Enhancing photocatalytic activity of titania materials by using porous structures and the addition of gold nanoparticles
Xingdong
Wang
a and
Rachel A.
Caruso
*ab
aCSIRO Materials Science and Engineering, Private Bag 33, Clayton South, Vic 3169, Australia
bParticulate Fluids Processing Centre, School of Chemistry, The University of Melbourne, Melbourne, Vic 3010, Australia. E-mail: rcaruso@unimelb.edu.au; Fax: +61 3 9347 5180; Tel: +61 3 8344 7146
First published on 26th October 2010
Abstract
Titanium dioxide is a photocatalyst that has attracted considerable attention for tackling pollution in liquid or gaseous environments. Titania has the benefits of high stability and low toxicity, it is abundant and therefore is relatively cheap. However, intrinsic issues in the material, in particular the recombination between the photon induced electron and hole pair, the wide band gap (∼3.2 eV), and the associated issues of nanoparticle separation (generally nanoparticle samples are required to achieve high surface areas) have hampered the full potential of this photocatalytic (PC) material. Here, recent progress in producing porous titania materials, the addition of gold nanoparticles (Au NPs) to the titania and the coupling of these two approaches to improve the PC properties are reviewed. Incorporating porosity within the titania material affords large surface areas without associated nanoparticulate separation issues, and increased accessibility for the organic pollutant to the active sites on the titania, thereby enhancing PC activity. Au NPs act as electron sinks to enhance the charge separation between the e−/h+ produced on photon absorption, hence improving the quantum yield of superoxide radicals, resulting in improved PC activity. Further enhancement can be achieved by coupling the porous structure of the TiO2 and the addition of Au NPs.
 Xingdong Wang | Dr Xingdong Wang conducted her PhD, M.S. and B.S. at The University of Melbourne (Australia), Fudan University (China), and Sichuan University (China), respectively. She is currently a Postdoctoral Fellow in the Materials Science and Engineering division of CSIRO, Australia. Xingdong's research interests include templating techniques, sol–gel chemistry, synthesis and engineering of nanoparticles and porous noble metal modified TiO2, and photocatalytic applications in environmental remediation. |
 Rachel A. Caruso | Dr Rachel A. Caruso received her B.Sc. and PhD from The University of Melbourne, Australia before working at the Hahn Meitner Institute and the Max Planck Institute for Colloids and Interfaces. She is currently an ARC Future Fellow with research groups in the School of Chemistry at The University of Melbourne and in the Materials Science and Engineering division of CSIRO working on advanced porous materials. |
Introduction
Since 1972, when Fujishima and Honda first reported the titanium dioxide catalyzed photolysis of water into hydrogen under ultraviolet (UV) light,1TiO2 based materials have attracted growing interest in the materials field. Titania has found promising applications in areas including photocatalysis, photovoltaics, electrochromics and sensors.2TiO2 is regarded as the most efficient and environmentally benign photocatalyst, and has been used widely for the photodegradation of a broad range of pollutants in both water and air.2 Photoactivity occurs when photons with an energy no less than the band gap of titania are absorbed, resulting in an electron in the valence band being excited to the conduction band, thereby forming an electron/hole (e−/h+) pair (a super reductant and oxidant, respectively). However, there are issues that restrict the photoactivity of TiO2 materials: (i) the low quantum yield (∼ 10%) due primarily to the recombination of e−/h+ pairs;2 (ii) the wide band gap energy of TiO2 (3.2 eV for anatase and 3.0 eV for rutile) that requires high energy UV light (achievable from only 5% of sunlight) to induce the excitation; and (iii) the low mass transport rates between the active centres of TiO2 photocatalyst and the organic pollutant. To enhance the PC efficiency, much effort has been devoted to the modification of the TiO2 materials by both physical and chemical means. Recent review papers focus on different aspects of these advancements: For example, Chen et al.2 and Zhang et al.3 wrote broad reviews on the improvement of PC activity of semiconductors (e.g., TiO2, ZnO, SnO2, WO3, Fe2O3, and CdS). Tada et al.4 reviewed the design and application of noble metal nanoparticle loaded titania for high efficiency photocatalysis. Corma et al.5 reviewed the enhanced PC activity of TiO2via purely controlling the spatial structuring and particle size. Pan et al.6 have reported on various synthesis strategies for preparing porous photocatalysts with defined structures.Here, recent progress in improving PC activity by introducing porosity or Au NPs to TiO2 materials is highlighted. These approaches will be discussed individually and then the benefits of combining porosity and Au NPs in the TiO2 materials will be discussed.
Preparation of porous titania materials
The addition of porosity in TiO2 based photocatalysts is an important means of enhancing the PC activity by improving mass transfer within the system, as compared to a bulk material.6–8 This is due to the increased surface accessibility, leading to increased PC reaction centres within the porous materials, and the enhanced surface area resulting in higher adsorption of the organic pollutant.8 Several strategies have been applied to produce porous TiO2 materials: soft templating, hard templating, and template-free approaches, see Fig. 1 for some examples. | ||
| Fig. 1 Images of porous TiO2 prepared using (a) soft templating;12 (b) hard templating;17 and (c) template-free24 methods. Reproduced and adopted with permission from ref. 12 (Copyright © 2008 American Chemical Society), 17 (Copyright © 2007 American Chemical Society) and 24 (Copyright © 2007 Wiley), respectively. | ||
A soft template generally has a relatively mobile structure, and is often the self-assembly of molecules. Surfactant micelles, vesicles, bi- or tri-block co-polymers, ionic liquids, and biomacromolecules are commonly used as soft templates in the preparation of mesoporous titania materials.7,9–11 The soft templating method is often carried out in a one pot synthesis and allows modulation of the pore structure. However, the soft template approach usually leads to TiO2 with low thermal stability and poor crystallinity (i.e. amorphous or semi-crystalline framework),12 which is not suitable for PC applications. Therefore, hard templates with more rigid structure are typically used to prepare crystalline TiO2 materials with specific pore structures and morphologies.13–17 Frequently used hard templates for TiO2 synthesis include mesoporous silica materials,14,15carbon nanotubes,18 porous alumina,19 colloidal crystals,8 and polymeric beads.20 For instance, Dong et al.21 have applied mesoporous carbon spheres as a template to prepare TiO2 spheres with homogeneous mesopores. Caruso and co-workers have applied porous polymer spheres,20 organic filter membranes (e.g. cellulose acetate membrane)22 and agarose gels23 as templates to prepare macroporous TiO2 materials with spherical, membrane and monolithic structures, respectively. Three-dimensionally ordered macroporous (3DOM) TiO2 materials have also been prepared by using latex arrays as templates.8 In these preparations, a chemically (e.g., through etching) or physically (e.g., through calcination) removable template is required.
Template-free techniques have also been developed to prepare porous networks.24–27 Yu et al.24 prepared bimodal macro-/mesoporous TiO2 materials using the microphase-separated regions of TiO2 particles and water/alcohol channels, where the final material properties could be tuned by adjusting the calcination temperature. Aggregated anatase crystals form a mesoporous structure either as a result of the space of recrystallization (through the transformation from a less dense amorphous phase to a more dense crystalline phase), and/or due to entrapped gas or via a multi-step formation mechanism.26,27 Disordered macropores can result from the space between microspherical agglomerates of TiO2 NPs that themselves contain interparticulate mesopores.28
Photocatalytic studies using porous titania materials
For structured TiO2 materials, improvements in photoactivity are influenced by the template dimension, which correlates to the pore size in the final material. For example, the first order rate constant, k, of 3DOM TiO2 materials demonstrated around three (k = 0.033 min−1, 1 μm polystyrene latex as template) to four (k = 0.042 min−1, 0.5 μm polystyrene latex as template) times higher activity for the degradation of methylene blue in the liquid phase than counterpart nonporous bulk materials (k = 0.009 min−1) prepared without template.8The preparation conditions, such as the synthesis time and calcination temperature significantly influence the PC activity of porous TiO2,24,26,29 as a result of effects on material properties including the pore structure, the surface area, and the crystal size and crystal phase of TiO2.7,24,25 For instance, when the calcination temperature increased from 300 °C to 800 °C for the hierarchical meso-/macroporous TiO2 materials, the pore structure was gradually damaged, the crystal size increased with a corresponding decrease in surface area, and the crystal phase transformed from anatase to rutile when the calcination temperature was higher than 600 °C.24 This hierarchical meso-/macroporous TiO2 had a maximum PC activity for the oxidation of acetone in the gas phase (around twice that of a commercial titania, Degussa P25, which is commonly used as a standard) when the material was calcined under 300 °C. The activity then decreased as the calcination temperature increased, due to destruction of the porous structure and the decrease in surface area, from 206 to 0.02 m2 g−1.24 Improved crystallinity, generally as a result of the higher calcination temperature or longer hydrothermal or calcination time,7,24,25 enhances PC activity and may compensate for the loss of activity caused by pore structure degradation.
A hierarchically micro-/mesoporous titania film showed increased catalytic activity; increases of ∼30–40% and 60–70% for mineralizing gaseous acetaldehyde and liquid phase phenol, respectively.28 This improvement is a result of the enhanced diffusion of the reactants within the photocatalyst, due to the porous channels in the material.
Addition of Au nanoparticles to TiO2
Doping or the deposition of metal or metal oxide nanoparticles in TiO2 materials has been widely studied2 to combat limitations due to the wide band gap (3.0–3.2 eV) and the high percentage of excited electron and hole recombinations which decrease PC activity. The Au NPs localize the conduction band electrons; this was experimentally determined by using time resolved microwave conductivity measurements, which showed that the presence of Au NPs significantly reduced the life time of mobile electrons generated by photoexitation.30 The electrons are then transferred to highly oxidative species, such as adsorbed O2 (an electron acceptor), to from reactive oxygen radicals that can decompose organic pollutants.31,32 The PC activity was remarkably increased after introducing Au NPs, as the charge separation between the excited e− and h+ was increased, and therefore improved the quantum yield.33–35Au NPs can be added to TiO2 using one of four main strategies, as outlined below.1. Preformed Au NPs can be deposited on preformed TiO2 substrates. Material properties, such as particle size, morphology and surface functional groups, of both the Au and TiO2 support can be tailored via individual synthesis procedures, with metal agglomeration within the support being avoided by preparation at a low temperature.36 The preparation and properties of Au NPs with different particle sizes and stabilizers have been comprehensively reviewed by Astruc et al.37 For the preformed TiO2, commercial Degussa P25 has been commonly used, or TiO2 synthesized by the sol–gel method. Au/TiO2 composites can then be prepared by simply mixing preformed Au NPs with TiO2 NPs.38 To ensure strong interaction between the Au and TiO2, methods such as surface functionalization36 and electrophoretic deposition39 have been utilized to attach the Au onto the TiO2 particle surface.
2. Another approach is to deposit Au precursors onto preformed TiO2 substrates. Various methods such as deposition precipitation (DP),40 impregnation,41 photoreduction,42,43 and chemical reduction,44 have been employed to deposit then reduce the metal precursor on the TiO2 substrates. The DP method to deposit gold nanoparticles onto TiO2 supports was established by Haruta et al.40 and is one of the most successful wet-chemical methods used to deposit highly dispersed Au NPs on TiO2.45 The hydroxide species on the TiO2 surface are important to obtain firmly deposited Au NPs with a particle size less than 5 nm.40 The Au particle size can be tailored by (i) adjusting the pH or initial Au concentration in the synthesis solution;40,46 (ii) changing the calcination time and temperature to achieve a particle size of 3–13 nm;45,47 or (iii) prolonging the soaking time from 1–20 h giving a particle diameter of 2–7 nm.48 It was noted that the Au/TiO2 composites prepared by the DP method were more PC active if the composites were not calcined at temperatures above 300 °C, which is a typical step for DP synthesis.40 To study the effect of the preparation conditions on the material properties and PC activity of the Au/TiO2, Dozzi et al.45 adopted the DP method to deposit Au NPs on TiO2. Two different procedures were employed to induce Au precipitation (conventional DP and modified DP using urea as a precipitating agent, denoted as DPU). Two different reduction routes, thermal (the DP and DPU) or chemical (using for example, NaBH4 or H2) were used to reduce Au (III) to Au (0). The resultant particle size of the Au was approximately 2–4 nm and 8–10 nm for the chemical and thermal routes, respectively (Fig. 2).45
 | ||
| Fig. 2 HR-TEM images of Au/TiO2 prepared using the deposition precipitation method with: (a) 1 wt % Au using NaBH4 reduction; (b) 1 wt % Au using H2reduction (c) 0.5 wt % Au using traditional DP thermal treatment; and (d) 0.5 wt % Au using modified DP-urea thermal treatment.45 Reproduced with permission from ref. 45. Copyright © 2009 Royal Society of Chemistry. | ||
The photoreduction method has been widely studied for depositing Au NPs on TiO2 as the size of the Au NPs can be kept ≤10 nm.31,34,42,49,50 In photoreduction (or photodeposition) the metal cations are reduced by the photogenerated electrons in TiO2, which requires the removal of O2 from the system by purging the solution with N2 or Ar.51 A hole scavenger, commonly methanol, is also needed to consume photogenerated holes.51 The metal particle size can be adjusted by controlling the photoreduction time or solution pH.34 Patterned TiO2 films with both superhydrophilic and hydrophobic regions have been used as a molecular microtemplate to selectively photodeposit various nanosized metal particles, including Au, onto the hydrophilic sites of the TiO2 to form well-structured microelectrode arrays.52
Very promising results with a Au particle size <3 nm have been obtained by combining chemisorption, to initially adsorb the Au ions onto the TiO2 surface, with photoreduction.42 Building on this strategy, Amal et al.53 changed the conditions of photoreduction to a nonaqueous system. This allowed highly dispersed Au NPs (average diameter of 4 nm) to be controllably deposited on TiO2 avoiding the high temperature treatment required in the DP method which results in thermal sintering. In addition, the nonaqueous photoreduction prevented the undesirable particle-coarsening associated with the aqueous phase method.
The impregnation method, commonly used to deposit Au onto the surface of TiO2, involves several steps:36 Soaking the TiO2 in a metal salt precursor solution, drying, thermal decomposition of precursor salt to metal oxide, and reduction of the metal oxide to metal particles. Because of uneven precursor-solution loading caused by gravitational settlement and trapping among the TiO2 particles, metal particles generated from this conventional process are normally polydisperse in size.36 Therefore, it is generally considered that this impregnation method is inferior to the DP technique due to the larger and less uniform Au particles being produced during impregnation, although some highly distributed Au particles of 2–4 nm in size were demonstrated by Liet al.41
A slight modification to the impregnation method, the chemical reduction method, loads Au onto the surface of TiO2 by using, for example, a citrate reduction44 or H2reduction.54 Recently, Au NPs were loaded onto the surface of citrate-functionalized TiO2 nanocrystallites via an in situreduction: The Au particle size and content were controlled by altering the amount of the functional citrate group and the Au precursor type, and even the contact time between the Au precursor and the TiO2 materials.55 For example, the Au particle size decreased from 38 nm to 19 nm (HAuCl4 as precursor) or from 49 nm to 30 nm (Au(en)2Cl3 as precursor) with an increase in the citrate functionalisation (Ti/citrate ratio = 100:20) on the TiO2 surface. The Au particle size was further decreased to ∼9 nm when the contact time decreased from 3 h to 30 min.55,56
Physical irradiation methods, such as synchrotron X-ray irradiation, microwave heating, and sonication have emerged recently to reduce Au on TiO2 supports.32,33,57 The advantage of the synchrotron method was that the resultant Au NPs had a uniform size distribution of 2–5 nm and a much shorter time (e.g. exposure time of 1–15 min) was required to obtain fully reduced Au NPs, in comparison with conventional chemical or photochemical methods.32
3. In this strategy, the TiO2 is synthesised by sol–gel chemistry, in the presence of Au NPs. This method has been scantily researched, possibly due to enlargement of metal particles during fabrication of the TiO2 materials (as crystallisation of TiO2 generally requires heating). Pradhan et al.58 prepared the snowman-like heterodimer Au/TiO2 with enhanced PC activity towards the photooxidation of methanol into formaldehyde compared with pure TiO2 alone. The gold Janus nanoparticle with a size of 2 nm in diameter was preformed using the Brust protocol,59 then the Au particle surface was protected on one hemisphere by a monolayer of hydrophobic 1-hexanethiolates, and the other hemisphere by hydrophilic 2-(2-mercaptoethoxy) ethanol. The surface sol–gel process occurred on the hydrophilic hemisphere resulting in TiO2 in an anatase phase.58 This was the first example of Au/TiO2 prepared with this approach without any heat treatment, and with a well-defined crystalline phase.
