A “tool box” for rational design of degradable controlled release formulations
Sam N.
Rothstein
ab and
Steven R.
Little
*abcde
aDepartment of Chemical Engineering, University of Pittsburgh, PA, USA
bMcGowan Institute for Regenerative Medicine, University of Pittsburgh, PA, USA
cDepartment of Bioengineering, University of Pittsburgh, PA, USA
dDepartment of Immunology, University of Pittsburgh, PA, USA
eBenedum Hall Room 1249, 3700 O’Hara Street, Pittsburgh, PA 15261, USA. E-mail: srlittle@pitt.edu; Fax: +1 412 383 5210; Tel: +1 412-624-9614
First published on 3rd September 2010
Abstract
Controlled release technology could provide a universal solution to the problems of patient compliance and sub-optimal dosing that often plague modern pharmaceuticals. Yet, harnessing this potential requires the ability to design drug delivery formulations which satisfy specific dosing schedules. This review intends to portray how material properties, processing methods and mathematical models can serve as effective tools for rationally tuning the duration and rate of drug release from biodegradable polymer matrices.
 Sam N. Rothstein | Sam Rothstein graduated with honors from Bucknell University in 2005, earning a BS in Chemical Engineering. He is presently a pre-doctoral fellow in the laboratory of Dr Steven R. Little at the University of Pittsburgh. His thesis research focuses on methods for rationally designing controlled release medications and the associated advances are being commercialized as part of a new pharmaceuticals company. |
 Steven R. Little | Dr Steven Little received his PhD in Chemical Engineering at the Massachusetts Institute of Technology in 2005. Subsequently, he served as a post-doctoral associate in the laboratories of Robert Langer, ScD. He currently holds the position of Assistant Professor and Bicentennial Alumni Faculty Fellow at the University of Pittsburgh in the Departments of Chemical Engineering, Bioengineering, Immunology, and The McGowan Institute for Regenerative Medicine. His laboratory focuses on advanced drug-delivery strategies for immunotherapeutics and tissue engineering. |
1. Introduction
Biodegradable controlled release technology holds potential to resolve patient compliance issues and adverse effects that account for 10% to 14% of hospitalizations and $136 billion in annual medical expenses.1 The reason for such potential is that controlled release formulations can (in concept) autonomously regulate the local and systemic administration rate of practically any drug while, at the same time, resorbing harmlessly inside the body. However, the reason that the field has not yet realized this enormous potential is that (in practice) generating a specific controlled release formulation (i.e. one customized to the dosing schedule demanded by any given medication) is extremely time consuming and costly. As such, only 11 biodegradable, controlled release formulations have reached the market.2 In each case, the associated drugs have clear compliance risks (elevating the need for extended dosing) or wide therapeutic windows (relaxing the requirement for stringent control over release). Yet, these medications represent just a small fraction of drugs that could be improved with properly designed controlled release formulations. By one estimate, at least 90% of the top 100 best selling prescription medications could further advance patient quality of life if they offered reduced dosing frequency.3,4 From this viewpoint, the field can be said to have broad therapeutic applicability, but comparatively limited implementation.Over the past two decades, a number of studies have aimed to increase the efficiency of designing degradable drug delivery formulations. The earliest studies sought to identify key physical properties of the polymer matrices that determine release behavior.5,6 Twenty years later, researchers are still experimenting with a variety of new formulation compositions, polymer chemistries and processing conditions in an attempt to tune this release behavior.7–10 Over time, a number of system properties and processing conditions have emerged as potential tools for tuning the kinetics of delivery systems.4,11 However, a standardized method of tuning remains elusive due to the complexity that arises when factors, such as polymer chemistry, alter several properties that determine release simultaneously, such as matrix crystallinity, pH, degradation rate and hydrophobicity.8
To better evaluate the complex mechanics of release, researchers have also attempted to implement mathematical models. For a number of years these models have been applied to experimentally acquired release data as a means of assessing their validity.12,13 The earliest of these fitted descriptions often deviated significantly from data or focused exclusively on initial or final release.14–16 More recently, models are beginning to accurately describe full complex release profiles, in many cases predicting aspects of the delivery kinetics without the need for optimization or “fitting”.17–20 However, subsequent implementation remains a rare occurrence because many of these models still require parameter values that must be calculated anew from experimentally acquired release data for each drug or polymer system considered.
Overall, the combined efforts of mathematical and experimental studies have produced a wealth of data on the various behaviors of biodegradable polymer matrices. Some recent reviews have sought to summarize advances in mathematical modeling, cataloging the nuances of their approaches (empirical, Monte Carlo, mechanistic, etc.).13,17 Still others have focused on documenting experimental techniques, such as nano/micro technologies for macromolecule delivery or the loading and release of small molecules.4,11 Ultimately, mathematical models and heuristic, empirical methods are simply two different approaches to achieve the same ultimate goal: obtaining a desired release behavior for a given drug with minimal time and cost. Mathematical models quantify release mechanics and relate them to tunable system properties, while empirical studies uncover tunable system properties and relate them to quantifiable release mechanics. Keeping these perspectives in mind, the wealth of data on biodegradable matrix controlled release can be distilled down into a number of “design tools” (or system properties and processing conditions that can be used to predicatively tune release). As controlled release formulations often present complex release profiles, it is likely that more than one design tool will be required to achieve total control over delivery kinetics.
