DOI:
10.1039/C0JM01284J
(Paper)
J. Mater. Chem., 2011,
21, 125-133
An atomistic simulation of the liquid-crystalline phases of sexithiophene
Received
2nd May 2010
, Accepted 5th August 2010
First published on 28th September 2010
Abstract
We have investigated, using atomistic molecular dynamics simulations, the high temperature molecular organization of the linear oligothiophene α-sexithienyl (T6), well known for its organic electronics applications. We have found a smectic and a nematic liquid crystalline phase in the same temperature range where they had been experimentally reported but not fully characterized. We have microscopically characterized the phases and connected the change of mesophase and of orientational and positional order to variations in the T6 conformation and effective shape. T6 phases obtained by rapid cooling from the ordered melts have also been simulated.
1 Introduction
Oligothiophenes represent a class of materials of considerable interest for organic electronics1 as semiconductors of high mobility2,3 in organic thin film transistors and organic photovoltaic cells.4 In this context an important role as p-type materials is played by their conjugated nature that can confer good charge (hole) mobility between molecular units when they are packed in an appropriate morphology. An ideal situation to maximize the electronic overlap between neighboring molecules could be represented by a defect-less crystal with aromatic units closely packed,5 while the reality consists, even in cases where such a crystalline phase exists, of a reduced mobility due to grain boundaries.6,7 It is thus of interest to consider the possibility of liquid crystalline phases formed by oligothiophenes, since these could be expected to ensure good parallelism between the molecules with the advantage of annealing out defects due to their fluidity.8,9
In order to explore the possibility of oligothiophene liquid crystal (LC) morphologies, α-sexithiophene (or α-sexithienyl, T6, Fig. 1) is particularly interesting for a theoretical study. In fact, on one hand its relatively simple structure without alkyl chains makes it relatively easy to model. On the other, a few experimental studies10,11 indicate that T6 forms at least a LC phase at high temperature, above 585 K, even if the nematic or smectic nature of this mesophase has been questioned. The material itself, beyond being a prototype for more complex substituted oligothiophenes, is not only of academic interest. Indeed good mobilities for an organic semiconductor in the range ∼![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.15 cm2/Vs in single crystal12 and 0.01–0.03 cm2/Vs in thin film and high current on/off ratios (∼104) have been reported.3,13–15 T6 mobility has also been widely studied in thin films16 and T6 films have been examined with several techniques such as IR,17 AFM,18 XRD19 to determine their molecular organization. Other investigations have dealt with the possibility of aligning T6 in a nematic host measuring its polarized photoluminescence20 or its structure in solution using NMR.21
0.15 cm2/Vs in single crystal12 and 0.01–0.03 cm2/Vs in thin film and high current on/off ratios (∼104) have been reported.3,13–15 T6 mobility has also been widely studied in thin films16 and T6 films have been examined with several techniques such as IR,17 AFM,18 XRD19 to determine their molecular organization. Other investigations have dealt with the possibility of aligning T6 in a nematic host measuring its polarized photoluminescence20 or its structure in solution using NMR.21
 |
| | Fig. 1 Chemical sketch of the α-sexithiophene molecule (T6) with the molecular reference frame. The three different typologies of thiophene–thiophene torsion are indicated as φ1, φ2, φ3. | |
The crystalline phases of T6 have also been characterized and different polymorphs have been found according to the preparation conditions. In particular a “low temperature” (LT) process with a prolonged sublimation at low pressure and T comprised between 493 K and 513 K produces a monoclinic (P21/n) structure with Z = 4, i. e. 4 molecules per unit cell,22 while a “high temperature” (HT) controlled melt growth (T between 553 K and 588 K) leads to a P21/a structure with Z = 2.23 Both structures are available from the Cambridge Crystallographic Data Centre.24 Much less attention has been paid to the mesogenic behaviour of sexithiophene. In fact, after a pioneering study by Taliani et al.,10 where it was reported that LT T6 forms a nematic liquid crystal phase when heated above 585 K, and the clearing point was not observed for temperatures up to 623 K, only Destri et al.11 observed the formation of a mesophase from HT T6 crystal heated up to 578 K, but they were not able to assess whether it was a nematic or a smectic one. To the best of our knowledge the high temperature liquid crystal phase of the sexithiophene was not experimentally investigated any further, even if recently similar all-aromatic compounds have attracted some interest for their mesogenic25,26 and semiconducting behaviour.27
Since a lot of effort has been devoted to bulk crystals and thin films, it is surprising that the nature of the liquid crystal phases of T6 and their properties have not been fully examined, e.g. in view of optimizing the intermolecular alignment. Part of the reason can be connected to the difficulty of performing detailed experimental investigations at temperatures around or above 600 K. However, theoretical studies of the mesophases are also lacking, while it has recently been shown28–32 that predictive atomistic simulation can reproduce the nematic–isotropic phase transition temperature of low molecular mass LC such as a cinnamates28 or cyanobiphenyl homologue series29–31 to within a few degrees and predict a variety of physical properties such as density, orientational order, and NMR dipolar couplings to a few percent error.
In this paper we thus wish to provide an atomistic model of T6, taking into account its flexibility through soft thiophene–thiophene torsional potentials, and use it to perform molecular dynamics simulations over a wide set of temperatures to determine its mesophases. We aim at comparing, where possible, simulated and experimental values as well as predicting static and dynamic observables susceptible to experimental determination, such as order parameters and diffusion coefficients. We also investigate the changes in the effective molecular shape that take place in correspondence of the different phases.