Recently, heterostructures with metal cores and semiconductor shells have become strong candidates for photocatalysis due to the chemical stability of the metal within the shell and the charge transfer between metal cores and semiconductors.60 Wu et al.61 prepared core-shell Au/TiO2 NPs with the TiO2 having a truncated wedge-shaped morphology using a hydrothermal method. The Au NPs were prepared by sodium citrate reduction, and then TiF4 was used as a titania precursor that underwent hydrothermal treatment at 180 °C for 48 h. The final Au/TiO2 product had a Au particle size of 37.5 nm and anatase crystallite size of 110 nm in length.61 The F− concentration played an important role in the morphological evolution of TiO2, which not only facilitated the formation of a wedge-like TiO2 shell, but also contributed to the truncated crystal (004) facets.61
4. In the last approach, the Au NPs and TiO2 are generated simultaneously. Recently, a simple and green method to produce M/TiO2 (M = Au, Pt, Ru or RuPt) used the reductive titanium(III) oxide as a support to simultaneously reduce the metal precursor to metal.62 The particle size (0.5–2.5 nm) and content of Au was simply tuned by the total amount and the mass ratio of the Au precursors added in two steps. Due to rapid nucleation on titanium(III) oxide, a large portion of the nuclei were formed on the surface of the support after the first batch of precursor injection. The Au precursor added in the second injection step resulted in growth of the seeds to NPs.62 Interestingly, monodisperse Au/TiO2 microspheres could be produced by simply introducing a trace amount of the Au precursor, tetrachloroauric acid (HAuCl4), as a stabilizing agent during TiO2 synthesis.63 The sphere diameter was tuned from 587 nm to 392 nm when varying the amount of HAuCl4 from 0.35 to 1.12 mol %. During the annealing process, the AuCl4− decomposed to form Au atoms which migrated together to form Au NPs with a particle diameter of 7 nm. The Au NPs in this system acted as an electron acceptor under UV light and electron donor in the visible light range.
Among these four methods, the second synthesis strategy (using preformed porous TiO2 materials) has been most researched since it can produce the Au/TiO2 materials with enhanced control of the gold particle size and loading, and also a more homogenous Au distribution and stronger Au attachment to the TiO2 support are observed, although there are agglomeration issues with the Au NPs during the synthesis. The other three synthesis strategies introduce some specific advantages to the materials: (1) the first synthesis strategy increased the attachment of Au NPs to the TiO2 by surface functionalization or physical methods; (2) the third synthesis method can produce unique morphologies such as the Au core/TiO2 shell structures, and (3) the fourth method is relatively facile.
Photocatalytic studies using Au/TiO2 materials
The PC activity of titania is remarkably increased after introducing Au NPs: The PC activity of 1 wt % Au in a Au/TiO2 material prepared by the photoreduction method at pH 7 for the degradation of oxalic acid in the liquid phase was two times higher than that of the reference support (commercial P25).34 The PC activity of a 1% Au in Au/TiO2 material using the flame spray pyrolysis method was ∼30 times that of a similarly prepared TiO2 without Au toward H2 production from methanol reforming, and was 50 times higher than Degussa P25.64 The PC activity of 2 wt % Au in Au/TiO2 inactivated the LoVo cancer cells more rapidly (2.5×) than pure TiO2 under the same reaction conditions.65The PC activity can be correlated to the Au particle size, Au loading amount, and preparation method.34,43,53 Smaller Au particles have demonstrated more effective PC activity.34,58 However, the Au core/TiO2 shell structure gave enhanced PC activity even with large (37.5 nm) Au cores for both UV and visible light irradiation towards the oxidative degradation of gaseous acetaldehyde, higher than Degussa P25.61 Often there is an optimum Au loading amount for enhancing the PC activity (between 0.5–2 wt % Au/TiO2, the exact value being dependent on other factors such as synthesis conditions),46 after which the loaded Au acts as an e−/h+ recombination centre, as electrons accumulate on the metal surface to form an electric field attracting the holes. Alternatively, too many Au clusters may inhibit photon adsorption by the TiO2.35 The preparation method also influences the PC activity by changing the properties of the materials.34 The 1 wt % Au/TiO2 prepared by the photoreduction method outperformed the DP method in photocatalytic degradation of oxalic acid in the liquid phase, even though the Au particle sizes were similar in diameter (5 nm).34 For the DP method, when using different approaches to reduce Au NPs, the resultant Au/TiO2 materials showed obvious differences in PC activity for the oxidative degradation of two organic molecules (i.e., azo dye Acid Red 1 and formic acid) in the liquid phase, which may be attributed to differences in the Au particle size.45 The Fermi energy of the Au NPs had a close relationship with the PC activity:47 PC activity for the photocatalyzed reduction of 2,2′-dipyridyl disulfide and nitrobenzene in the liquid phase increased with increase in Fermi energy as the Au NP particle size increased from 3–13 nm.
Combining porosity and the addition of Au nanoparticles in TiO2 structures
To further optimize the PC activity of TiO2, a recent trend is to combine the porous structure of titania with the incorporation of Au NPs. Four approaches have been applied in this kind of material synthesis: Incorporate preformed Au NPs in porous TiO2; grow Au NPs in porous TiO2; form porous TiO2 in presence of Au NPs; or prepare porous TiO2 and Au simultaneously. Examples of these approaches are now given.To ensure the strong attachment between the preformed Au NP and the TiO2 network, physical assistance is often applied to this approach. Chen et al. prepared mesoporous TiO2 thin film by a DC magnetron sputtering technique followed by hydrothermal-oxidation of Ti to TiO2 with a H2O2 (30%) solution.66 During this oxidation process, pores are probably generated in the gel formed due to the O2 produced from the decomposition of H2O2. The Au NPs preformed by citrate reduction were then embedded into this mesoporous TiO2 by a capillary-precipitation method.66 Alternatively, an electrophoretic method has been used to deposit preformed Au NPs onto the preformed perpendicular meso-channels of the TiO2 film, prepared by spin-coating a sol–gel solution containing P123 as a template.67
The second approach is to add Au NPs by infiltrating Au precursors into porous TiO2 materials, followed by a reducing reaction such as a chemical reduction, photoreduction or heat treatment. 46,68–70 For example, Au modified TiO2 nanotubes were prepared by deposition of HAuCl4 at pH 7 or 3, followed by photoreduction.68,69 This approach was also applied to incorporate Au into 3DOM TiO2 materials.8Au NPs of various content and size were deposited onto porous TiO2 networks (prepared by templating agarose gels) using the DP method.46 In another example, a low temperature synthesis route was developed using an anionic surfactant as a pore directing agent to produce tunable mesoporous crystalline transition metal oxides, including TiO2. The calcined mesoporous titania was used as a support onto which Au NPs were deposited using the deposition–precipitation method at pH 10.12
Another interesting study to prepare Au/TiO2 used two approaches: (i) the conventional impregnation technique coupled with chemical reduction by NaBH4 and then modified with an S-group to avoid agglomeration of Au NPs; and (ii) pulsed cathodic electrodeposition.70 The former method leads to mesoporous films, red in colour because of the spherical Au NPs smaller than 5 nm in size (prolonging the impregnation time from 5 min to 180 min, resulted in a particle size of 8–10 nm). The latter approach represented a considerably easier and faster synthetic method which resulted in dendritic Au nanostructures that replicated the pore system.70 Progressive nucleation and then growth of the Au occurred with the applied potential pulses, producing a blue film due to the larger Au particle size.70
To increase the Au content in the TiO2 material, highly dispersed Au NPs were synthesised on the porous TiO2 support using the urea-deposition precipitation method (see Fig. 3 for the synthesis scheme).71 The periodic mesoporous TiO2 support was prepared by using triblock copolymer Pluronic F127 as a template, and dip coating to form a thin film on both sides of glass substrates. After heat treatment at 350 °C for 4 h, this preformed mesoporous TiO2 was soaked in the Au precursor solution containing urea at room temperature. The positive surface charge of the TiO2 (isoelectric point of TiO2: 6.6) afforded adsorption of the negatively charged Au hydroxides produced by the reaction of HAuCl4 with the OH− slowly released by urea in the aqueous solution. The slow release of OH− in the DP-urea methods is an advantage over the traditional DP method. This allowed a gradual and homogenous addition of hydroxide ions throughout the whole solution, and thus a local increase in pH value and precipitation of metal hydroxide in solution could be prevented.72,73 After reduction of Au (III) to Au (0) during heating, a very high amount (53.3 wt %) of Au was incorporated in the mesoporous channels. The Au particle diameters were 6 nm due to the confinement effect of mesoporous channels.71
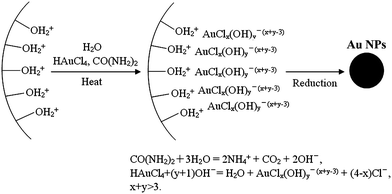 | ||
| Fig. 3 Schematic representation for the synthesis of Au NPs incorporated into porous TiO2 (TiO2 depicted as the curved charged surface) by the DP-urea method. Note that the values of x and y were real-time dependent on the pH value of the solutions, and that the sum of them was higher than three during synthesis using the deposition precipitation urea method.71 Reproduced with permission from ref. 71. Copyright © 2009 Royal Society of Chemistry. | ||
In the third approach, porous TiO2 was formed in the presence of preformed Au NPs. Commonly, TiO2 was hydrolyzed in templates pre-incorporated with Au NPs. For instance, porous TiO2 was prepared using structured agarose gels containing Au NPs as a template (the agarose gel served as both a support and reducing agent for Au NP formation).74 Hierarchically structured Au/TiO2 nanotubes have also been reported by a one-pot approach using a natural cellulosic substance as a template.75 The prepared composites can have a Au loading as high as 40 wt %.75 Recently, a hydrothermal treatment was applied to a mixture of a preformed Au colloid solution prepared using the citrate reduction method and tetrabutyl titanate at 180 °C for 7 h, which led to mesoporous Au/TiO2 beads with a pore size centered at approximately 7 nm.76 The final Au particle size remained as small as 6-7 nm after hydrothermal treatment.
The fourth approach is the simultaneous preparation of both the Au nanoparticle and titania porous network. One example to embed Au within a TiO2 framework was to utilize a multicomponent assembly approach, where the Au and titania precursors were added together with a surfactant template for one-pot synthesis.77 In this case, the Au content was typically below 2.0 mol %, with a Au particle size of approximately 12 nm. When above 2.0 mol %, the Au particle size became too large and was expelled from the framework.
Li et al. encapsulated Au NPs in TiO2 spheres producing core-shell structures using consecutive solvothermal and hydrothermal treatments.78 The encapsulation of Au NPs could be described in terms of alcoholysis-induced assembly, involving the aggregation of the TiO2 building clusters into spheres and their subsequent reaction, dissolution and re-deposition processes, together with the Au3+in situreduction. The combination of solvo- and hydrothermal treatment is essential to form the uniform Au particles within the mesoporous sphere structure. Fig. 4 shows that all the Au/TiO2 precursors transformed from solid microspheres into core-shell microspheres after solvo- and hydrothermal treatment. Solvothermal treatment alone only produced Au particles that were irregular in shape and size due to agglomeration. However, using only hydrothermal treatment produced solid Au/TiO2 microspheres instead of a mesoporous structure.78
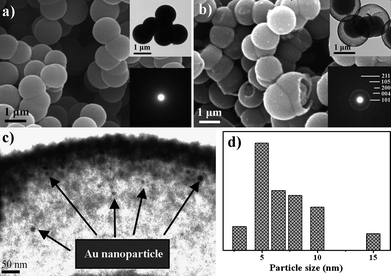 | ||
| Fig. 4 (a) SEM image of the Au/TiO2 materials before hydrothermal treatment. (b–d) SEM, TEM and Au size distribution pattern of the Au/TiO2 after solvo- and hydrothermal treatment. The insets in (a) and (b) are the TEM and selected area electron diffraction images.78 Reproduced with permission from ref. 78. Copyright © 2009 Royal Society of Chemistry. | ||
Photocatalytic studies using porous Au/TiO2 materials
The PC activity for such dual-modified (porosity and Au NP deposition) materials showed superior performance to those of either non-porous and/or non-metal modified corresponding materials.77,79 The combined effects of the enlargement of surface area, which offers more accessible active sites to allow organic reagent adsorption and degradation,77,79 and the “electron-sink” properties offered by Au deposition, to improve the charge separation of excited e−/h+ thereby increasing the quantum yield, led to the higher PC activity for the oxidation of NO in the gaseous phase (Fig. 5).70 In another example, the Au deposition (2 wt %, particle size of ∼2 nm) can further improve the PC activity by ∼40% for the decomposition of methylene blue in the liquid phase with respect to the porous control sample without Au modification.46 PC decomposition of phenol in the liquid phase can be enhanced over three times when 0.5 mol % Au was deposited in mesoporous TiO2 as compared with pure TiO2 (see Fig. 6).77 Similar to the Au-modified TiO2 materials, the enhancement of PC activity for these materials after introducing porosity depends on a range of factors, such as the Au size, dispersion and content, and the crystal phase and crystal size of TiO2, and the preparation method.46,77,79 | ||
| Fig. 5 Comparison of PC activities of pristine and Au-loaded mesoporous TiO2 film as well as Photosan thin layer, a commercial colour product for air pollutant degradation.70 Reproduced and adopted with permission from ref. 70. Copyright © 2009 American Chemical Society. | ||
![Representative TEM images of 0.5 mol % Au/TiO2 along (a) [100] and (b) [110] planes of a hexagonally mesoporous TiO2. (c) The PC activity of Au/TiO2 nanocomposites containing 0–5% Au in phenol-oxidation and chromium-reduction reactions.77 Reproduced and adopted with permission from ref. 77. Copyright © 2007 American Chemical Society.](/image/article/2011/JM/c0jm02620d/c0jm02620d-f6.gif) | ||
| Fig. 6 Representative TEM images of 0.5 mol % Au/TiO2 along (a) [100] and (b) [110] planes of a hexagonally mesoporous TiO2. (c) The PC activity of Au/TiO2 nanocomposites containing 0–5% Au in phenol-oxidation and chromium-reduction reactions.77 Reproduced and adopted with permission from ref. 77. Copyright © 2007 American Chemical Society. | ||
Conclusion and outlook
The PC activity of TiO2 is determined by accessibility of the active sites, light-harvesting efficiency, effectiveness of the photogenerated electron/hole, and the rate of electron/hole recombination. Introducing porosity and loading Au NPs in the TiO2 structure improved the PC performance. Either parameter, porosity or Au NP loading, significantly enhances the photoactivity compared with corresponding reference materials. By combining the two parameters in the one material, further enhancement was observed, though no synergetic effect has been reported. In future, more effort needs to be devoted to optimizing this approach to design advanced photocatalysts, taking full advantage of the merits brought about by the individual modifications: Improved pollutant diffusion through the porous structure and localization of photoexcited electrons. This could be realized by improved control of the porosity, such as producing hierarchically interconnected porous structures, increasing the thermal stability of the pore walls, increasing the homogeneity and mono-dispersibility of the macropore, and designing the composite material with preferable morphology (e.g., membrane or film) for environmental remediation. Along with the ability to exhibit further control on the Au NP–dispersibility, thermal stability, particle size and amount deposited by employing the advantages of Au NP growth on preformed porous TiO2 structures and further exploring the merits of the other synthesis strategies.Acknowledgements
RAC acknowledges an Australian Research Council Future Fellowship (FT0990583).References
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS.
- H. J. Zhang, G. H. Chen and D. W. Bahnemann, J. Mater. Chem., 2009, 19, 5089–5121 RSC.
- H. Tada, T. Kiyonaga and S. Naya, Chem. Soc. Rev., 2009, 38, 1849–1858 RSC.
- C. Aprile, A. Corma and H. Garcia, Phys. Chem. Chem. Phys., 2008, 10, 769–783 RSC.
- J. H. Pan, H. Q. Dou, Z. G. Xiong, C. Xu, J. Z. Ma and X. S. Zhao, J. Mater. Chem., 2010, 20, 4512–4528 RSC.
- Y. Wang, Z. H. Jiang and F. J. Yang, Materials Science and Engineering B-Solid State Materials for Advanced Technology, 2006, 128, 229–233 Search PubMed.
- M. Srinivasan and T. White, Environ. Sci. Technol., 2007, 41, 4405–4409 CrossRef CAS.
- E. L. Crepaldi, G. Soler-Illia, D. Grosso, F. Cagnol, F. Ribot and C. Sanchez, J. Am. Chem. Soc., 2003, 125, 9770–9786 CrossRef CAS.
- S. D. Miao, Z. J. Miao, Z. M. Liu, B. X. Han, H. Zhang and J. Zhang, Microporous Mesoporous Mater., 2006, 95, 26–30 CrossRef CAS.
- S. Yuan, Q. R. Sheng, J. L. Zhang, H. Yamashita and D. N. He, Microporous Mesoporous Mater., 2008, 110, 501–507 CrossRef CAS.
- D. H. Wang, Z. Ma, S. Dai, J. Liu, Z. M. Nie, M. H. Engelhard, Q. S. Huo, C. M. Wang and R. Kou, J. Phys. Chem. C, 2008, 112, 13499–13509 CrossRef CAS.
- Y. D. Xia and R. Mokaya, J. Mater. Chem., 2005, 15, 3126–3131 RSC.
- W. B. Yue, X. X. Xu, J. T. S. Irvine, P. S. Attidekou, C. Liu, H. Y. He, D. Y. Zhao and W. Z. Zhou, Chem. Mater., 2009, 21, 2540–2546 CrossRef CAS.
- L. Zhao and J. G. Yu, J. Colloid Interface Sci., 2006, 304, 84–91 CrossRef CAS.
- Y. Kondo, H. Yoshikawa, K. Awaga, M. Murayama, T. Mori, K. Sunada, S. Bandow and S. Iijima, Langmuir, 2008, 24, 547–550 CrossRef CAS.
- J. I. L. Chen, G. von Freymann, V. Kitaev and G. A. Ozin, J. Am. Chem. Soc., 2007, 129, 1196–1202 CrossRef CAS.
- W. G. Fan and L. Gao, Chem. Lett., 2006, 35, 670–671 CrossRef CAS.
- L. K. Tan, M. A. S. Chong and H. Gao, J. Phys. Chem. C, 2008, 112, 69–73 CrossRef CAS.
- U. Meyer, A. Larsson, H. P. Hentze and R. A. Caruso, Adv. Mater., 2002, 14, 1768 CrossRef CAS.
- A. G. Dong, N. Ren, Y. Tang, Y. J. Wang, Y. H. Zhang, W. M. Hua and Z. Gao, J. Am. Chem. Soc., 2003, 125, 4976–4977 CrossRef CAS.
- R. A. Caruso and J. H. Schattka, Adv. Mater., 2000, 12, 1921 CAS.
- J. F. Zhou, M. F. Zhou and R. A. Caruso, Langmuir, 2006, 22, 3332–3336 CrossRef CAS.
- J. G. Yu, Y. R. Su and B. Cheng, Adv. Funct. Mater., 2007, 17, 1984–1990 CrossRef CAS.
- J. G. Yu, L. J. Zhang, B. Cheng and Y. R. Su, J. Phys. Chem. C, 2007, 111, 10582–10589 CrossRef CAS.
- J. G. Yu, G. H. Wang, B. Cheng and M. H. Zhou, Appl. Catal., B, 2007, 69, 171–180 CrossRef CAS.
- B. Liu and H. C. Zeng, Chem. Mater., 2008, 20, 2711–2718 CrossRef CAS.
- Y. Zhao, X. T. Zhang, J. Zhai, J. L. He, L. Jiang, Z. Y. Liu, S. Nishimoto, T. Murakami, A. Fujishima and D. B. Zhu, Appl. Catal., B, 2008, 83, 24–29 CrossRef CAS.
- T. Y. Peng, D. Zhao, K. Dai, W. Shi and K. Hirao, J. Phys. Chem. B, 2005, 109, 4947–4952 CrossRef CAS.
- J. T. Carneiro, T. J. Savenije and G. Mul, Phys. Chem. Chem. Phys., 2009, 11, 2708–2714 RSC.
- A. V. Rupa, D. Divakar and T. Sivakumar, Catal. Lett., 2009, 132, 259–267 CrossRef CAS.
- C. J. Liu, T. Y. Yang, C. H. Wang, C. C. Chien, S. T. Chen, C. L. Wang, W. H. Leng, Y. Hwu, H. M. Lin, Y. C. Lee, C. L. Cheng, J. H. Je and G. Margaritondo, Mater. Chem. Phys., 2009, 117, 74–79 CrossRef CAS.
- Y. Mizukoshi, Y. Makise, T. Shuto, J. W. Hu, A. Tominaga, S. Shironita and S. Tanabe, Ultrason. Sonochem., 2007, 14, 387–392 CrossRef CAS.
- V. Iliev, D. Tomova, L. Bilyarska and G. Tyuliev, J. Mol. Catal. A: Chem., 2007, 263, 32–38 CrossRef CAS.
- J. Xu, Y. Sun, Y. M. Zhao, J. J. Huang, C. M. Chen and Z. Y. Jiang, Int. J. Photoenergy, 2007, 1–7 CrossRef.
- J. Li and H. C. Zeng, Chem. Mater., 2006, 18, 4270–4277 CrossRef CAS.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- L. C. Du, A. Furube, K. Yamamoto, K. Hara, R. Katoh and M. Tachiya, J. Phys. Chem. C, 2009, 113, 6454–6462 CrossRef CAS.
- N. Chandrasekharan and P. V. Kamat, Nano Lett., 2001, 1, 67–70 CrossRef CAS.
- M. Haruta, Catal. Today, 1997, 36, 153–166 CrossRef CAS.
- W. C. Li, M. Comotti and F. Schuth, J. Catal., 2006, 237, 190–196 CrossRef CAS.
- T. Soejima, H. Tada, T. Kawahara and S. Ito, Langmuir, 2002, 18, 4191–4194 CrossRef CAS.
- C. Yogi, K. Kojima, T. Takai and N. Wada, J. Mater. Sci., 2009, 44, 821–827 CrossRef CAS.
- M. C. Hidalgo, M. Maicu, J. A. Navio and G. Colon, J. Phys. Chem. C, 2009, 113, 12840–12847 CrossRef CAS.
- M. V. Dozzi, L. Prati, P. Canton and E. Selli, Phys. Chem. Chem. Phys., 2009, 11, 7171–7180 RSC.
- X. D. Wang, D. R. G. Mitchell, K. Prince, A. J. Atanacio and R. A. Caruso, Chem. Mater., 2008, 20, 3917–3926 CrossRef CAS.
- T. Kiyonaga, M. Fujii, T. Akita, H. Kobayashi and H. Tada, Phys. Chem. Chem. Phys., 2008, 10, 6553–6561 RSC.
- M. A. Elmoula, E. Panaitescu, M. Phan, D. Yin, C. Richter, L. H. Lewis and L. Menon, J. Mater. Chem., 2009, 19, 4483–4487 RSC.
- S. C. Chan and M. A. Barteau, Langmuir, 2005, 21, 5588–5595 CrossRef CAS.
- M. J. Uddin, F. Cesano, D. Scarano, F. Bonino, G. Agostini, G. Spoto, S. Bordiga and A. Zecchina, J. Photochem. Photobiol., A, 2008, 199, 64–72 CrossRef CAS.
- S. L. Lee, J. Scott, K. Chiang and R. Amal, J. Nanopart. Res., 2009, 11, 209–219 CrossRef CAS.
- X. G. Li, Y. Tian, P. P. Xia, Y. P. Luo and Q. Rui, Anal. Chem., 2009, 81, 8249–8255 CrossRef CAS.
- R. Kydd, J. Scott, W. Y. Teoh, K. Chiang and R. Amal, Langmuir, 2010, 26, 2099–2106 CrossRef CAS.
- J. J. Zou, Y. P. Zhang and C. J. Liu, Langmuir, 2006, 22, 11388–11394 CrossRef CAS.
- V. Mendez, V. Caps and S. Daniele, Chem. Commun., 2009, 3116–3118 RSC.
- A. Kafizas, S. Kellici, J. A. Darr and I. P. Parkin, J. Photochem. Photobiol., A, 2009, 204, 183–190 CrossRef CAS.
- S. Shironita, T. Takasaki, T. Kamegawa, K. Mori and H. Yamashita, Catal. Lett., 2009, 129, 404–407 CrossRef CAS.
- S. Pradhan, D. Ghosh and S. W. Chen, ACS Appl. Mater. Interfaces, 2009, 1, 2060–2065 Search PubMed.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CrossRef CAS.
- X. F. Wu, H. Y. Song, J. M. Yoon, Y. T. Yu and Y. F. Chen, Langmuir, 2009, 25, 6438–6447 CrossRef CAS.
- Y. Xie, K. L. Ding, Z. M. Liu, R. T. Tao, Z. Y. Sun, H. Y. Zhang and G. M. An, J. Am. Chem. Soc., 2009, 131, 6648–6649 CrossRef CAS.
- P. Wang, T. F. Xie, H. Y. Li, L. A. Peng, Y. Zhang, T. S. Wu, S. Pang, Y. F. Zhao and D. J. Wanga, Chem.–Eur. J., 2009, 15, 4366–4372 CrossRef CAS.
- G. L. Chiarello, E. Selli and L. Forni, Appl. Catal., B, 2008, 84, 332–339 CrossRef CAS.
- J. Xu, Y. Sun, Y. M. Zhao, J. J. Huang, C. M. Chen and Z. Y. Jiang, International Journal of Photoenergy, 2007, Search PubMed article ID 97308, 1–7.
- Z. Y. Chen, Y. Hu, T. C. Liu, C. L. Huang and T. S. Jeng, Thin Solid Films, 2009, 517, 4998–5000 CrossRef CAS.
- M. N. Patel, R. D. Williams, R. A. May, H. Uchida, K. J. Stevenson and K. P. Johnston, Chem. Mater., 2008, 20, 6029–6040 CrossRef CAS.
- B. L. Zhu, K. R. Li, Y. F. Feng, S. M. Zhang, S. H. Wu and W. P. Huang, Catal. Lett., 2007, 118, 55–58 CrossRef CAS.
- Q. Zhao, M. Li, J. Y. Chu, T. S. Jiang and H. B. Yin, Appl. Surf. Sci., 2009, 255, 3773–3778 CrossRef CAS.
- I. Bannat, K. Wessels, T. Oekermann, J. Rathousky, D. Bahnemann and M. Wark, Chem. Mater., 2009, 21, 1645–1653 CrossRef CAS.
- F. M. Cui, Z. L. Hua, C. Y. Wei, J. Q. Li, Z. Gao and J. L. Shi, J. Mater. Chem., 2009, 19, 7632–7637 RSC.
- R. Zanella, S. Giorgio, C. H. Shin, C. R. Henry and C. Louis, J. Catal., 2004, 222, 357–367 CrossRef CAS.
- R. Zanella, S. Giorgio, C. R. Henry and C. Louis, J. Phys. Chem. B, 2002, 106, 7634–7642 CrossRef.
- X. D. Wang, C. E. Egan, M. F. Zhou, K. Prince, D. R. G. Mitchell and R. A. Caruso, Chem. Commun., 2007, 3060–3062 RSC.
- J. G. Huang, T. Kunitake and S. Onoue, Chem. Commun., 2004, 1008–1009 RSC.
- J. G. Yu, L. Yue, S. W. Liu, B. B. Huang and X. Y. Zhang, J. Colloid Interface Sci., 2009, 334, 58–64 CrossRef CAS.
- H. X. Li, Z. F. Bian, J. Zhu, Y. N. Huo, H. Li and Y. F. Lu, J. Am. Chem. Soc., 2007, 129, 4538–4539 CrossRef CAS.
- Z. F. Bian, J. Zhu, F. L. Cao, Y. F. Lu and H. X. Li, Chem. Commun., 2009, 3789–3791 RSC.
- H. B. Yi, T. Y. Peng, D. N. Ke, D. Ke, L. Zan and C. H. Yan, Int. J. Hydrogen Energy, 2008, 33, 672–678 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |