynamic vacuum conditions (pressure maintained at 10−5 mbar) and then dosed with CO under increasing pressure (0.08–4 mbar). The FTIR spectra were recorded after each dosage. In the FTIR spectra corresponding to GO and RGO materials before CO adsorption (see Fig. 1a) bands typical of aromatic graphene sheets can be observed at 1468 and 1591 cm−1, together with bands associated to the following functional groups: (i) hydroxyl groups interacting with water (the broad band at 3400 cm−1), (ii) carbonyl groups (1720 cm−1), (iii) C–OH groups (1227 cm−1), and (iv) epoxy groups in the range of 1100–1300 cm−1. The FTIR band at 1635 cm−1 is assigned to physisorbed water. GO and RGO FTIR difference spectra (with respect to the corresponding (R)GO background spectra before the adsorption of CO) after 0.08 and at 4 mbar with CO doses at −176 °C are shown in Fig. 1. After CO adsorption over the GO sample (Fig. 1b) five bands appear in the FTIR spectrum with increasing intensity as the amount of CO dosage increases: (1) two broad bands centered at 1090 and 1173 cm−1 in the region of the ether and the epoxide vibrations (1000–1300 cm−1), (2) two bands at 1463 and 1594 cm−1 in the typical region of skeletal vibrations of the sp2graphene network, and (3) one band at around 1627 cm−1 in the region of highly activated CO molecules in a tilted configuration. This last band could also be associated to physisorbed H2O, but its high stability toward evacuation rules out this assignation. No bands ascribed to CO interacting with oxygen functional groups are evidenced. CO adsorption was also investigated over the reduced graphene oxide (RGO) sample, Fig. 1c. In this case, four bands (centered at 1110, 1217, 1458 and 1592 cm−1) of smaller intensity than for the GO sample are observed, indicating a smaller amount of active sites available for CO interaction. No FTIR band associated to CO tilted adsorption complexes was observed. Indeed, less defective graphene sheets might be expected from the higher content of sp2-hybridized C atoms (∼10%) observed by XPS in the reduced sample, and the smaller amount of oxygen functional groups (C/O 5.1 in RGOversus 1.8 in GO), see Table 1.
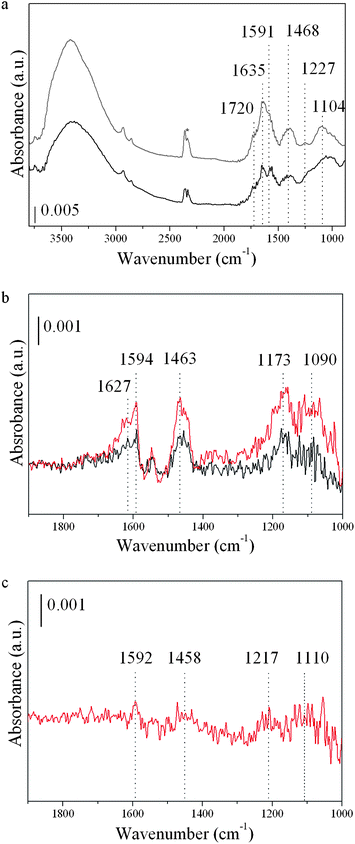 | ||
| Fig. 1 FTIR spectra of graphene oxide (GO) and partially reduced graphene oxide (RGO) samples. (a) Spectra of GO (grey) and RGO (black) samples, before CO adsorption takes place. (*) CO2 due to band background subtraction. (b) Difference spectra (with respect to the GO background) after CO adsorption at −176 °C on GO. CO doses of 0.08 (black) and 4 (red) mbar. (c) Difference spectrum (with respect to the RGO background) after CO adsorption (dose of 4 mbar) on RGO sample. | ||
Theoretical investigations have been previously used to provide an atomistic view of small molecules (such as CO, N2, H2O, NH3 or CO2) interacting with the C sp2 network and also with defective graphene sheets.40–43 Here, a series of periodic models were selected to investigate CO adsorption on GO and RGO sheets at the density functional theory (DFT) level, including intermolecular (dispersion) interactions. Ideal or non-defective graphene domains in the GO and RGO sheets are expected to be chemically inert regions, and consequently CO adsorption should occur in the defective areas, that is, in the proximity of holes resulting from carbon vacancies in the network and/or in functionalized regions. From the optimized structure of graphite (space groupP63/mmc, cell parameters ap = 2.45 Å, cp = 6.64 Å and γ = 120° and unit cell composition C4), a fixed-volume supercell containing one graphene sheet (cell parameter a = 19.60 Å, with a distance of 20 Å between periodic images) was obtained with a composition of C128 (referred to as model G). Computational details can be found as the ESI†. CO adsorption was investigated to be taking place on an ideal (non-defective) graphene domain and in four defective environments: (1) in the proximity of a single-vacancy defect (denoted G1V model), (2) near a vacancy-pair (named G2V model), (3) in an area with a hole in the graphene sheet generated by the removal of four C atoms (referred to as GH model), and (4) on an OH functionalized GH sheet (labeled GOH). Periodic DFT optimized structures of the five graphene models used (G, G1V, G2V, GH and GOH) are depicted in Fig. 2. For each environment investigated, different initial CO molecule locations were considered and the calculated properties of the CO adsorption complexes on the graphene models are reported in Table 2 and the most stable ones are depicted in Fig. 3. In all cases CO adsorption led to the formation of a new C–C bond between the C atom belonging to the CO molecule and a carbon atom in the network, this process being barrier-less and exothermic, with the release of more than 200 kJ mol−1. C–C bond formation led to a 5- or 6-membered ring closure, see Fig. 3g–k. Thus, the incorporation of a C atom initially belonging to the CO molecule into the graphene structure allows the reconstruction of the sp2 network on the adjacent C atoms of the graphene sheet, marked with black squares in Fig. 3. Moreover, after CO adsorption had taken place, a variety of oxo-species such as epoxides (Fig. 3b), carbonyls (Fig. 3c, e and f) or cyclic ethers (Fig. 3d) were observed, with varying C–O bond activation. The calculated harmonic CO frequency in the epoxide complex is 1252 cm−1, and 1307 cm−1 in the case of the cyclic ether, see Table 2. These two species can be associated to the bands in the range of 1100–1300 cm−1 observed in the FTIR spectra of the GO and RGO samples, see Fig. 1. The calculated CO vibration frequencies for highly activated carbonyl adsorption species (C–O bond distances extend from 0.08 to 0.10 Å) are in the 1600–1690 cm−1 range, and correlate with the experimentally observed band centered at 1627 cm−1.
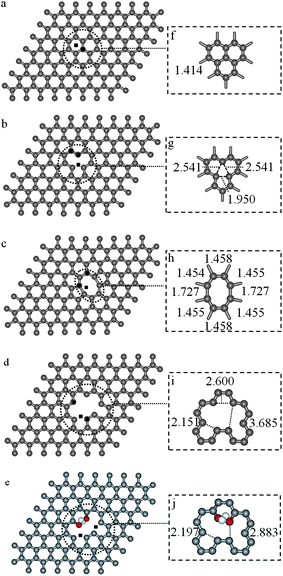 | ||
| Fig. 2 Periodic DFT optimized geometries for the graphene models: (a) a perfect graphene sheet, G (geometry details, inset f), (b) reconstructed single-vacancy graphene sheet, G1V (vacancy site inset g), (c) graphene sheet with a vacancy pair, G2V (inset h, eight membered ring reconstructed), (d) graphene sheet with a hole, GH (vacancy area, inset i), and (e) functionalized GH sheet, GOH (geometry details, inset j). Initial CO positions are marked as black squares in parts (a–e). Distances in Å. H, C and O atoms are depicted in white, grey and red, respectively. | ||
| Graphene a | Initial locationb | Complexc | ΔEPW91d/kJ mol−1 | E D3 e/kJ mol−1 | ΔEPW91–D3f/kJ mol−1 | R g/Å | r CO h/Å | ω i/cm−1 |
|---|---|---|---|---|---|---|---|---|
| a Model used to investigate CO adsorption. b Initial location of CO molecule as labelled in Fig. S7†. The orientation of the CO molecule was taken mainly with the C atom closer to the graphene plane (C-down); italics are used to show initial O-downwards approach to the graphene plane. c Adsorption complex formed, where N means non-interacting CO complex, E refers to epoxide formation, T denotes a tilted CO adsorption complex, C means that a cyclic ether complex has been formed, and sp2 is used to indicate when CO adsorption leads to the reconstruction of the graphene network. d Interaction energy calculated at the periodic PW91 level for the process CO(g) + S → (CO)/S (1), where S represents the graphene-like model. e Intermolecular (dispersion) correction using eqn (1) at the D3 level. f Interaction energy calculated at the periodic PW91–D3 level using eqn (1). PW91–D3 energies (EPW91–D3) were calculated adding the intermolecular (dispersion) correction energy (ED3) to the electronic PW91 energies (EPW91), EPW91–D3 = EPW91 + ED3. g Distance between the graphene C atoms and the atoms of the CO molecule; distances involving O atoms are italicised. h CO bond distance. i Harmonic frequencies involving C and/or O atoms of the CO molecule. All the intermediate structures were identified as stationary points of order zero. The calculated harmonic C–O frequency for the gas phase CO molecule at the same level of theory is 2128 cm−1, whereas the experimental value is 2143 cm−1. | ||||||||
| G | 1 | N | −1.4 | −7.1 | −8.5 | 3.739 | 1.142 | 2128 |
| G1V | 1 | E, sp2 | −568.9 | −13.0 | −581.8 | 1.461, 1.461 | 1.460 | 1252 |
| 1 | N | −4.0 | −7.1 | −11.0 | 3.393 | 1.143 | ||
| 2 | N | −2.7 | −7.1 | −9.7 | 4.097 | 1.142 | ||
| 3 | N | −2.4 | −7.1 | −9.5 | 4.425 | 1.142 | ||
| G2V | 1 | T, sp2 | −209.6 | −14.8 | −224.4 | 1.475, 1.475 | 1.232 | 1659 |
| 1 | C, sp2 | −51.5 | −14.4 | −65.9 | 1.421, 1.421, 2.527, 2.505 | 1.418 | 242–1307 | |
| 2 | T | 20.0 | −13.9 | +6.1 | 1.427, 1.488 | 1.229 | 1659 | |
| 3 | T | 105.2 | −7.1 | +98.1 | 1.427 | 1.160 | ||
| GH | 1 (2) | C, sp2 | −685.1 | −14.1 | −699.2 | 1.426, 1.429, 1.371 | 1.376 | 1079–1260 |
| 3 | T, sp2 | −440.9 | −15.6 | −456.5 | 1.543, 1.543 | 1.220 | 1688 | |
| GOH | 1 | T, sp2 | −407.9 | −16.7 | −424.7 | 1.522, 1.483 | 1.239 | 1603 |
| 2 | T, sp2 | −389.4 | −16.7 | −406.2 | 1.486, 1.492 | 1.255 | 1543 |
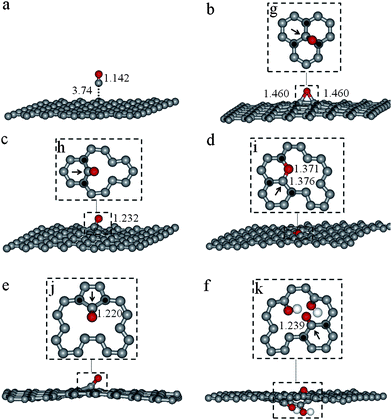 | ||
| Fig. 3 Periodic DFT optimized geometries for CO adsorption complexes on: (a) a perfect graphene sheet (G), (b) single-vacancy graphene sheet (G1V), (c) vacancy-pair graphene sheet (G2V), (d) graphene sheet with a hole (GH), (e) GH with the CO molecule in a different initial location, and (f) functionalized GH sheet (GOH). Details of the CO adsorption complexes (in parts b–f) are shown in the insets, parts g–k, respectively. A black arrow has been used to indicate the C atoms in the network that initially belonged to the CO molecule. Distances in Å. H, C and O atoms are depicted in white, grey and red, respectively. | ||
The CO adsorption complexes formed on the defective graphene sheets can be explained in terms of the electronic structure of graphene (prior to CO adsorption) and steric hindrance effects on the CO molecule. On one hand, the presence of defects in the graphene sheet causes the highest occupied and/or the lowest unoccupied bands to be located around the defective area, the electron density protruding outside the graphene basal plane as can be observed in Fig. 4 where band decomposed charge density for all of the graphene sheets investigated is depicted. Overlapping between the graphene bands and the CO molecule orbitals is therefore more likely in the regions around the defects, and will determine the CO adsorption site. On the other hand, the final structure of the CO adsorption complex formed will also be influenced by the local environment around the CO adsorption site. Thus, CO adsorption over a graphene sheet with a hole (GH system, Fig. 2d) leads to the formation of a cyclic ether (with the C and the O atoms of the CO molecule in the graphene basal plane) due to the unoccupied space at the vacancy hole (see Fig. 3d); whereas a carbonyl complex with the CO molecule tilted outside of the basal plane is observed (see Fig. 3f) at the same site when hydroxyl groups are located opposite the CO molecule, see GOH model in Fig. 2e.
![Highest occupied bands (parts a–e) and lowest unoccupied bands (part f–j) viewed along the [100] (parts a–j) and [001] (parts k–t) directions of: (i) a perfect graphene sheet, G (top); (ii) a reconstructed single-vacancy graphene sheet, G1V (second row); (iii) a vacancy-pair graphene sheet, G2V (middle); (iv) a graphene sheet with a hole, GH (fourth row); and (v) a functionalized GH sheet, GOH (bottom). Optimized geometry given for the sake of clarity. For the coloring scheme see Fig. S7. Charge density surface (yellow colour) represented by a constant value of 0.002 e Å−2.](/image/article/2012/JM/c1jm14514b/c1jm14514b-f4.gif) | ||
| Fig. 4 Highest occupied bands (parts a–e) and lowest unoccupied bands (part f–j) viewed along the [100] (parts a–j) and [001] (parts k–t) directions of: (i) a perfect graphene sheet, G (top); (ii) a reconstructed single-vacancy graphene sheet, G1V (second row); (iii) a vacancy-pair graphene sheet, G2V (middle); (iv) a graphene sheet with a hole, GH (fourth row); and (v) a functionalized GH sheet, GOH (bottom). Optimized geometry given for the sake of clarity. For the coloring scheme see Fig. S7†. Charge density surface (yellow colour) represented by a constant value of 0.002 e Å−2. | ||
Our calculations show that when CO adsorption takes place on the graphene sheets, the C atom belonging to the CO molecule interacts strongly with the under-coordinated C atoms of the support, in a barrier-less and highly exothermic process, restoring their sp2 hybridization (see Fig. 3), irrespective of the final oxo-species that is formed. This would explain the appearance of the experimental FTIR bands centered at 1463 and 1594 cm−1 (see Fig. 1), associated to the graphene sp2 network vibrations, observed in the different spectra. The immense release of energy and the absence of an activation barrier for the C–C bond formation process after CO adsorption would explain why the sp2 reconstruction takes place at such a low temperature, −176 °C. According to the DFT calculations the reactivity of single vacancies is high enough to induce CO dissociation at low temperature. The experimental FTIR bands centered at 1090, 1110, 1173, 1217 and 1627 cm−1 observed in the difference spectra (Fig. 1) would arise from the variety of oxo-species formed after CO adsorption, such as the epoxides, cyclic ethers and carbonyl groups predicted from the DFT calculations.
In essence, our results demonstrate the high reactivity of under-coordinated carbon atoms even at low temperatures such as −176 °C. The formation of C–C bonds between the graphene and the C atoms of the CO molecule allows the reconstruction of the sp2graphene network, where a variety of oxo-complexes are also formed. In the graphene oxide (GO) there is a higher content of active sites (non-sp2-hybridized C atoms) compared with the partially reduced graphene oxide (RGO), explaining the larger amount of CO adsorbed on the GO sample. Moreover, the nature of the oxo-species formed after CO adsorption depends on the structure of the material prior to adsorption. We consider this result to be important not only in the broad context of acquiring a better understanding of the material and its reactivity, but also because it helps to improve the quality of the graphene sp2 network.
Acknowledgements
The authors thank the Spanish Science and Innovation Ministry (Consolider Ingenio 2010, Project MULTICAT, ref. CSD2009-00050) for their financial support, and Red Española de Supercomputación (RES) and Centre de Càlcul de la Universitat de València for offering computing facilities and technical assistance. P. A. and A. P. thank the Spanish Science and Innovation Ministry (Ramón y Cajal and Juan de la Cierva programs) and C. B. thanks FICYT for their grants. Special thanks to Pablo Ares from Nanotec Electronica™ for AFM contribution.References
- A. K. Geim, Science, 2009, 324, 1530–1534 CrossRef CAS.
- C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef.
- G. R. Hotopan, V. Hoeye, V. Antuna, C. Diaz, F. Garcia, L.-H. Andrés, P. Alvarez and R. Menéndez, Prog. Electromagn. Res., 2011, 118, 57–69 CrossRef.
- R. Camblor, S. Hoeye, G. Hotopan, C. Vazquez, M. Fernandez, F. Las-Heras, P. Alvarez and R. Menéndez, J. Electromagn. Waves, 2011, 25, 1921–1929 CrossRef.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS.
- M. Allen, V. Tung and R. Kaner, Chem. Rev., 2010, 110, 132–145 CrossRef CAS.
- C. Berger, Z. M. Song, T. B. Li, X. B. Li, A. Y. Ogbazghi, R. Feng, Z. T. Dai, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, J. Phys. Chem. B, 2004, 108, 19912–19916 CrossRef CAS.
- M. Muller, C. Kubel and K. Mullen, Chem.–Eur. J., 1998, 4, 2099–2109 CrossRef CAS.
- S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- W. Cai, R. Piner, F. Stadermann, S. Park, M. Shaibat, Y. Ishii, D. Yang, A. Velamakanni, S. An, M. Stoller, J. An, D. Chen and R. Ruoff, Science, 2008, 321, 1815–1817 CrossRef CAS.
- A. Lerf, H. He, M. Forster and J. Klinowski, J. Phys. Chem. B, 1998, 102, 4477–4482 CrossRef CAS.
- T. Nakajima and Y. Matsuo, Carbon, 1994, 32, 469–475 CrossRef CAS.
- D. Pacilé, J. C. Meyer, A. Fraile Rodríguez, M. Papagno, C. Gómez-Navarro, R. S. Sundaram, M. Burghard, K. Kern, C. Carbone and U. Kaiser, Carbon, 2011, 49, 966–972 CrossRef.
- T. Szabó, O. Berkesi, P. Forgó, K. Josepovits, Y. Sanakis, D. Petridis and I. Dékány, Chem. Mater., 2006, 18, 2740–2749 CrossRef.
- T. Szabo, O. Berkesi and I. Dekany, Carbon, 2005, 43, 3186–3189 CrossRef CAS.
- C. Botas, P. Álvarez, C. Blanco, R. Santamaría, M. Granda, P. Ares, F. Rodríguez-Reinoso and R. Menéndez, Carbon, 2011, 50, 275–282 CrossRef.
- Z.-S. Wu, W. Ren, L. Gao, B. Liu, C. Jiang and H.-M. Cheng, Carbon, 2009, 47, 493–499 CrossRef CAS.
- M. Inagaki, Y. A. Kim and M. Endo, J. Mater. Chem., 2011, 21, 3280–3294 RSC.
- C. Mattevi, G. Eda, S. Agnoli, S. Miller, A. Mkhoyan, O. Celik, D. Mastrogiovanni, G. Granozzi, E. Garfunkel and M. Chhowalla, Adv. Funct. Mater., 2009, 19, 2577–2583 CrossRef CAS.
- A. Bagri, C. Mattevi, M. Acik, Y. Chabal, M. Chhowalla and V. Shenoy, Nat. Chem., 2010, 2, 581–587 CrossRef CAS.
- W. Gao, L. Alemany, L. Ci and P. Ajayan, Nat. Chem., 2009, 1, 403–408 CrossRef CAS.
- P. Alvarez, M. Granda, J. Sutil, R. Menendez, J. J. Fernandez, J. A. Vina, T. J. Morgan, M. Millan, A. A. Herod and R. Kandiyoti, Energy Fuels, 2008, 22, 4077–4086 CrossRef CAS.
- R. A. Forrest and H. Marsh, Carbon, 1977, 15, 348–349 CrossRef CAS.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS.
- A. J. Patil, J. L. Vickery, T. B. Scott and S. Mann, Adv. Mater., 2009, 21, 3159 CrossRef CAS.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS.
- H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso, D. H. Adamson, R. K. Prud'homme, R. Car, D. A. Saville and I. A. Aksay, J. Phys. Chem. B, 2006, 110, 8535–8539 CrossRef CAS.
- C. Gomez-Navarro, R. T. Weitz, A. M. Bittner, M. Scolari, A. Mews, M. Burghard and K. Kern, Nano Lett., 2007, 7, 3499–3503 CrossRef CAS.
- S. Stankovich, R. D. Piner, X. Q. Chen, N. Q. Wu, S. T. Nguyen and R. S. Ruoff, J. Mater. Chem., 2006, 16, 155–158 RSC.
- A. Ismach, C. Druzgalski, S. Penwell, A. Schwartzberg, M. Zheng, A. Javey, J. Bokor and Y. G. Zhang, Nano Lett., 2010, 10, 1542–1548 CrossRef CAS.
- M. A. Pimenta, G. Dresselhaus, M. S. Dresselhaus, L. G. Cancado, A. Jorio and R. Saito, Phys. Chem. Chem. Phys., 2007, 9, 1276–1291 RSC.
- M. Shafiei, P. G. Spizzirri, R. Arsat, J. Yu, J. du Plessis, S. Dubin, R. B. Kaner, K. Kalantar-Zadeh and W. Wlodarski, J. Phys. Chem. C, 2010, 114, 13796–13801 CAS.
- T. C. Chiang and F. Seitz, Ann. Phys., 2001, 10, 61–74 CrossRef CAS.
- C. Kozlowski and P. M. A. Sherwood, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 2099–2107 RSC.
- D. Yang, A. Velamakanni, G. Bozoklu, S. Park, M. Stoller, R. D. Piner, S. Stankovich, I. Jung, D. A. Field, C. A. Ventrice and R. S. Ruoff, Carbon, 2009, 47, 145–152 CrossRef CAS.
- Y. Geng, S. J. Wang and J.K. Kim, J. Colloid Interface Sci., 2009, 336, 592–598 CrossRef CAS.
- K. R. Lee, K. U. Lee, J. W. Lee, B. T. Ahn and S. I. Woo, Electrochem. Commun., 2010, 12, 1052–1055 CrossRef CAS.
- O. Leenaerts, B. Partoens and F. M. Peeters, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 125416 CrossRef.
- P. Cabrera-Sanfelix, J. Phys. Chem. A, 2009, 113, 493–498 CrossRef CAS.
- B. Sanyal, P. Eriksson, U. Jansson and H. Grennberg, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 113409 CrossRef.
- F. Banhart, J. Kotakoski and A. V. Krasheninnikov, ACS Nano, 2011, 5, 26–41 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1jm14514b |
| This journal is © The Royal Society of Chemistry 2012 |