2 Force field derivation and testing
The accuracy and predictive capability of a simulation study strongly depends on the quality of the description of the molecular geometry and of intermolecular interactions, which in atomistic simulations are introduced through the molecular mechanics force field (FF) expression and parameters employed. The task of modeling liquid crystal molecules33 is particularly challenging as the different mesophases are generated by a subtle entropy–enthalpy balance which is reflected in a strong dependence of the transition temperatures on small changes in the FF details, e.g. van der Waals parameters31 and also on the system size as it will be shown in the following.
In this study we have relied on well-established parameters (AMBER force field34–37), and complying with a standard procedure37 we have complemented them with atomic partial charges and thiophene–thiophene torsional potentials obtained with quantum chemistry methods. In particular atomic charges have been derived with the electrostatic potential fitting method38 from Gaussian0339 calculations performed at the equilibrium geometry of T6 optimized at the B3LYP//cc-pVTZ level of theory.
The issue of the inter-ring torsional potential and of the attendant equilibrium geometry of oligothienyls has been investigated for a long time,40,41 and their dependence on the surrounding medium also examined.42 The two different sexithiophene (T6) X-ray crystal structures mentioned earlier present perfectly flat geometries: this can be easily understood considering the crystalline packing of the structures and its flattening effect on dihedral angles as also seen for polyphenyls and other oligothiophenes.41,43 2,2′ Dithienyl (T2) has been studied as a solute in condensed LC phases by 1H NMR21 and a detailed maximum entropy analysis44 has shown a preference for a torsional angle of 180°. On the other hand quantum chemical calculations on T2,45–47 subsequently used by Raos and Marcon for crystal cell simulation of T4 and T6,48 suggest that the minima energy geometry for T2 in a vacuum is at a dihedral angle of about 145° (Fig. 2), rather than 180°.
![Torsional potential U(φ) for thiophene–thiophene rotation for dithiophene and sexithiophene calculated at different levels: T2 MP2/aug-cc-pVTZ (red line) and B3LYP//aug-cc-pVTZ (blue dotted line) from reference [46]; T2 MP2/cc-pVTZ (blue squares); T6 terminal dihedral φ3 with B3LYP//cc-pVDZ (black dotted line and empty circles) T6 central dihedral φ1 with B3LYP//cc-pVDZ (black empty squares) and B3LYP//cc-pVTZ (empty cyan triangles).](/image/article/2011/JM/c0jm01284j/c0jm01284j-f2.gif) |
| | Fig. 2 Torsional potential U(φ) for thiophene–thiophene rotation for dithiophene and sexithiophene calculated at different levels: T2 MP2/aug-cc-pVTZ (red line) and B3LYP//aug-cc-pVTZ (blue dotted line) from reference [46]; T2 MP2/cc-pVTZ (blue squares); T6 terminal dihedral φ3 with B3LYP//cc-pVDZ (black dotted line and empty circles) T6 central dihedral φ1 with B3LYP//cc-pVDZ (black empty squares) and B3LYP//cc-pVTZ (empty cyan triangles). | |
However, the barrier profile obtained theoretically was also shown to depend on the level of the Quantum Mechanics (QM) calculations.46,48 To keep into account this issue, in our modelling of T6, aimed at determining the geometry, torsional barriers and charge distribution, we performed several optimizations with different methods and basis sets. First we obtained optimized structures with the Hartree–Fock and B3LYP density functional approaches using the STO-3G, 3-21G, 6-31G, cc-pVDZ and cc-pVTZ basis sets. In all cases the equilibrium angle was found to be 180 degrees, different from dithiophene.46,48 We proceeded then to relaxed scans of the central torsional angle U(φ1) of T6 with B3LYP//cc-pVDZ and cc-pVTZ calculations (Fig. 2, black empty squares and cyan triangles). These confirmed the flat geometry for T6, in agreement also with B3LYP/6-31G** calculations on T6 and other fluoroarene-oligothiophene semiconductors.49 The potential energy surface is indeed very shallow around the minima, both for the SS-anti (φ1 = 180°) and for the SS-syn conformation (φ1 ≈ 22°), which is predicted to be less stable by only 0.68 kcal mol−1, hence relatively easily populated by increasing temperature. The anti–syn barrier is located at about 90 degrees and needs about 3.7 kcal mol−1 to be overcome, while the syn–syn barrier at 0 degree is found to be very weak (approximately 0.1 kcal mol−1 above the most stable SS-syn geometry). We have evaluated also the energy profile for the terminal thienyl–thienyl torsion φ3 (Fig. 2, black circles), finding it rather similar to the φ1 curve with the exception of the anti–syn barrier which decreases to 3.2 kcal mol−1.
In the simulation, we used the U(φ) = U(φ1) potential for describing all the thienyl–thienyl dihedrals, independently from their position along the T6 molecule. To introduce correctly within the force field the ab initio potential it is necessary to evaluate the torsional contributions of the other force field terms and subtract them from the QM profile.50,51 These contributions UC(φ1) have been calculated from the logarithm of the nonbonded torsional angle distribution PC(φ1) obtained from a separate MD simulation of an isolated T6 molecule at 600 K, in which the explicit FF torsional potential term UFF(φi) (fourth column in Table 1) was fixed to zero. The difference between the QM and the nonbonded torsional potential UFF(φ1) = UVTZ(φ1) + kBT ln PC(φ1) has then been fitted with a truncated Fourier series expansion that has been used as the new effective FF torsional potential, whose parameters are reported in Table 1.
Table 1 Fitting parameters for the T6 torsional potentials expressed as a Fourier series U(φi)=∑8n = 1Kncos(nφi): central thiophene–thiophene dihedrals calculated at B3LYP//cc-pVTZ level (φ1 VTZ), end dihedral calculated at B3LYP//cc-pVDZ level (φ3 VDZ), central dihedral as implemented in the molecular mechanics force field after the removal of the non-bonded contribution (φ1 FF)
|
K
n
/kcal mol−1 |
| n |
φ
1 VTZ |
φ
3 VDZ |
φ
1 FF |
| 1 |
+0.295 |
+0.338 |
+0.465 |
| 2 |
−1.669 |
−1.340 |
−2.333 |
| 3 |
+0.090 |
+0.084 |
+0.131 |
| 4 |
+0.502 |
+0.487 |
+0.387 |
| 5 |
0.000 |
+0.020 |
−0.001 |
| 6 |
−0.026 |
−0.018 |
−0.048 |
| 7 |
+0.016 |
0.000 |
0.000 |
| 8 |
+0.030 |
0.000 |
0.000 |
For a first validation of the force field we verified, as in reference [48], its capability of reproducing the crystalline cells of the two different monoclinic polymorphs of T6. We built the different crystallographic cells with the specific tool in Mercury 1.4.2 software,52 and through the replica of a single cell in three dimensions we obtained two small T6 crystals, used as starting configuration for Molecular Dynamics simulations. The simulated LT T6 crystal is a 3 × 6 × 3 replica of the single cell (108 molecules), while the HT T6 cell is replicated 1 × 5 × 7 on each single cell dimension (140 molecules). The LT and HT initial samples used in the MD simulation were equilibrated with the NAMD53 software with simulation time 10 ns and temperatures set to 292 K and 295 K respectively. As shown in Table 2, the properties of the simulated cells are comparable with the experimental ones, and despite the relative simplicity of our potential energy function, the force field demonstrates performances comparable to the ones specifically obtained by Marcon et al.48 and Della Valle et al.54 for these crystalline phases.
Table 2 Simulation results for LT and HT polymorphs of T6
| T6 |
LT polymorph |
HT polymorph |
| expa |
simb |
simc |
simd |
expe |
simb |
simc |
|
Data from reference [22].
data from our simulation.
data from reference [54].
data from reference [48].
data from reference [23].
|
|
ρ/g cm−3 |
1.553 |
1.509 |
1.579 |
1.515 |
1.55 |
1.52 |
1.61 |
| a (Å) |
44.708 |
45.650 |
45.001 |
45.509 |
9.1404 |
8.401 |
8.248 |
| b (Å) |
7.851 |
7.804 |
7.682 |
7.972 |
5.6843 |
5.983 |
5.798 |
| c (Å) |
6.029 |
6.113 |
6.023 |
5.974 |
20.672 |
21.527 |
21.499 |
|
α (°) |
90 |
89.70 |
90 |
90 |
90 |
90.23 |
90 |
|
β (°) |
90.76 |
89.52 |
89.97 |
88.43 |
97.78 |
93.86 |
95.81 |
|
γ (°) |
90 |
89.79 |
90 |
90 |
90 |
90.10 |
90 |
|
T/K |
292 |
292 |
292 |
298 |
295.65 |
295 |
295 |
3 Results and discussion
3.1 Liquid crystal phases
Our first aim is to investigate if anisotropic phases exist between the crystalline and the isotropic one and to classify them. To this end we started heating progressively a LT sample composed of 140 molecules at T > 570 K up to 700 K with a typical production time of 60 ns and a time step of 1 fs. Every sample was equilibrated by simulations in NPT condition (P = 1 atm) using periodic boundary conditions (PBC), Berendsen thermostat and anisotropic Berendsen barostat.55 Electrostatic contributions were evaluated with the Particle Mesh Ewald method56 using a mesh of 1.2 Å and 3rd order splines; the scaling factors for intramolecular 1–4 electrostatic and Lennard Jones interactions were set to 5/6 and to 1/2 respectively.36 It is important to remember here that the FF torsional parameters reported in Table 1 are valid only for this choice of the 1–4 scaling factors.
For these N = 140 molecules samples we found a large (∼20%) change in density at T ∼ 580 K, possibly corresponding to a crystal to smectic transition. However we had difficulty in assessing the smectic character and even in locating the isotropisation temperature, due to the small sample size. For a more detailed investigation we thus prepared and employed larger samples of N = 1120 T6 molecules (49280 atoms) by replicating the small samples at each temperature twice along each dimension and re-equilibrating these for 5 ns; we then performed production runs for further 20 ns. In addition we heated the larger sample at 650 K to even higher temperatures to locate the nematic–isotropic transition temperature, reaching a total production time of ∼20 ns. For all these runs we have characterized the phases obtained in terms of density, order parameter, radial distribution function, energy and translational diffusion coefficient, and studied their variations with temperature. Starting with the density as a function of temperature in Fig. 3 we see evidences of several phase transitions: a first large jump of about 0.2 g cm−3 between 575 K and 580 K, and at higher temperatures an almost monotonical decrease with two small humps around T = 605 K and 665 K. To characterize the nature of the phases we proceeded to examine the orientational order parameter 〈P2〉, that measures the average degree of alignment of a molecular axis u along the preferred direction n (the director):
| | 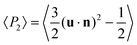 | (1) |
 |
| | Fig. 3 Density of N = 140 and N = 1120 T6 samples as a function of temperature, showing that density is essentially unchanged by the increase in sample size. | |
The angular brackets indicate here an average over all the molecules in the sample to give an instantaneous P2(t) and a time average of this. The choice of the reference molecular direction u is not unique and here we have chosen for the prolate molecule T6 the eigenvector of the instantaneous T6 inertia tensor corresponding to the lowest eigenvalue. Following a well-established procedure in LC simulations57 the scalar order parameter P2 at time t can be determined for each configuration by setting up and diagonalizing the ordering matrix, Q:
| |  | (2) |
where
ui(
t) is the axis of molecule
i,
I is the identity matrix and the sum runs over all the
N molecules of the sample. The instantaneous order parameter
P2(
t) corresponds to the largest eigenvalue of
Q(
t), while the related eigenvector is the instantaneous director
n(
t). Examining the ensemble-averaged orientational order parameter 〈
P2〉, (
Fig. 4) we see two small but appreciable jumps approximately located at 577.5 K and 607.5 K and a more significant and well defined drop between 665 K and 670 K, where the order parameter reaches effectively isotropic values (〈
P2〉 < 0.2) and the clearing transition occurs. It is also interesting to note the system-size effects which determine that these transitions, except for the first one at 577.5 K, are less pronounced and appear at a temperature of about 15 K higher for the small sample than in the larger one.
 |
| | Fig. 4 Orientational order parameter for the N = 140 and N = 1120 T6 samples as a function of temperature, showing the importance of a sufficiently large system size for locating the phase transitions. The snapshots of the different phases correspond, from the left to the right, to simulations at 575, 590, 650 and 685 K respectively. Molecules are color-coded according to the orientation of their reference axis u with respect to the phase director n, ranging from blue (parallel) to red (perpendicular). | |
From the analysis of the density and order parameter temperature trends, three different phase transitions then appear: the one at lower temperature corresponds to the melting of the crystal into a less dense and ordered phase; this phase eventually changes to a slightly less ordered one at about 607.5 K and finally at 665 K (clearing temperature) an isotropic phase appears. To better characterize the nature of the phases obtained, especially the ones intermediate between the crystal and the isotropic one, a knowledge of the positional order and structure can be helpful and we have accessed it as is customary, through the calculation of the radial distribution function (rdf):
| |  | (3) |
where
rij is the distance between T6 centers of mass of molecules
i and
j,
N is the total number of molecules,
V is the volume. From the rdf plots (
Fig. 5) we note, as expected, a very structured distribution corresponding to the crystal phase at 575 K, while at 590 K the sample presents a typical smectic
LC phase rdf. In fact, in this phase we observe the presence of various peaks indicating significant positional order at long range and suggesting the existence of layers. Increasing the temperature we see instead at 625 K and 675 K that the rdf is more liquid-like and only one or two short range peaks, representing correlated shells of molecules, are visible: considering that at 625 K we have also orientational order (
Fig. 4) it is then possible to classify the corresponding phase as a nematic one. We verified that in all ordered phases the short range peak at about 5 Å corresponds to first neighboring molecules in herringbone configuration, with the molecular planes not lying parallel but making an angle of about 60 degrees. The phase that appears at the highest temperature is instead devoid of orientational and positional order—only the first peak of the rdf survives—consistent with an isotropic liquid phase.
 |
| | Fig. 5 Comparison of the radial distribution function of T6 in the different phases: in the main plot, smectic A (cyan) versus nematic (blue) and isotropic (red dashed line); in the inset, smectic (cyan) versus crystal phase (black). | |
Simulations allow us to compute enthalpies as a function of temperature via the textbook equation 〈H(T)〉 = 〈U(T)〉 + p〈V(T)〉, where 〈U(T)〉 and 〈V(T)〉 are the average value of the force field energy and of the box volume at a given temperature and p is the external pressure. From the calculated values of enthalpy we estimated the transition enthalpies (ΔHtrs) and entropies (ΔStrs), reported in Table 3. We notice that the transition entropy value is significant for the crystal–smectic transition corresponding to a strong first order character, while for the other two transitions it is within our experimental uncertainty. These values have been compared with the experimental data present in literature for substituted liquid crystalline oligothiophenes,58,59 finding a qualitative agreement with experiment for these different but related compounds. Moreover, the available experimental data for T6 crystal–mesophase transition enthalpy,11 10–11 kcal mol−1 measured with DSC, is in good agreement with our simulated value of 12.1 kcal mol−1.
Table 3 Values of simulated phase transition temperatures (Ttrs, in K), enthalpies (ΔHtrs, in kcal mol−1) and entropies (ΔStrs, in cal/(mol K)) for sexithiophene
| Transition |
T
trs
|
ΔHtrs |
ΔStrs |
| I–N |
667.5 |
0.4 |
−0.5 |
| N–SmA |
607.5 |
0.2 |
−0.3 |
| SmA–K |
577.5 |
12.1 |
−20.8 |
3.2 Characterization of the smectic phase
The presence of the layered molecular organization typical of smectics is confirmed by the typical sinusoidal trend of the density distribution function ρ(z), where z is the projection of the intermolecular vector along the normal to the layers. The scaled ρ(z) can be expanded in a Fourier series:60–62| | | ρ(z) = ρ0[1 + 〈τ1〉 cos(qSz) + … + 〈τn〉 cos(nqSz)] | (4) |
where ρ0 is the average density, 〈τi〉 = 〈cos(nqSz)〉 are the smectic positional order parameters,  is the wavefactor of the density wave, d the interlayer distance. From Fig. 6 we can notice that indeed the linear density ρ(z) from T = 580 K to T = 605 K follows the oscillatory behaviour predicted by eqn (4), characteristic of a smectic A phase.62 To obtain an estimate of the first non-vanishing smectic parameter 〈τ1〉 and of the layer spacing d we fitted the ρ(z) shown in Fig. 6 with eqn (4) truncated at the first two terms, obtaining the values reported in Table 4. The low root mean square errors of the fit indicate that the first order approximation is very good for this specific system and that the fit parameters are physically meaningful. The layer spacing appears to be constant with temperature, as indicated also by the positions of the peaks of ρ(z) in Fig. 6, and its value of 26.2 Å is comparable with the SS-trans molecular length of 26.5 Å. The good agreement between the periodicity of ρ(z) and the molecular size indicates a negligible interdigitation between the layers. Finally the smectic order parameter 〈τ1〉 decreases gradually with temperature, confirming a second order or very weak first order transition from the smectic to the nematic phase already indicated by the low transition enthalpy in Table 3. A further confirmation of the fluidity and of the anisotropic nature of the high temperature phases obtained can be verified by the translational diffusion tensor components:
is the wavefactor of the density wave, d the interlayer distance. From Fig. 6 we can notice that indeed the linear density ρ(z) from T = 580 K to T = 605 K follows the oscillatory behaviour predicted by eqn (4), characteristic of a smectic A phase.62 To obtain an estimate of the first non-vanishing smectic parameter 〈τ1〉 and of the layer spacing d we fitted the ρ(z) shown in Fig. 6 with eqn (4) truncated at the first two terms, obtaining the values reported in Table 4. The low root mean square errors of the fit indicate that the first order approximation is very good for this specific system and that the fit parameters are physically meaningful. The layer spacing appears to be constant with temperature, as indicated also by the positions of the peaks of ρ(z) in Fig. 6, and its value of 26.2 Å is comparable with the SS-trans molecular length of 26.5 Å. The good agreement between the periodicity of ρ(z) and the molecular size indicates a negligible interdigitation between the layers. Finally the smectic order parameter 〈τ1〉 decreases gradually with temperature, confirming a second order or very weak first order transition from the smectic to the nematic phase already indicated by the low transition enthalpy in Table 3. A further confirmation of the fluidity and of the anisotropic nature of the high temperature phases obtained can be verified by the translational diffusion tensor components:| |  | (5) |
where ri is the component along the axis i = x,y,z of the director frame (the eigenvectors of Q, eqn (2)) of the molecular center of mass position vector for each molecule. The limit for t→∞ in eqn (5) has been in practice approximated with the value for t = 5 ns, and the diffusion coefficients parallel and perpendicular to the director shown in Fig. 7, correspond respectively to Dzz and (Dxx + Dyy)/2, while Diso is calculated as (Dxx + Dyy + Dzz)/3.
 |
| | Fig. 6 Scaled density probability along the alignment direction z showing the existence of smectic layers and the decrease of spatial correlation with increasing temperature. | |
Table 4 Smectic order parameter τ1 and interlayer distance d with relative standard deviation obtained by fitting the density distribution functions with the first two terms on the right hand side of eqn (4)
|
T/K |
τ
1
|
d/Å |
rms |
| 580 |
0.82 |
26.2 |
0.06 |
| 585 |
0.79 |
26.2 |
0.05 |
| 590 |
0.63 |
26.2 |
0.03 |
| 595 |
0.51 |
26.2 |
0.02 |
| 600 |
0.21 |
26.2 |
0.00 |
| 605 |
0.03 |
26.4 |
0.00 |
 |
| | Fig. 7 Translational diffusion coefficients as functions of temperature for the N = 1120 samples: isotropic (Diso = (Dxx + Dyy + Dzz)/3, red squares), perpendicular (D⊥ = (Dxx + Dyy)/2, green circles) and parallel (D|| = Dzz, blue triangles) to the phase director. | |
As expected for a solid phase, the diffusion coefficient is negligible below the melting temperature, and increases with small but clearly visible jumps at the K–SmA, SmA–N and N–I transitions. In particular it exhibits a faster diffusion in the direction parallel to the phase director, not only in the nematic but also in the smectic phase. This dealing, somehow unexpected within the folklore of a smectic as a collection of independent two dimensional layers, but instead typical of the nematic phase, has been nevertheless reported in other smectic A phases, such as the double layered smectic phase exhibited by octyl-cyano-biphenyls63 and the smectic phases produced by suspensions of rod-like viruses.64 Such a behaviour has been related to the extent of positional order and the rigidity of the smectic layers and, maybe more interestingly, to the order of the smectic–nematic phase transition which is generally present at higher temperature:65,66 if this transition is weak (like here), the smectic is believed to exhibit a “nematic-like” diffusional behavior, as seems the case for T6.
To verify that the positional order is not a legacy of the original crystal starting configuration and more generally the thermodynamic stability of the smectic phase, we have attempted also to prepare a smectic sample by cooling from a nematic state point. We thus started from a previously equilibrated configuration at 610 K and we cooled instantaneously to the temperature of 600 K, hence below the smectic–nematic transition temperature obtained by heating runs. After 20 ns, smectic order was achieved, demonstrating that the smectic–nematic transition is reversible and not monotropic and confirming the satisfactory equilibration of previous heating runs. In fact the two simulations show very similar ρ(z), orientational order parameter, density and diffusion coefficients (Table 5).
Table 5 Density, order parameters and translational diffusion coefficients values confirm the reversibility of the smectic–nematic transition for a sample of 1120 T6 molecules at 600 K obtained from heating and cooling runs
| |
ρ (g/cm3) |
〈P2〉 |
〈τ1〉 |
d/Å |
D
⊥ (m2/s) |
D
|| (m2/s) |
| heating |
1.15 ± 0.01 |
0.83 ± 0.02 |
0.21 |
26.3 |
9 × 10−10 |
25 × 10−10 |
| cooling |
1.15 ± 0.01 |
0.82 ± 0.02 |
0.18 |
26.2 |
9 × 10−10 |
25 × 10−10 |
3.3 Molecular shape
The molecular shape is an important parameter in understanding LC phase stability and the change in transition temperatures induced by small chemical modifications.28,31 For instance we have shown by atomistic simulations that the odd–even effect in the transition temperature of the homologue series of cinnamate28 and n-alkyl cyanobiphenyl series31 is related to the effective average molecular length/breadth ratio: its increase leads to an enhancement of the ordered phase stability and thus of the nematic–isotropic transition temperature. The difficulty in using this concept as a predictive tool lies in the need for employing the actual shape of the molecule and in the fact that this will be determined by several possible conformations, differently populated as temperature changes. In T6 the effect is particularly interesting given the relatively weak inter-ring torsional barrier (Fig. 2) and that an occasional syn–anti conversion around one of the bonds connecting two thiophene moieties can lead to a significant geometrical change, and affect the relaxation energies of the neutral and charged molecules which are so relevant for the charge transport on this class of organic semiconductors.67 Indeed in Fig. 8 we note that the probability of finding thiophene molecules with central dihedral angle values different from 180° (linear planar shape) increases with temperature, and that at high T almost all torsional angles become populated.
 |
| | Fig. 8 Distribution of the dihedral φ1 between the central rings of the simulated phases. The average cosine of the angle versus temperature is plotted in the inset. | |
For purely geometrical reasons, given that the thiophene–thiophene bonds are not parallel to the long molecular axis, the torsional disorder is expected to reduce the molecular dimensions. To quantify them we consider the sides rl (l = x, y, z) of the minimal rectangular box containing the molecule rotated in its inertial frame as molecular size indicators, so the average molecular length is defined as 〈rz〉 and molecular breadth as 〈(rx + ry)/2〉.28,31 The average molecular length (Fig. 9, left), having a value of ∼27 Å in the crystal, suddenly lowers to ∼26.6 Å at the smectic transition, and thereafter continues to decrease with a marked temperature dependence, clearly related to the average value of the cosine of central thiophene–thiophene dihedral (inset of Fig. 8). The distribution of the aspect ratio (length/breadth, Fig. 9, right) reflects this behaviour, highlighting the conformational change from a linear to a bent shape. In fact the most probable conformation (peak of the distribution) shifts towards lower values, and the distribution itself gets wider with increasing temperature. In the crystal phase the aspect ratio is ∼4.5; on increasing the temperature a contraction of the effective size anisotropy and a broadening of the distribution is observed.
 |
| | Fig. 9 T6 average length at the simulated temperatures (left) and distribution of molecular length to breadth ratio for the simulated phases (right). The space filling representation of three conformers extracted from the simulation is also shown. | |
The conformational disorder has as further consequences the breaking of the molecular symmetry and the increase of the molecular dipole: this effect, which here is induced by the temperature increase only, has also been attributed as the driving force for T6 adsorption energy on clean gold surfaces.68 In Fig. 10 we report the average value of dipole moment of T6 molecule as a function of the simulated temperatures and we note that this quantity nearly doubles at the crystal–smectic transition. The prevalent component is the one perpendicular to the long molecular axis (||μ||⊥, about 1 Debye in the high temperature phases), suggesting a negative dielectric anisotropy and susceptivity both in the smectic and in the nematic phases, and the possibility of aligning T6 liquid crystalline phases with external electric fields.
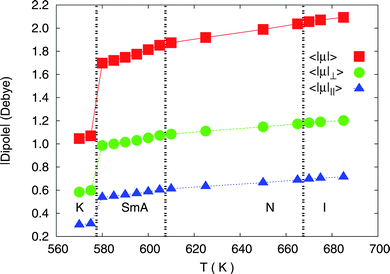 |
| | Fig. 10 Average of the absolute value of total molecular dipole |μ| (red squares), and of its components perpendicular (|μ|⊥, green circles) and parallel (|μ|||, blue triangles) to the long molecular axes, as a function of temperature for the N = 1120 samples. | |
3.4 Cooling liquid crystalline T6 at room temperature
Given that T6 would be inevitably employed in potential organic electronic applications at room temperature, the bent conformations just discussed seem difficult to reconcile with the flat conformations that would be optimal for high mobility. We have thus simulated T6 cooling and examined if the bent structures can be trapped by glass formation, and to test this the hard way we have chosen a very fast cooling that should enhance the conformational trapping. We have cooled from the high temperature LC phases of T6 directly to room temperature (298 K) using as starting configurations the equilibrated samples respectively in nematic (from T6 simulated at 650 K) and in the smectic phase (from T6 at 590 K); the samples have been equilibrated for 40 ns and the observables have been calculated during a further 10 ns run. Both samples reached well ordered configurations with very similar properties (see Table 6), even if less packed and more disordered than the original crystal structure, as can be noticed by comparing the radial distribution functions in Fig. 11, and from the lower value of the density (1.44 of the “glassy” samples vs. 1.53 g cm−3 for the crystal). Nevertheless the snapshots (Fig. 11) reveal a substantial rearrangement of the molecules in a crystal-like packing, suggesting that with more realistic (i.e. slower) cooling rates well formed crystals could be obtained starting from T6 LC phases; the fact that the conformational disorder obtained is very low (〈cos φ1〉 = 0.97) and comparable to the one of the crystal, strengthens this hypothesis.
 |
| | Fig. 11 Radial distribution function of the cooled structures with respect to the crystal phase simulated at 570 K (red). The trends of the new morphologies (green and blue) are similar. The initial and final snapshots of cooling simulation at room temperature obtained from the nematic (top) and smectic phase (bottom) are shown as inset. | |
Table 6 Density, order parameter and molecular shape properties for solid samples obtained by cooling at room temperature (298 K) a smectic (SmA →RT) and a nematic configuration (N →RT), compared with crystal phase results at the same temperature
|
T/K |
ρ (g/cm3) |
〈P2〉 |
〈cos φ1〉 |
length/Å |
length/breath |
| 298 (SmA →RT) |
1.44 |
0.97 |
0.95 |
26.99 |
4.50 |
| 298 (N →RT) |
1.44 |
0.97 |
0.94 |
27.03 |
4.58 |
| 292 (K) |
1.51 |
1.00 |
0.97 |
27.12 |
4.84 |
4 Conclusions
The high temperature phase behaviour of sexithiophene was studied with atomistic Molecular Dynamics simulations, by performing progressive heating of a crystalline sample of 1120 molecules of the “low temperature” polymorph. We detected a phase transition from crystal to liquid crystal at about 580 K, in agreement with experiment;2,10,11 the latter phase shows positional order and can be classified as a smectic A. The transition is associated with a relevant density variation (from 1.4 to 1.2 g cm−3) and to strong conformational changes of T6, namely the molecule in the liquid crystal phase easily assumes a bent shape, deviating from the planar structure typical of the crystal phase. The reversibility of the smectic–nematic transition was assessed as we obtained smectic ordering also by cooling a nematic sample. At 610 K we found another LC phase, nematic, that becomes isotropic at 670 K. We predict values for the order parameters, diffusion coefficients and the average molecular dipole of T6—connected to molecular deformations —as a function of temperature and we hope these findings will stimulate future experimental determinations.
Acknowledgements
The research leading to these results has received funding from the European Commission through the projects MODECOM (FP6 NMP CT-2006-016434), ONE-P (FP7 NMP CT-2008-212311) and MINOTOR (FP7 NMP CT-2009-228424). We acknowledge also INSTM (http://www.instm.it) and CINECA supercomputing center (http://www.cineca.it) for cofunding the access to computational resources through a “High Performance Computing” grant.
References
- D. Fichou, J. Mater. Chem., 2000, 10, 571 RSC.
- H. E. Katz, L. Torsi and A. Dodabalapur, Chem. Mater., 1995, 7, 2235 CrossRef CAS.
- G. Horowitz, Adv. Mater., 1998, 10, 365 CrossRef CAS.
- J. Sakai, T. Taima, T. Yamanari and K. Saito, Sol. Energy Mater. Sol. Cells, 2009, 93, 1149 CrossRef CAS.
- J. Cornil, J.-P. Calbert, D. Beljonne, R. Silbey and J.-L. Brédas, Adv. Mater., 2000, 21, 978 CrossRef.
- H. E. Katz and Z. N. Bao, J. Phys. Chem. B, 1999, 104, 671.
- A. J. Lovinger, D. D. Davis, A. Dodabalapur, H. E. Katz and L. Torsi, Macromolecules, 1996, 29, 4952 CrossRef CAS.
- I. Mcculloch, M. Heeney, C. Bailey, K. Genevicius, I. Macdonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. M. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328 CrossRef CAS.
- M. M. Ling and Z. N. Bao, Chem. Mater., 2004, 16, 4824 CrossRef CAS.
- C. Taliani, R. Zamboni, G. Ruani, S. Rossini and R. Lazzaroni, J. Mol. Electron., 1990, 6, 225 CAS.
- S. Destri, M. Mascherpa and W. Porzio, Adv. Mater., 1993, 5, 43 CrossRef CAS.
- G. Horowitz, M. E. Hajlaoui and R. Hajlaoui, J. Appl. Phys., 2000, 87, 4456 CrossRef CAS.
- F. Dinelli, M. Murgia, P. Levy, M. Cavallini, F. Biscarini and D. M. de Leeuw, Phys. Rev. Lett., 2004, 92, 116802 CrossRef.
- C. D. Dimitrakopoulos and D. J. Mascaro, IBM J. Res. Dev., 2001, 45, 11 CrossRef CAS.
-
M. E. Rose, Handbook of Oligo- and Polythiophenes, Academic, New York, 1998 Search PubMed.
- F. Biscarini, P. Samorì, A. Lauria, P. Ostoja, R. Zamboni, C. Taliani, P. Viville, R. Lazzaroni and J. L. Brédas, Thin Solid Films, 1996, 284, 439 CrossRef.
- M. Kramer and V. Hoffman, Opt. Mater., 1998, 9, 65 CrossRef CAS.
- F. Biscarini, R. Zamboni, P. Samorì, P. Ostoja and C. Taliani, Phys. Rev. B: Condens. Matter, 1995, 52, 14868 CrossRef CAS.
- B. Servet, S. Ries, M. Trotel, P. Alnot, G. Horowitz and F. Garnier, Adv. Mater., 1993, 5, 461 CrossRef CAS.
- N. S. Sariciftci, U. Lemmer, D. Vacar, A. J. Heeger and R. A. I. Janssen, Adv. Mater., 1996, 8, 651 CrossRef CAS.
- L. C. Terbeek, D. S. Zimmerman and E. E. Burnell, Mol. Phys., 1991, 74, 1027 CrossRef CAS.
- G. Horowitz, B. Bachet, A. Yassar, P. Lang, F. Demanze, J. L. Fave and F. Garnier, Chem. Mater., 1995, 7, 1337 CrossRef CAS.
- T. Siegrist, R. M. Fleming, R. C. Haddon, R. A. Laudise, A. J. Lovinger, H. E. Katz, P. Bridenbaugh and D. D. Davis, J. Mol. Res., 1995, 10 Search PubMed.
- Cambridge Crystallographic Data Centre, Cambridge Crystallographic Data Centre, http://www.ccdc.cam.ac.uk/.
- N. A. Zafiropoulos, E.-J. Choi, T. J. Dingemans, W. Lin and E. T. Samulski, Chem. Mater., 2008, 20, 3821 CrossRef CAS.
- S. Kuiper, W. F. Jager, T. J. Dingemans and S. J. Picken, Liq. Cryst., 2009, 36, 389 CrossRef CAS.
- M.-H. Yoon, A. Facchetti, C. E. Stern and T. J. Marks, J. Am. Chem. Soc., 2006, 128, 5792 CrossRef CAS.
- R. Berardi, L. Muccioli and C. Zannoni, ChemPhysChem, 2004, 5, 104 CrossRef CAS.
- L. De Gaetani, G. Prampolini and A. Tani, J. Phys. Chem. B, 2007, 111, 7473 CrossRef CAS.
- M. Cifelli, L. De Gaetani, G. Prampolini and A. Tani, J. Phys. Chem. B, 2008, 112, 9777 CrossRef CAS.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, ChemPhysChem, 2009, 10, 125 CrossRef CAS.
- L. De Gaetani and G. Prampolini, Soft Matter, 2009, 5, 3517–3526 RSC.
- M. R. Wilson, Int. Rev. Phys. Chem., 2005, 24, 421 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. A. Case, U. C. Singh, C. Ghio, G. Alagona, S. Profeta and P. Weiner, J. Am. Chem. Soc., 1984, 106, 765 CrossRef CAS.
- S. J. Weiner, P. A. Kollman, D. T. Nguyen and D. A. Case, J. Comput. Chem., 1986, 7, 230 CrossRef CAS.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179 CrossRef CAS.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comput. Chem., 1990, 11, 431 CrossRef CAS.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision C.02), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. L. Brédas, G. B. Street, B. Thèmans and J. M. André, J. Chem. Phys., 1985, 83, 1323 CrossRef CAS.
- O. A. Gus'kova, P. G. Khalatur and A. R. Khokhlov, Macromol. Theory Simul., 2009, 18, 219 CrossRef CAS and references therein.
- F. Rodriguez-Ropero, J. Casanovas and C. Aleman, Chem. Phys. Lett., 2005, 416, 331 CrossRef CAS.
- D. R. Ferro, W. Porzio, S. Destri, M. Ragazzi and S. Bruckner, Macromol. Theory Simul., 1997, 6, 713 CrossRef CAS.
- R. Berardi, F. Spinozzi and C. Zannoni, Liq. Cryst., 1994, 16, 381 CrossRef CAS.
- C. Alemán, V. M. Domingo, L. Fajarí, L. Juliá and A. Karpfen, J. Org. Chem., 1998, 63, 1041 CrossRef CAS.
- G. Raos, A. Famulari and V. Marcon, Chem. Phys. Lett., 2003, 379, 364 CrossRef CAS.
- J. C. Sancho-Garcia and J. Cornil, J. Chem. Phys., 2004, 121, 3096 CrossRef CAS.
- V. Marcon and G. Raos, J. Phys. Chem. B, 2004, 108, 18053 CrossRef CAS.
- S. E. Koh, C. Risko, D. A. da Silva, O. Kwon, A. Facchetti, J.-L. Brédas, T. J. Marks and M. A. Ratner, Adv. Funct. Mater., 2008, 18, 332–340 CrossRef CAS.
- R. Berardi, G. Cainelli, P. Galletti, D. Giacomini, A. Gualandi, L. Muccioli and C. Zannoni, J. Am. Chem. Soc., 2005, 127, 10699 CrossRef CAS.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, Chem. Phys. Chem., 2010, 11, 1018 CAS.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. K. Kessler, C. F. Macrae, P. McGabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 389 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS.
- A. Brillante, I. Bilotti, F. Biscarini, R. G. Della Valle and E. Venuti, Chem. Phys., 2006, 328, 125 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, A. Di Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. A. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 101, 8577 CrossRef CAS.
-
C. Zannoni, in The Molecular Physics of Liquid Crystals, ed. G. R. Luckhurst and G. W. Gray, Academic Press, London, 1979, ch. 3, pp. 51–83 Search PubMed.
- M. Melucci, F. Favaretto, M. Gazzano, N. Camaioni, P. Maccamagni, P. Ostoja, M. Monari and G. Barbarella, Chem.–Eur. J., 2007, 13, 10046 CrossRef CAS.
- J. Leroy, N. Boucher, M. Sferrazza and Y. H. Geerts, Eur. J. Org. Chem., 2007, 1256 CrossRef CAS.
- W. L. McMillan, Phys. Rev. A: At., Mol., Opt. Phys., 1972, 6, 936 CrossRef CAS.
-
P. G. de Gennes, The Physics of Liquid Crystals, Clarendon Press, Oxford, 1974 Search PubMed.
- R. G. Marguta, E. M. del Rio and E. de Miguel, J. Phys.: Condens. Matter, 2006, 18, 10335 CrossRef CAS.
- S. V. Dvinskikh, I. Furó, H. Zimmermann and A. Maliniak, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2002, 65, 061701 CrossRef CAS.
- M. P. Lettinga and E. Grelet, Phys. Rev. Lett., 2007, 99, 197802 CrossRef.
- O. Oishi and S. Miyajima, J. Magn. Reson., 2003, 160, 74 CrossRef CAS.
- M. Cifelli and C. A. Veracini, Phys. Chem. Chem. Phys., 2005, 7, 3412 RSC.
- D. A. da Silva, V. Coropceanu, D. Fichou, N. E. Gruhn, T. G. Bill, J. Gierschner, J. Cornil and J.-L. Brédas, Philos. Trans. R. Soc. London, Ser. A, 2007, 365, 1435 CrossRef CAS.
- N. Sändig, F. Biscarini and F. Zerbetto, J. Phys. Chem. C, 2008, 112, 19516 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.