In this review, we will present an empirical and model-based “tool-box” containing a broad range of conditions that can be used to adjust the release behavior of biodegradable polymer matrices in various ways. We will focus primarily on the most widely studied biodegradable polymeric materials (i.e. polyesters, polyanhydrides, poly(ortho esters), etc.) given their prevalence in the literature, long history of use, and similarity of fundamental release behavior (encompassing both the underlying mechanisms and resulting kinetics). Although the techniques that can be used to control release in these systems are generally applicable to any size, shape, and orientation of a degradable matrix, we will most often refer to one of the most commonly reported configurations of these matrices, a spherical particulate system (Fig. 1). Regardless, these degradable systems are presently understood to control drug release via fundamental phenomena of diffusion (passive movement of drug), degradation (breakdown of the polymer matrix), erosion (mass loss from the polymer matrix) and dissolution (the solubilization of drug).8 By analysing the attempts to control these fundamental system phenomena and documenting their effects on release behavior, we can, correspondingly, identify a set of tools for rationally designing custom release behavior.
 | ||
| Fig. 1 Microscopy images of particle matrix cross-sections and exteriors (inset). (A) In homogenous systems (like single emulsion microparticles) the drug resides in-phase with the polymer matrix (adapted with permission from ref. 12). (B) In contiguous systems (like double or water-in-oil-in-water emulsion microparticles), the drug resides in pockets that are separated from the polymer matrix (unpublished data). | ||
2. Experimental studies
Studies have empirically varied independent system properties (such as matrix size, degradation rate or polymer molecular weight) or processing conditions (such as emulsion type, solution osmolarity, or solvent choice) and documented their effects with in vitro assays. Each of these experimental variations can be evaluated for its potential as a design tool that a scientist or engineer can use to tune release behavior.As a general rule, drug delivery vehicles can be tuned to provide a specific rate and duration of release independently. However, biodegradable matrices have been commonly observed to produce up to three distinct phases: (1) an initial phase (a.k.a. “initial burst”) that is typically categorized by the rapid delivery of drug upon hydration, (2) a “lag phase” marked by a near-zero rate of release for some period of time and (3) a “final phase” where measurable release resumes, typically in a Fickian fashion. Therefore, it would be useful to classify tools by their suitability for tuning the magnitude and/or duration of each individual phase in order to gain complete control over release (Fig. 2).
 | ||
| Fig. 2 Tri-phasic release profile depicting: (A) initial burst, (B) lag phase and (C) final release. Note that any one of these phases may or may not be apparent depending upon the properties of the release system. | ||
2.1. Initial burst
Numerous studies have addressed the “initial burst” and a summary of findings have been the focus of two reviews in the past decade.21,22 Both of these reviews discuss hypothetical mechanisms of “burst” release and potential strategies for preventing or eliminating it. Maintaining the theme of this review, we will focus on the impact that individual “design tools” have on the magnitude and/or kinetics of the initial burst.2.1.1.1. Dispersion of drug in the polymer matrix. Several studies have succeeded in creating dramatic reductions in burst magnitude by forcing hydrophilic proteins to disperse in the hydrophobic polymer matrix. For example, Fu et al. eliminated the initial release of a water-soluble protein (human glial-cell line derived neurotrophic factor) by using an ionic surfactant to dissolve the hydrophilic protein in-phase with the polymer.23 In contrast, an equivalent double emulsion formulation (with polymer entrapping pockets of aqueous protein) produced more than 70% initial release.23 A similar approach was adopted to eliminate the burst release of insulin using PEGylation, which aided the dissolution of the protein in dichloromethane.24 Practically, partitioning experiments can be used to determine if other agents will dissolve/disperse in the same phase as the polymer (e.g. organic phase) with the aid of surfactants or other modifications.25 Since this design tool simply involves the dispersion of drug and polymer in a matrix, it should readily apply to any number of systems.
Interestingly, less predictable results are observed when a cosolvent is used to stabilize hydrophilic drugs in the same phase as the hydrophobic polymer matrix.26,27 Using this approach on insulin-loaded PLGA microparticles reduced burst magnitude from 65 to 20%.27 However, when applied to another protein (granulocyte colony stimulating factor), this technique actually increased the magnitude of the initial burst.26 Yet both of these studies produce single emulsion systems by dissolving a protein in the cosolvent, dimethylsulfoxide before it mixing with a polymer–dichloromethane solution. It is unknown as to the source of the disparity, but it may be possible that this process may cause protein molecules to aggregate into a separate phase, giving rise to a measurable burst magnitude.
2.1.1.2. Manipulation of osmotic pressure. Subtle changes in burst magnitude have also been achieved by changing the osmotic pressure during the processing of systems that are intentionally fabricated with an internal aqueous phase (e.g. double (water-in-oil-in-water) emulsion systems, see Fig. 1B). Accordingly, Jiang et al. tuned the initial burst of bovine serum albumin (BSA) to values between 30% and 80% of total release by adding salt or sucrose to the outermost aqueous phase during microparticle fabrication.28 The same technique has been used in other macromolecule-loaded systems to reduce the magnitude of the initial burst.29–32 Within each study, the reduction in burst magnitude was proportional the amount of NaCl added to the outermost aqueous phase (i.e. the strength of the osmotic pressure gradient driving water out of the microparticles). Because osmosis is a fundamental process, this design tool should extend to a wide range of contiguous systems, and is particularly important to account for in systems where the drug itself dramatically affects the osmotic pressure (such as plasmid DNA).33,34
2.1.1.3. Manipulation of matrix size. A number of studies have also varied matrix size to tune the magnitude of burst release in double emulsion systems.29,35–37 While this property is particularly easy to tune during fabrication, its effectiveness at controlling the magnitude of initial release varies from agent to agent. For example, initial release of the small molecule, lidocaine, from polymeric microparticles dropped 30% as particle size increased 10 fold.35 This trend is echoed over a larger size span by polyanhydride particles loaded with butorphanol.37 However, the release of insulin was more sensitive to changes in particle size, dropping 35% in magnitude from just a 3 fold change in particle size29 (Fig. 3). Limitations arise as matrix size is reduced to below 5 µm because such small bodies are readily cleared in vivo by the reticuloendothelial system (RES, consisting of phagocytic cells like macrophages)38 or above 500 µm in diameter as matrix hydration begins to effect the kinetics of the initial burst.19,39 However, for median sizes, this method presents an effective approach to tuning the magnitude of initial burst release.
 | ||
| Fig. 3 Comparison of burst magnitudes (% total release) from double emulsion microparticles when varying size. Analysis was conducted on data from three different studies: small molecule lidocaine,24insulin protein25 and melittin peptide.26 Initial burst magnitude of each system was affected to different extents by changes in matrix size. | ||
2.1.1.4. Manipulation of drug loading. Discrete changes in initial burst magnitude have also been produced by altering the drug loading. Working with a variety of model proteins (lysozyme, carbonic anhydrase, and alcohol dehydrogenase), Sandor et al. noted that decreasing drug loading from 7 to 1 wt% reduced the initial burst from a high of roughly 80% to just 15–40% of total release.40 Equally dramatic reductions in burst magnitude have been observed following changes in peptide loading.29,36 Studies on small molecule release from polyester and polyanhydride implants have also reported similar trends.41–45 Limitations to this technique do arise at low loadings (lower payload) or high loading (breakdown of matrix structure, e.g. percolation).46 However, the simplicity and broad applicability of this tool still make very attractive for inducing measurable changes to burst magnitude.
2.1.1.5. Manipulation of initial porosity. In some cases, controlled release matrices are fabricated to be porous instead of solid, which can noticeably influence the magnitude of the initial burst. One of the most common ways to produce such a system is to increase the ratio of drug phase to polymer phase. In fact a number of studies have even correlated scanning electron microscopy images of microparticles porosity with dramatic changes burst magnitude.32,36,47,48 Further, this relation holds true for small molecule, peptide and protein release data, provided that the pore size is large enough to allow for free-diffusion of the agent.49 Another approach to altering initial porosity has been to change the concentration of stabilizer or surfactant used in processing. In such studies, altering the poy(vinyl) alcohol or poloxamer concentration inversely changed burst magnitude on the order of 10–20%.47,50 However, several reports note that stabilizer concentration also impacted matrix size which may have also accounted for observed changes as described in Section 2.1.1.3.36,51 Finally, post-fabrication pore-closing procedures, where matrices are subject to thermal or solvent-based annealing, have also successfully decreased the magnitude of the initial burst in porous matrices.32,47 Hence, depending on the initial state of a polymer matrix (porous or solid) altering processing conditions to change its porosity can be an effective way of either increasing or decreasing the magnitude of the initial burst.
2.1.2.1. Controlling drug dissolution rate. One way to influence burst release kinetics is to alter the encapsulated agent's dissolution rate. This has been accomplished by co-encapsulating an agent with a variety of excipients.52,53 Experimenting with different cyclodextrin excipients, Wang et al. were able to tune the duration of initial release of β-lapachone (a hydrophobic chemotherapeutic) to values between 1 week and 1 day by complexing it with hydrophilic cyclodextrin of varying size.52 This approach to increasing burst rate should also apply to other hydrophobic small molecules that readily complex with cyclodextrin or other hydrophilic agents.22
Interestingly, reports describing the use of excipients to decrease the rate of early release (rendering hydrophilic molecules more hydrophobic) are absent from the literature, possibly because evidence suggests that these types of systems exhibit little to no initial burst.23 Further, a study intending to reduce dissolution rate by switching from amorphous to crystalline drug reported a similar change in burst magnitude, but not kinetics.54 However, studies comparing agents with different intrinsic dissolution rates have noted a correlation to burst kinetics in polyanhydride implants.55,56 This suggests that methods for reducing an agent's dissolution rate could slow its burst release. However, until such methods are realized, excipients remain a reliable tool for increasing burst release rate of hydrophobic agents.
2.1.2.2. Effect of radial drug distribution. A number of different fabrication methods have been used to control the radial distribution of drug within biodegradable polymer matrices, thereby altering their initial burst kinetics. Such heterogeneous distributions have been achieved with double-walled microparticles which are formed by using multiple immiscible solvents to separate polymers of differing solubility into core and shell phases. These systems consistently show reduced protein burst kinetics when the drug is trapped in the matrix core rather than when it is in the shell or loaded throughout.57,58 Further, the extent of this reduction is proportional to the thickness of the shell separating the drug-loaded core from the outside environment.57 Coated implants (tablets, discs, or spheres) made from polyesters or polyanhydrides have produced similar results.59–62 Mixed results were observed in some small molecule loaded matrices, which could be explained by the preferential partitioning of such agents into the coating shell instead of the matrix core.63–66 Fortunately, studies have reported control over the radial distribution of small molecules through an electrospray fabrication process.67–69Piroxicam and rhodamine loaded microparticles produced by this method showed significantly slower initial release kinetics when drug was concentrated at the matrix core than when it was distributed closer the particle surface69 (Fig. 4). This technique was also recently applied to macromolecule loaded (rhodamine–BSA or FITC–dextran), double emulsion microparticles, but only the magnitude of the initial burst was altered,70 as discussed in Section 2.1.1.3. Between electrospray fabricated microparticles and systems such as double-walled particles or implants, radial drug distribution can successfully modified for a diverse array of active agents.
2.2. Lag phase
Following the initial burst, a lag (or pause in release) may occur before the remaining encapsulated drug is released. By definition, this phase lacks measurable kinetics, but may possess significant duration. However, particularly slow initial release or, conversely, early onset of final release may serve to disguise this phase. Hence, the duration of the lag phase should be defined as time elapsed between day 0 (while the competing burst phase is occurring) and the onset of final release (a time denoting the resumption of drug delivery).2.2.1.1. Setting of initial polymer molecular weight. Many studies have shown that the duration of the lag phase can be altered by varying the polymer's initial molecular weight (Mw). For example, Friess and Schlapp induced a 10 day lag phase in gentamicin loaded microparticles by switching from a 13.5 kDa PLGA to a higher molecular weight of 36.2 kDa.71 Comparable results have been reported for small molecule loaded polyester implants.72 This relation also holds true in peptide loaded microparticles, as well.73–75Macromolecule release data from different studies also confirm a clear trend between lag phase duration and the molecular weight of the PLGA matrix (Fig. 5).23,40,75–80 This fundamental trend is only preserved within a given class of agents, suggesting that effect of polymer molecular weight is dependent, at least in part, upon some property (or properties) of the encapsulated agent.71,73,79
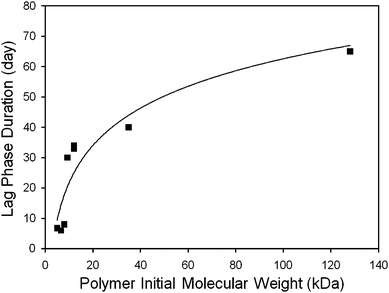 | ||
| Fig. 5 Relationship between polymer molecular weight and lag duration in macromolecule releasing systems. The lag phase duration (squares) was determined by analyzing release data from a number of different studies.25,27,42,77–81 The results fit a power expression (line, r2 = 0.8565). | ||
2.2.1.2. Controlling polymer degradation rate. Another well-documented tool that can be used to alter the duration of the lag phase is the polymer's degradation rate. For copolymers, this is simply controlled by altering the ratio of the two monomers, with degradation rate typically being inversely proportional to hydrophobicity of the resulting polymer chain. For example, in work by Cui et al. where 9.5 kDa of 50
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 PLGA microparticles produced a melittin release profile with an 8 day lag phase, while 10 kDa of 75:25 PLGA microparticles presented a 14 day lag phase.36 Similarly, Wang et al. tested ethacrynic acid loaded films of 110 kDa 50:50 PLGA and PLA which produced lags of 1 or 6 days, respectively.81 This trend has also been echoed by polyanhydride copolymer microparticles loaded with BSA,82PLGA and PLA fibers loaded with BSA80 as well as other polyester implants loaded with small molecule drugs.83,84 The consistent performance of the polymer degradation rate and initial molecular weight as tools for controlling lag phase duration suggests that the two may act in concert via a common property such as polymer lifespan.
:50 PLGA microparticles produced a melittin release profile with an 8 day lag phase, while 10 kDa of 75:25 PLGA microparticles presented a 14 day lag phase.36 Similarly, Wang et al. tested ethacrynic acid loaded films of 110 kDa 50:50 PLGA and PLA which produced lags of 1 or 6 days, respectively.81 This trend has also been echoed by polyanhydride copolymer microparticles loaded with BSA,82PLGA and PLA fibers loaded with BSA80 as well as other polyester implants loaded with small molecule drugs.83,84 The consistent performance of the polymer degradation rate and initial molecular weight as tools for controlling lag phase duration suggests that the two may act in concert via a common property such as polymer lifespan.
2.2.1.3. Use of catalytic excipients. When a specific polymer chemistry or molecular weight is desired (and therefore not accessible as a tool to tune release), the degradation rate and, in turn, duration of the lag phase can also be modified by using an excipient. This is evident in one study where proteinase K increased the degradation rate of PLA fibers eliminating all lag from the release of paclitaxel and doxorubicin.85 This enzyme will also catalyze the degradation of L-lactic linkages in PLGA copolymers and consequently should shorten the duration of lag phase in said systems as well.86 An anhydride (acid) has also been used as a catalyst to hasten the degradation of poly(ortho ester) matrices, completely eliminating a 2 day lag phase.87 This mechanism should apply equally well to polyester or polyanhydride matrices whose hydrolysis is also effected by the presence of acid.39 Future work is needed to determine if acidic excipients will cause measurable damage to encapsulated peptides or proteins.
2.2.1.4. Post fabrication irradiation. The duration of the lag phase can also be reduced by γ-irradiation.88,89 The most dramatic changes produced by this method were reported for the release of progesterone from PLA microspheres, where 100 kGY of radiation reduced the lag duration from nearly 200 days (prior to exposure) to just 50 days.89 Working with small molecule loaded PLGA microspheres, Faisant et al. also reported a similar trend when 5-fluorouracil loaded PLGA microparticles were irradiated (4–33 kGy).88 These changes in lag duration can likely be attributed to the cleavage of polymer chains in the encapsulating matrix (e.g.reduction in initial polymer molecular weight), a phenomenon whose effects are described earlier in this section. Interestingly, both studies also noted an increase in burst rate, phenomena not observed with other degradation-based methods of altering the duration of the lag phase. While this method for altering lag phase should apply to a wide range of polyester matrices, its utility may be limited because: (1) it simultaneously alters the kinetics of the initial burst, (2) it appears to be only capable of shortening the lag phase (not lengthening it) and (3) it may degrade sensitive agents such as peptides and proteins.
2.3. Final release phase
Control over the final release phase can help to extend drug delivery or even determine how pronounced the effects of the initial burst and lag phase will be on the overall release profile. Because this phase is responsible for the delivery of the remainder of drug in the polymer matrix, its magnitude is (by definition) predetermined by the magnitudes of the prior release phases. However, the kinetics of this phase can still be readily tuned by several different methods.2.3.1.1. Use of polymer blends. Firstly, the rate of final release can be reduced (or its duration extended) by blending together like polymers.71,73,90–93 For example, by adding together equal measures of 36.2 kDa PLGA and 13.5 kDa PLGA, Friess and Schlapp were able to extend the final release of gentamicin (small molecule) from just 3 days to 7 days.71 Similar mixtures have also been used to extended the release of peptides and proteins.73,91 This tool has even used to sustain protein (lysozyme) release from polyanhydride microspheres.93 Interestingly enough, this method of reducing the final release rate extends directly from methods for tuning the lag phase duration (which also marks the time until final release). For instance, mixing together polymers with different lag phases could stagger the onset of final release, yielding an overall slower final release rate than either polymer could achieve alone. Hence this technique for modulating the rate of final release should prove effective for any matrix system with an adjustable lag phase.
2.3.1.2. Control viacopolymer ratio. Data also suggest that duration of final release in polyester systems is dependent on the copolymer ratio. Studies on melittin microparticles show that poly(lactic-co-glycolic) acid composed of equal amounts of each monomer (50
:50 PLGA) completes final release in just 2 weeks, while 75:25 PLGA takes 3 weeks to deliver nearly the same amount of peptide.36 A similar observation can be made for the ethacrynic acid loaded films.81 (Note: in all aforementioned cases altering copolymer ratio also adjusted the duration of the lag phase via the polymer degradation rate, as described in Section 2.2.1.2.) While, this effect appears consistent across different PLGA copolymers, further research is needed to better understand its mechanism. Tuning common factors such as polydispersity in the polymer molecular weight or semicrystallinity of the polymer matrix (which lead to variance in the polymer lifespan94) may produce comparable effects in other polymer types.95
2.4. Summary of experimental results
Many methods for tuning the release kinetics of biodegradable polymer matrices have been tested in carefully designed experiments on a variety of different drugs. The result is a set of independent methods for tuning the magnitude or kinetics of the initial burst, the duration of the lag phase and the rate of final release. When used in combination, these design tools can produce release profiles ranging pure the Fickian diffusion to complex tri-phasic behaviors. In the latter case, when phases are distinct, it is often clear how to apply the experimental-based design tools discussed thus far. However, if phases are indistinct from one another, mathematical models are often helpful for determining the best way to control release behavior.3. Mathematical models
In 1961, the Higuchi equation set a new standard for design by permitting diffusion and solubility-limited release to be tuned predictably through experimental system properties.96 This equation predicted that the cumulative release of drug from planar films of cream or ointment would be proportional to the square root of time provided that a “core” of drug remained above its solubility limit. Although this model was originally designed for non-degradable and non-swellable systems, many different matrices also produce diffusion/solubility-limited release provided that other processes which influence release (e.g.degradation, erosion, etc.) occur at a much slower rate than dissolution and diffusion84,97–101 For such systems, the Higuchi equation quantifies how agent solubility and matrix geometry effect release, making it one of the first mathematical model-based “tools” for controlled release. Over the past 50 years, mathematical models have continued to quantify the fundamental mechanics of release uncovered in experimental studies. Although past reviews12,13 have adequately described the differences in mathematics and paradigms of existing models, analysis of their implementation (e.g. the description or prediction of experimentally acquired release data) is needed to evaluate each model's utility as a design tool.In order to serve as an effective design tool, a mathematical model must provide a means of predicting how changes in system properties will affect the release of a given drug. This is an important distinction, as many models, through regression, will fit tri-phasic release data, while only predicting how one or two system properties will affect release. As most system properties only alter a single aspect or phase of release kinetics, this would limit a model's ability to tailor release kinetics. On the other end of the spectrum, models accounting for too many system properties can be difficult to implement as a number of different parameter values would be required for their successful solution. Thus, as design tools, models can be categorized by the phases of release that they effectively tune and then be evaluated for their accuracy, applicability and ease of implementation.
3.1. Models for tuning initial burst release
Zhang et al. have derived a detailed model for describing mono-, bi-, and tri-phasic protein release profiles.105 To account for this diversity in release behavior, this model actually contains three different versions of its core equations optimized to approximate a diverse range of experimentally observed erosion behavior. Each version of the model's equations was tested on release data from systems with different erosion profiles. By fitting the model first to mass loss (erosion) data, the most appropriate version of its equations was determined and values were computed for erosion rate constants. Then release data were described by optimizing values for the initial tortuosity and dissolution rate constant. Values for the remaining system properties (agent solubility limit, initial diffusivity, microparticle radius, drug loading, initial tortuosity and initial porosity) were taken from the literature. Because sensitivity analysis shows that the erosion mechanism can have a dramatic effect on release kinetics, matrix-specific properties that are likely to effect erosion (e.g. microparticle radius, initial porosity or initial tortuosity) may prove a difficult means of precisely altering release. Fortunately, this model still accounts for agent-specific system properties such as agent loading and solubility which can be used to tune the magnitude and kinetics of the initial burst, respectively (Sections 2.1.1.3 and 0).
3.2. Models for both burst release and the lag phase
3.3. Models for tuning tri-phasic release
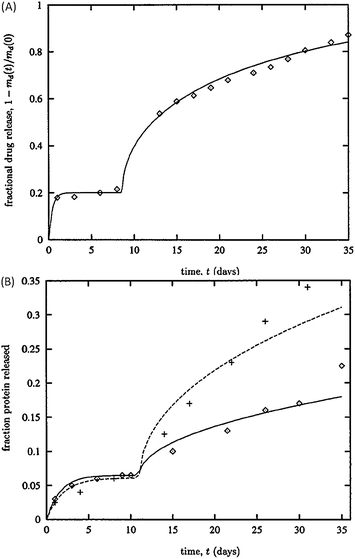 | ||
| Fig. 6 Predictions of protein release from PLGA and PLA microparticles. (A) Model's prediction (solid line) compares favourably with glycoprotein release data (diamonds). (B) Estimations of tetanus toxin release (solid line) capture the initial burst and lag phase of the data from PLGA (crosses) and PLA (diamonds) microparticles. Reproduced with permission of ref. 108. | ||
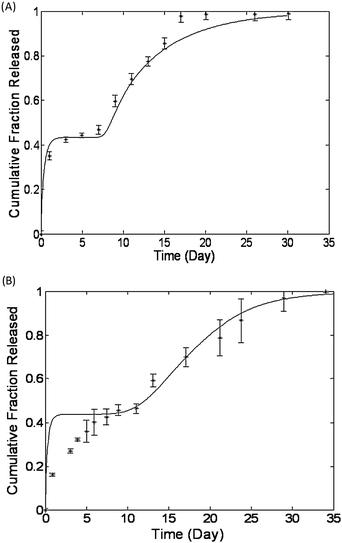 | ||
| Fig. 7 Regression free predictions of release data from Rothstein et al. (A) Melittin peptide release from PLGA microparticles and (B) bovine serum albumin protein release from polyanhydride microparticles. In each case the model's prediction appear as a solid line and the experimentally acquired release data are represented with asterisks. Reproduced with permission of ref. 19. | ||
Recently, the model by Rothstein et al. has been extended to handle matrix implants and hydrophobic agents. This required additional equations accounting for matrix hydration and drug dissolution kinetics, but did not require adjustments for the model's empirical correlations. The new model was tested successfully with regression free predictions of drug release from 2 polyanhydride and 2 poly(ortho ester) implants. However, the mechanisms (hydration, dissolution, degradation, erosion and diffusion) used in this model are fundamental to PLGA and PLA matrices as well.8 Predictions for such implants can be made from just the 4 readily attainable parameters (degradation rate, matrix size, polymer initial molecular weight and agent molecular weight) or alternatively 6 parameters (adding on values for agent dissolution rate and maximum solubility) if release is limited by dissolution as well.
3.4. Summary of mathematical models
Mathematical models have successfully described release data from a number of different biodegradable matrix formulations. As design tools these models predict anywhere from 1 to 3 phases of release. A few of these models18,19,108 can even be solved without requiring regression to a system's in vitro release data. By predicting how tunable system properties effect each phase of release, these models make real the possibility of rationally designing the release kinetics of biodegradable polymer matrices.4. Conclusions
The literature presents a number of different approaches to controlling drug release from polyester and polyanhydride matrices. Empirical studies of release have produce methods for independently tuning the different aspects of the standard tri-phasic release profile. Mathematical models are beginning to successfully correlate quantitative predictions of release kinetics to physically tunable properties and conditions. Between empirical and mathematical methods a number of different tools have been developed to provide the means for precisely tuning the release kinetics of a broad range of active agents.Challenges in drug delivery still exist, such as the design of systems with drug–materials interactions, or with rapid changes release kinetics, and it is important to continue the development of experimental and theoretical tools to handle these increasingly complex scenarios as well as new agents and polymers. Such tools may prove essential for a number of applications requiring precise temporo-spatial control, including the delivery of growth factors for regenerative medicine and the delivery of cytokines or chemokines for immunotherapy. Furthermore, to better incorporate controlled release behavior into new therapeutics, tools will also be needed for predicting in vivo performance of hydrolysable polymer matrices in various ways. Fortunately new techniques such as live animal imaging may make it easier to measure and compare in vivo release kinetics, helping researchers understand how in vitro design tools might be applied to precisely control the concentration of drug in a specific physiological compartment over time. With continued advances to the design “tool-box”, future scientists and engineers may someday tailor controlled release formulations to provide specific dosing kinetics to any given physiological compartment by simply selecting correct materials and processing methods. This would pave the way to not only to the broad-scale production of custom release systems for any application, but perhaps even to patient-specific, or “individualized”, controlled release systems that can be accompany individualized medicine in the future.
Notes and references
- P. Clinton, Pharmaceut. Exec., 2005, 25, 21–21 Search PubMed.
- V. R. Sinha and A. Trehan, J. Controlled Release, 2003, 90, 261–280 CrossRef CAS.
- A. Humphreys and R. Mayer, MedAdNews, 2007, 13, 33–38 Search PubMed.
- R. C. Mundargi, V. R. Babu, V. Rangaswamy, P. Patel and T. M. Aminabhavi, J. Controlled Release, 2008, 125, 193–209 CrossRef CAS.
- I. M. Sanders, J. S. Kent and G. I. McRae, J. Pharm. Sci., 1984, 73, 1294–1297 CrossRef.
- L. M. Sanders, B. A. Kell, G. I. McRae and G. W. Whitehead, J. Pharm. Sci., 1986, 75, 356–360 CrossRef CAS.
- S. Freiberg and X. X. Zhu, Int. J. Pharm., 2004, 282, 1–18 CrossRef CAS.
- F. Alexis, Polym. Int., 2005, 54, 36–46 CrossRef CAS.
- J. P. Jain, S. Modi, A. J. Domb and N. Kumar, J. Controlled Release, 2005, 103, 541–563 CrossRef CAS.
- S. Lakshmi, D. S. Katti and C. T. Laurencin, Adv. Drug Delivery Rev., 2003, 55, 467–482 CrossRef CAS.
- C. Wischke and S. P. Schwendeman, Int. J. Pharm., 2008, 364, 298–327 CrossRef CAS.
- J. Siepmann and A. Gopferich, Adv. Drug Delivery Rev., 2001, 48, 229–247 CrossRef CAS.
- D. Y. Arifin, L. Y. Lee and C. H. Wang, Adv. Drug Delivery Rev., 2006, 58, 1274–1325 CrossRef CAS.
- A. G. Thombre and K. J. Himmelstein, AIChE J., 1985, 31, 759–766 CrossRef CAS.
- A. Gopferich and R. Langer, AIChE J., 1995, 41, 2292–2299 CrossRef.
- R. Wada, S. H. Hyon and Y. Ikada, J. Controlled Release, 1995, 37, 151–160 CrossRef CAS.
- J. Siepmann and F. Siepmann, Int. J. Pharm., 2008, 364, 328–343 CrossRef CAS.
- S. N. Rothstein, W. J. Federspiel and S. R. Little, J. Mater. Chem., 2008, 18, 1873–1880 RSC.
- S. N. Rothstein, W. J. Federspiel and S. R. Little, Biomaterials, 2009, 30, 1657–1664 CrossRef CAS.
- N. S. Berchane, F. F. Jebrail and M. J. Andrews, Int. J. Pharm., 2010, 383, 81–88 CrossRef CAS.
- X. Huang and C. S. Brazel, J. Controlled Release, 2001, 73, 121–136 CrossRef CAS.
- S. D. Allison, Expert Opin. Drug Delivery, 2008, 5, 615–628 Search PubMed.
- K. Fu, R. Harrell, K. Zinski, C. Um, A. Jaklenec, J. Frazier, N. Lotan, P. Burke, A. M. Klibanov and R. Langer, J. Pharm. Sci., 2003, 92, 1582–1591 CrossRef CAS.
- K. D. Hinds, K. M. Campbell, K. M. Holland, D. H. Lewis, C. A. Piché and P. G. Schmidt, J. Controlled Release, 2005, 104, 447–460 CAS.
- L. E. Bromberg and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 143–147 CrossRef CAS.
- S. H. Choi and T. G. Park, Int. J. Pharm., 2006, 311, 223–228 CrossRef CAS.
- W. Bao, J. Zhou, J. Luo and D. Wu, J. Microencapsulation, 2006, 23, 471–479 CrossRef CAS.
- G. Jiang, B. C. Thanoo and P. P. DeLuca, Pharm. Dev. Technol., 2002, 7, 391–399 CrossRef CAS.
- R. Liu, S. S. Huang, Y. H. Wan, G. H. Ma and Z. G. Su, Colloids Surf., B, 2006, 51, 30–38 CrossRef CAS.
- A. L. Gomes dos Santos, A. Bochot, A. Doyle, N. Tsapis, J. Siepmann, F. Siepmann, J. Schmaler, M. Besnard, F. Behar-Cohen and E. Fattal, J. Controlled Release, 2006, 112, 369–381 CrossRef.
- J. M. Pean, M. C. Venier-Julienne, F. Boury, P. Menei, B. Denizot and J. P. Benoit, J. Controlled Release, 1998, 56, 175–187 CrossRef CAS.
- J. Kang and S. P. Schwendeman, Mol. Pharmaceutics, 2007, 4, 104–118 CrossRef CAS.
- S. R. Little, D. M. Lynn, S. V. Puram and R. Langer, J. Controlled Release, 2005, 107, 449–462 CrossRef CAS.
- S. R. Little, D. M. Lynn, Q. Ge, D. G. Anderson, S. V. Puram, J. Chen, H. N. Eisen and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9534–9539 CrossRef CAS.
- D. Klose, F. Siepmann, K. Elkharraz, S. Krenzlin and J. Siepmann, Int. J. Pharm., 2006, 314, 198–206 CrossRef CAS.
- F. Cui, D. Cun, A. Tao, M. Yang, K. Shi, M. Zhao and Y. Guan, J. Controlled Release, 2005, 107, 310–319 CrossRef CAS.
- H. C. Chang and L. C. Li, Drug Dev. Ind. Pharm., 2000, 26, 829–835 CrossRef CAS.
- J. A. Champion and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4930–4934 CrossRef CAS.
- F. Burkersroda, L. Schedl and A. Gopferich, Biomaterials, 2002, 23, 4221–4231 CrossRef CAS.
- M. Sandor, D. Enscore, P. Weston and E. Mathiowitz, J. Controlled Release, 2001, 76, 297–311 CrossRef CAS.
- M. J. Dorta, A. Santovena, M. Llabres and J. B. Farina, Int. J. Pharm., 2002, 248, 149–156 CrossRef CAS.
- D. B. Masters, C. B. Berde, S. Dutta, T. Turek and R. Langer, Pharm. Res., 1993, 10, 1527–1532 CrossRef CAS.
- H. Onishi, M. Takahashi and Y. Machida, Biol. Pharm. Bull., 2005, 28, 2011–2015 CrossRef CAS.
- M. Ramchandani and D. Robinson, J. Controlled Release, 1998, 54, 167–175 CrossRef CAS.
- P. B. Storm, J. L. Moriarity, B. Tyler, P. C. Burger, H. Brem and J. Weingart, J. Neuro-Oncol., 2002, 56, 209–217 CrossRef.
- T. Ehtezazi and C. Washington, J. Controlled Release, 2000, 68, 361–372 CrossRef CAS.
- H. K. Kim, H. J. Chung and T. G. Park, J. Controlled Release, 2006, 112, 167–174 CrossRef CAS.
- S. E. Bae, J. S. Son, K. Park and D. K. Han, J. Controlled Release, 2009, 133, 37–43 CrossRef CAS.
- N. K. Varde and D. W. Pack, J. Controlled Release, 2007, 124, 172–180 CrossRef CAS.
- Y. Y. Yang, T. S. Chung and N. Ping Ng, Biomaterials, 2001, 22, 231–241 CrossRef CAS.
- T. Feczkó, J. Tóth and J. Gyenis, Colloids Surf., A, 2008, 319, 188–195 CrossRef CAS.
- F. Wang, G. M. Saidel and J. Gao, J. Controlled Release, 2007, 119, 111–120 CrossRef CAS.
- Y. S. Nam, S. H. Song, J. Y. Choi and T. G. Park, Biotechnol. Bioeng., 2000, 70, 270–277 CrossRef CAS.
- A. Gèze, M. C. Venier-Julienne, D. Mathieu, R. Filmon, R. Phan-Tan-Luu and J. P. Benoit, Int. J. Pharm., 1999, 178, 257–268 CrossRef CAS.
- E. S. Park, M. Maniar and J. C. Shah, J. Controlled Release, 1998, 52, 179–189 CrossRef CAS.
- A. H. Shikani and A. J. Domb, Laryngoscope, 2000, 110, 907–917 CrossRef CAS.
- N. A. Rahman and E. Mathiowitz, J. Controlled Release, 2004, 94, 163–175 CrossRef CAS.
- L. E. Kokai, H. Tan, S. Jhunjhunwala, S. R. Little, J. W. Frank and K. G. Marra, J. Controlled Release, 2010, 141, 168–176 CrossRef CAS.
- A. Gopferich, J. Controlled Release, 1997, 44, 271–281 CrossRef.
- Z. Xiang, P. Sarazin and B. D. Favis, Biomacromolecules, 2009, 10, 2053–2066 CrossRef CAS.
- C. M. Negrin, A. Delgado, M. Llabres and C. Evora, J. Controlled Release, 2004, 95, 413–421 CrossRef CAS.
- D. Teomim, I. Fishbien, G. Golomb, L. Orloff, M. Mayberg and A. J. Domb, J. Controlled Release, 1999, 60, 129–142 CrossRef CAS.
- W. Zheng, Int. J. Pharm., 2009, 374, 90–95 CrossRef CAS.
- C. T. Eng, R. Lin and C. H. Wang, J. Colloid Interface Sci., 2005, 291, 135–143 CrossRef.
- E. J. Pollauf, K. K. Kim and D. W. Pack, J. Pharm. Sci., 2005, 94, 2013–2022 CrossRef CAS.
- C. Berkland, A. Cox, K. Kim and D. W. Pack, J. Biomed. Mater. Res., Part A, 2004, 70, 576–584.
- C. Berkland, K. Kim and D. W. Pack, J. Controlled Release, 2001, 73, 59–74 CrossRef CAS.
- C. Berkland, M. King, A. Cox, K. Kim and D. W. Pack, J. Controlled Release, 2002, 82, 137–147 CrossRef CAS.
- C. Berkland, K. Kim and D. W. Pack, Pharm. Res., 2003, 20, 1055–1062 CrossRef CAS.
- C. Berkland, E. Pollauf, C. Raman, R. Silverman, K. Kim and D. W. Pack, J. Pharm. Sci., 2007, 96, 1176–1191 CrossRef CAS.
- W. Friess and M. Schlapp, J. Pharm. Sci., 2002, 91, 845–855 CrossRef CAS.
- W. Vogelhuber, P. Rotunno, E. Magni, A. Gazzaniga, T. Spruß, G. Bernhardt, A. Buschauer and A. Gopferich, J. Controlled Release, 2001, 73, 75–88 CrossRef CAS.
- X. Luan and R. Bodmeier, J. Controlled Release, 2006, 110, 266–272 CrossRef CAS.
- R. Bhardwaj and J. Blanchard, J. Controlled Release, 1997, 45, 49–55 CrossRef CAS.
- M. Takenaga, Y. Yamaguchi, A. Kitagawa, Y. Ogawa, Y. Mizushima and R. Igarashi, J. Controlled Release, 2002, 79, 81–91 CrossRef CAS.
- G. Wei, G. J. Pettway, L. K. McCauley and P. X. Ma, Biomaterials, 2004, 25, 345–352 CrossRef CAS.
- C. Sturesson, P. Artursson, R. Ghaderi, K. Johansen, A. Mirazimi, I. Uhnoo, L. Svensson, A. C. Albertsson and J. Carlfors, J. Controlled Release, 1999, 59, 377–389 CrossRef CAS.
- J. L. Cleland, E. Duenas, A. Daugherty, M. Marian, J. Yang, M. Wilson, A. C. Celniker, A. Shahzamani, V. Quarmby, H. Chu, V. Mukku, A. Mac, M. Roussakis, N. Gillette, B. Boyd, D. Yeung, D. Brooks, Y. F. Maa, C. Hsu and A. J. S. Jones, J. Controlled Release, 1997, 49, 193–205 CrossRef CAS.
- S. Mao, J. Xu, C. Cai, O. Germershaus, A. Schaper and T. Kissel, Int. J. Pharm., 2007, 334, 137–148 CrossRef CAS.
- B. B. Crow, A. F. Borneman, D. L. Hawkins, G. M. Smith and K. D. Nelson, Tissue Eng., 2005, 11, 1077–1084 CrossRef CAS.
- Y. Wang, P. Challa, D. L. Epstein and F. Yuan, Biomaterials, 2004, 25, 4279–4285 CrossRef CAS.
- A. S. Determan, B. G. Trewyn, V. S.-Y. Lin, M. Nilsen-Hamilton and B. Narasimhan, J. Controlled Release, 2004, 100, 97–109 CrossRef CAS.
- H. Murakami, M. Kobayashi, H. Takeuchi and Y. Kawashima, J. Controlled Release, 2000, 67, 29–36 CrossRef CAS.
- D. Sendil, D. L. Wise and V. Hasirci, J. Biomater. Sci., Polym. Ed., 2002, 13, 1–15 CrossRef CAS.
- J. Zeng, L. Yang, Q. Liang, X. Zhang, H. Guan, X. Xu, X. Chen and X. Jing, J. Controlled Release, 2005, 105, 43–51 CrossRef CAS.
- M. S. Reeve, S. P. McCarthy, M. J. Downey and R. A. Gross, Macromolecules, 1994, 27, 825–831 CrossRef CAS.
- A. Joshi and K. J. Himmelstein, J. Controlled Release, 1991, 15, 95–104 CrossRef CAS.
- N. Faisant, J. Siepmann, P. Oury, V. Laffineur, E. Bruna, J. Haffner and J. P. Benoit, Int. J. Pharm., 2002, 242, 281–284 CrossRef CAS.
- S. Yoshioka, Y. Aso and S. Kojima, J. Controlled Release, 1995, 37, 263–267 CrossRef CAS.
- M. J. Blanco-Prieto, M. A. Campanero, K. Besseghir, F. Heimgatner and B. Gander, J. Controlled Release, 2004, 96, 437–448 CrossRef CAS.
- D. K. Pettit, J. R. Lawter, W. J. Huang, S. C. Pankey, N. S. Nightlinger, D. H. Lynch, J. A. C. L. Schuh, P. J. Morrissey and W. R. Gombotz, Pharm. Res., 1997, 14, 1422–1430 CrossRef CAS.
- S. Duvvuri, K. Gaurav Janoria and A. K. Mitra, Pharm. Res., 2006, 23, 215–223 CrossRef CAS.
- P. A. Thomas, T. Padmaja and M. G. Kulkarni, J. Controlled Release, 1997, 43, 273–281 CrossRef.
- G. Schliecker, C. Schmidt, S. Fuchs, R. Wombacher and T. Kissel, Int. J. Pharm., 2003, 266, 39–49 CrossRef CAS.
- A. S. Determan, J. R. Graham, K. A. Pfeiffer and B. Narasimhan, J. Microencapsulation, 2006, 23, 832–843 CrossRef CAS.
- T. Higuchi, J. Pharm. Sci., 1961, 50, 874–875 CrossRef CAS.
- N. Kunou, Y. Ogura, T. Yasukawa, H. Kimura, H. Miyamoto, Y. Honda and Y. Ikada, J. Controlled Release, 2000, 68, 263–271 CrossRef CAS.
- Y. S. Lee, J. P. Lowe, E. Gilby, S. Perera and S. P. Rigby, Int. J. Pharm., 2010, 383, 244–254 CrossRef CAS.
- H. Zhang, Y. Lu, G. Zhang, S. Gao, D. Sun and Y. Zhong, Int. J. Pharm., 2008, 351, 244–249 CrossRef CAS.
- U. Edlund and A. C. Albertsson, Adv. Polym. Sci., 2002, 157, 67–112 CAS.
- Y. Sun, J. Wang, X. Zhang, Z. Zhang, Y. Zheng, D. Chen and Q. Zhang, J. Controlled Release, 2008, 129, 192–199 CrossRef CAS.
- H. M. Wong, J. J. Wang and C. H. Wang, Ind. Eng. Chem. Res., 2001, 40, 933–948 CrossRef CAS.
- J. Siepmann, N. Faisant and J. P. Benoit, Pharm. Res., 2002, 19, 1885–1893 CrossRef CAS.
- J. Siepmann, K. Elkharraz, F. Siepmann and D. Klose, Biomacromolecules, 2005, 6, 2312–2319 CrossRef CAS.
- M. Zhang, Z. Yang, L. L. Chow and C. H. Wang, J. Pharm. Sci., 2003, 92, 2040–2056 CrossRef CAS.
- J. He, C. Zhong and J. Mi, Drug Delivery, 2005, 12, 251–259 CrossRef CAS.
- C. Raman, C. Berkland, K. Kim and D. W. Pack, J. Controlled Release, 2005, 103, 149–158 CrossRef CAS.
- R. P. Batycky, J. Hanes, R. Langer and D. A. Edwards, J. Pharm. Sci., 1997, 86, 1464–1477 CrossRef CAS.
- D. E. Zak, J. Stelling and F. J. Doyle Iii, Comput. Chem. Eng., 2005, 29, 663–673 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |