Biophysical alteration of the secretory track in β-cells due to molecular overcrowding: the relevance for diabetes
Constantin
Ionescu-Tirgoviste
a and
Florin
Despa
*b
aInstitute of Diabetes “N. Paulescu”, Bucharest, Romania
bDepartment of Pharmacology, University of California Davis, Davis, CA 95616. E-mail: fdespa@ucdavis.edu
First published on 21st December 2010
Abstract
Recent data demonstrate that accumulation of misfolded proteins within the early part of the secretory track of β-cells causes impaired insulin synthesis and development of diabetes. The molecular mechanism of this cellular dysfunction remains largely unknown. Using basic molecular principles and computer simulations, we suggested recently that hyperglycemic conditions can generate substantial molecular crowding effects in the secretory track of β-cells leading to significant alterations of the insulin biosynthesis capabilities. Here, we review the major molecular mechanisms that may be implicated in the alteration of insulin synthesis in susceptible β-cells. Steric repulsions and volume exclusion in the endoplasmic reticulum (ER) increase the propensity of misfolding of proinsulin (the precursor molecule of insulin). In addition, similar forces might act in the next secretory compartments (Golgi and vesicles) leading to (i) altered packaging of proinsulin in vesicles (ii) entrapment of proinsulin convertases and/or restricted accessibility for these convertases to the cleavage sites on the surface of the proinsulin and (iii) depressed kinetic rate of the transformation of the native proinsulin in active insulin and C-peptide. These concepts are expressed in simple mathematical terms relating the kinetic coefficient of proinsulin to insulin conversion to the levels of proinsulin misfolding and hyperglycemic stress. The present approach is useful for understanding molecular phenomena associated with the pathogenesis of diabetes. It also offers practical means for predicting the state of pancreatic β-cells from measurements of the insulin to proinsulin ratio in the blood. This is of immediate clinical relevance and may improve the diagnosis of diabetes.
Insight, innovation, integrationWe present a review of recent advances on understanding molecular mechanisms implicated in the alteration of insulin synthesis in susceptible β-cells and development of diabetes. Using basic molecular principles, computer simulations and published experimental data we suggest that hyperglycemic conditions can generate substantial molecular crowding effects in the secretory track of β-cells leading to significant alterations of the insulin biosynthesis capabilities. Our approach is highly innovative in the attempt to show that molecular crowding theory can be used to identify chemistry mechanisms involved in the β-cell pathology. We point out how this approach can help us to identify defects in translational mechanisms underlying the biosynthesis of insulin in β-cells and to elucidate the chemistry aspects of this secretory defect. We also discuss how effective computational models could prove very informative in a clinical setup for prediction and assessment of diabetic states in susceptible patients. |
1. Introduction
Insulin is the final product of a sequential biochemical process (see, for review, Steiner et al.1). Initially, the insulin mRNA is translated as a single chain precursor called preproinsulin (PP). PP undergoes a series of post-translational modifications which are required for progression to the active insulin (I). Removal of its signal peptide during insertion into the ER generates the proinsulin (P) molecule. Within the ER, the P molecules are folded in their native states, which are then exported to the Golgi apparatus and packaged in secretory vesicles. The successful conversion of proinsulin to insulin requires the detachment of the Cpeptide (CP) chain from the single-stranded polypeptideP. The process is mediated by PC2 and PC3, two endoproteases which are packaged together with the P molecules in the secretory vesicles.Following the dissociation of proinsulin to insulin and Cpeptide, the total macromolecular surface area exposed to the surrounding cellular environment becomes larger. Basic molecular principles demonstrate that equilibrium and kinetics of such reactions are susceptible to major changes due to steric repulsions and volume exclusion.2–10 Therefore, we hypothesized that the conversion of native proinsulin to insulin in a crowded environment has a depressed rate.8–10 The conversion rate can be decreased even further by the entrapment of the two convertases within the crowded secretory track and/or by restricting the accessibility to the cleavage sites on the surface of the proinsulin. Based on similar concepts of the molecular crowding theory, we argued that crowding forces can enhance the propensity of misfolding of the proinsulin precursor.8,9 These aspects may have some implications in the pathogenesis of diabetes and will be discussed in the context of several seminal experimental results reported recently.11–15
In β-cells, insulin represents a significant part of the total protein biosynthesized during functional stimulation by glucose.1 Therefore, the main contributors to molecular crowding within the secretory compartments of these cells are insulin and the precursor molecule, proinsulin. The common source of molecular crowding in β-cells may be the increased translational regulation of proinsulin during a sustained functional stimulation by glucose.16–18 While proinsulin molecular chain is rapidly formed in the ER, the conformational maturation of newly formed proinsulin requires a much longer time, of about 10 min.19 Therefore, the accumulation of proinsulin in the endoplasmic reticulum (ER) may quickly exceed the normal level. Inherently, placing an enormous burden on the ER enhances the probability of protein to misfold.8,9 Accumulation of misfolded proinsulin in the secretory track represents a source of crowding in beta cells. An increasing amount of experimental data11–15,20 indicates that dysfunctional β-cells, i.e.cells associated with the pathogenesis of diabetes, contain substantial amounts of misfolded proinsulin. The experiments revealed that (i) misfolded proinsulin accumulates in the ER, pre-Golgi intermediates and Golgi apparatus,11,15 (ii) the volume density in these compartments increases by 3–5-fold11 and (iii) misfolded proinsulin molecules are prone to aggregation12via hydrophobic interactions.21 All these data11–13,20,22,23 clearly indicate that misfolded proinsulin molecules and their aggregates represent a source of molecular crowding in susceptible β-cells. Other intracellular crowding agents, such as preamyloid oligomers have also been detected in dysfunctional β-cells.24 These toxic residues are formed by aggregation of amylin (also known as islet amyloid polypeptide; IAPP), a polypeptide cosecreted with insulin. IAPP amyloids represent a common feature in the pathogenesis of type 2 diabetes mellitus.25,26 We suggested that prolonged stimulation of the β-cells to increase the biosynthesis of insulin (and islet amyloid polypeptide; IAPP) can lead to a gradual impairment of the processing of their precursor molecules (proinsulin and proamylin) in the ER.1 Moreover, the increase of IAPP production can lead to formation of harmful IAPP preamyloid oligomers, which may accumulate in pancreatic islets24,27 and/or secreted in the blood.27 Increased secretion of amyloidogenic IAPP species in the blood can favor the formation of IAPP amyloids in the heart, causing major cardiac dysfunction.27
Experimental data suggest that molecular overcrowding may be a source of various stresses culminating in β-cell dysfunction.13,15–18,22,23,28–32 Recent data13,22 suggest that β-cells containing proinsulin aggregates are predisposed to the ER stress. Apparently, the ER stress may have major implications in β-cell dysfunction and development of diabetes.23,28–32 Therefore, a careful investigation of the chemical effect of crowding induced by the accumulation of toxic residues in β-cells can be useful for a better understanding of the molecular aspects involved in the pathogenesis of diabetes. This is precisely the focus of the present review. We discuss possible molecular mechanisms triggered by increased crowding effects that may interfere with the efficiency of β-cells to synthesize native proinsulin and insulin. We also summarize the mathematical approach connecting the efficiency of proinsulin to insulin conversion (r) to the state of molecular crowding in the secretory pathway.
Deciphering the consequences of the aberrant folding of proinsulin and the consequent accumulation of toxic residues in β-cells may have therapeutic implications,23,33 since besides improvement of the delivery of exogenous insulin, treatment should also aim at removing toxic residues, resuscitating injured cells and reestablishing the insulin synthesis to normal.34–36
2. Results and discussion
The efficiency of β-cells to convert proinsulin to insulin depends critically on the folding capabilities of the ER, including available space for protein folding and rapid clearance of misfolded proteins to avoid accumulation of toxic debris. It also depends on the efficient transport of folded proinsulin to the next secretory compartment (Golgi apparatus) and proper packing in vesicles. Our stochastic numerical simulations8–10 suggest that, under conditions in which the volume (ΔV) available for processing additional proinsulin molecules drops to zero (ΔV → 0), molecular crowding effects ensue, altering the insulin production. In Fig. 1, we predict the relative departure (Δr) of the kinetic coefficient of proinsulin to insulin conversion (r) under hyperglycemic stress from the value r0 corresponding to ideal proinsulin processing conditions,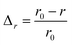 . Here, ideal secretory capabilities are defined as those conditions in which all proinsulin molecules are correctly folded and packed in vesicles. An upsurge of the proinsulin load (W ≫ ΔV) alters the efficiency of proinsulin to insulin conversion (Fig. 1).10 Here, W represents the volume occupied by a given amount (NP) of additional proinsulin molecules. Measurements37,38 of the proinsulin to insulin conversion in normal β-cells revealed high percentage for the conversion coefficient (r). However, the secretory function in β-cells cannot reach the ideal level (i.e. r < r0), as secretion of unprocessed proinsulin in the blood occurs frequently. Within circumstances assumed by the molecular crowding theory, r/r0 < 1 in normal β-cells is a consequence of the volume exclusion effects within the finite space of the secretory compartments.
. Here, ideal secretory capabilities are defined as those conditions in which all proinsulin molecules are correctly folded and packed in vesicles. An upsurge of the proinsulin load (W ≫ ΔV) alters the efficiency of proinsulin to insulin conversion (Fig. 1).10 Here, W represents the volume occupied by a given amount (NP) of additional proinsulin molecules. Measurements37,38 of the proinsulin to insulin conversion in normal β-cells revealed high percentage for the conversion coefficient (r). However, the secretory function in β-cells cannot reach the ideal level (i.e. r < r0), as secretion of unprocessed proinsulin in the blood occurs frequently. Within circumstances assumed by the molecular crowding theory, r/r0 < 1 in normal β-cells is a consequence of the volume exclusion effects within the finite space of the secretory compartments.
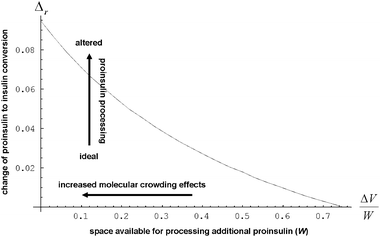 | ||
Fig. 1 The behavior of 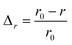 as a function of as a function of 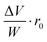 is assumed to describe the proinsulin to insulin conversion under ideal conditions in beta cells, in which the level of protein misfolding is very low (KP > 10) and the volume available in the secretory compartments for newly synthesized proinsulin is large ΔV ≫ W. For the present computation, we approximated the ratio between the molecular volumes of native and misfolded proinsulin by is assumed to describe the proinsulin to insulin conversion under ideal conditions in beta cells, in which the level of protein misfolding is very low (KP > 10) and the volume available in the secretory compartments for newly synthesized proinsulin is large ΔV ≫ W. For the present computation, we approximated the ratio between the molecular volumes of native and misfolded proinsulin by 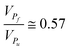 .12 We also assumed that the volume of a secretory compartment (i.e.vesicle) is much larger than that of proinsulin molecule .12 We also assumed that the volume of a secretory compartment (i.e.vesicle) is much larger than that of proinsulin molecule 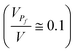 . We estimated based on scaled theory that the overall increase of the partial volume corresponding to the product molecules I and CP in comparison with the partial volume corresponding to the precursor molecule P is about 20%, so that VPf ≅ 1.2VI+CP. . We estimated based on scaled theory that the overall increase of the partial volume corresponding to the product molecules I and CP in comparison with the partial volume corresponding to the precursor molecule P is about 20%, so that VPf ≅ 1.2VI+CP. | ||
Dramatic volume exclusions affecting secretory function of β-cells can also be expected from the presence of “inert” bodies, such as islet amyloid polypeptide oligomers24,39–41 in the secretory pathway of proinsulin. From Fig. 1, we infer that the presence of such crowding agents in the end part of the secretory track, i.e. in vesicles,9,24 may affect the conversion of proinsulin to insulin.
In Fig. 2, we predict the departure of the kinetic coefficient of insulin synthesis in β-cells susceptible to significant accumulation of misfolded proinsulin molecules in the secretory track. In computations,8–10 we assumed that the population density number within the proinsulin misfolded states (CPm) is large. We can see that Δr increases gradually with the accumulation of misfolded proinsulin, CPm → CPf, where CPf stands for the population density number within the proinsulin normally folded states. We can notice that (CPm = CPf) represents the starting point of an asymptotic increase of the values of Δr, which corresponds to a marked decrease of the insulin production. Increased levels of misfolded proteins (CPm > CPf) can generate steric repulsions and volume exclusion to the limit at which the insulin biosynthesis will stop.10
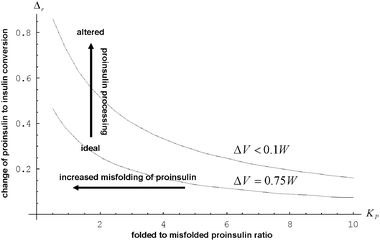 | ||
Fig. 2 The behavior of 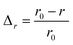 as a function of the fraction of misfolded proinsulin KP for ΔV = 0.75 W (lower curve) and for ΔV = 0 (upper curve), respectively. All other parameters remained at the same values as above. as a function of the fraction of misfolded proinsulin KP for ΔV = 0.75 W (lower curve) and for ΔV = 0 (upper curve), respectively. All other parameters remained at the same values as above. | ||
Experiments with proteins in solution demonstrated that the accumulation of large quantities of misfolded proteins (CPm > CPf) leads inherently to a rapid protein aggregation.42,43 It has been shown that aggregates of misfolded proinsulin may increase in time by incorporating other misfolded or native proinsulin.12,22 Misfolding of proinsulin and formation of protein aggregates compete with native proinsulin for available space in the local cellular environment.8,9 The molecular crowding approach8–10 allow us to investigate the efficiency of proinsulin to insulin conversion under extreme crowding conditions generated by severe hyperglycemic stress, which increases the proinsulin load (W→ΔV), and significant accumulation of misfolded proinsulin in the secretory track (CPm ≫ CPf). We can infer from Fig. 2 (see the upper curve) that such extremely adverse conditions for β-cell function could generate dramatic effects on the insulin output. For instance, a level of 50% protein denaturation (CPm = CPf), which is the (ex vivo) threshold for massive protein aggregation,42 can drastically decrease the synthesis of insulin (Δr > 65%).
These results reveal interesting chemistry features of the β-cell pathophysiology under hyperglycemic conditions.8,9 Thus, intense functional stimulation of β-cells by blood glucose can cause the overload of ER, which may trigger failure of protein folding capabilities8,9 and downstream pathogenic signaling cascades, including ER stress and apoptosis.22
3. Theoretical
Molecular crowding concepts2–4 are derived from the principle that an enhanced volume density of the local environment results in reduced configuration space and distribution of states (less entropy) of the reactant macromolecules in comparison with the macromolecular products. Therefore, the overall entropy loss is lower, which leads to more significant decrease in free energy and higher equilibrium constants for the reaction. This can have a major effect on all processes with a change in excluded volume, such as protein folding, unfolding and aggregation processes.5–10,43–46The effect of crowding on the behavior of the macromolecules of a certain type j in a multicomponent system can be assessed in terms of their apparent activity coefficient γj. γj measures the excess chemical potential of these macromolecules due to the interactions between a newly added macromoleculej in the local environment and all the other components. Using the scaled particle theory and hard particle approximation,47,48γj can be derived as a function of the average molecular volume of the species j (Vj), the average molecular volume of the crowding agent (Vk) and the volume fraction of the crowding agent (f)
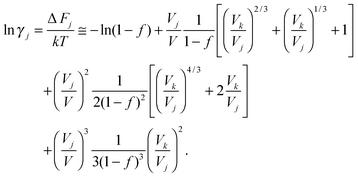 | (1) |
3.1 Kinetic coefficients of proinsulin to insulin conversion under crowding conditions
Let us consider a compartment of volume V within the end part of the secretory pathway of β-cells that contains NP proinsulin molecules. These molecules can be either in native or misfolded states (Fig. 3a). The kinetic coefficient of the proinsulin transition between native and misfolded states at equilibrium is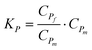 and CPf represent the population density numbers of the two species, which obey the following equation, CPm + CPf = 1. The molecular volume of a native P molecule is VPf and that corresponding to a misfolded one is VPm. We assume that the volume of the secretory compartment can be written as V ≅ NPVPmCPm + NpVPf + ΔV. ΔV represents the space available for adding new proinsulin molecules in the local environment which, in normal β-cells, is much larger than VPf, ΔV ≫ VPf. In contrast, β-cells under critical conditions accumulate toxic residues, which will quickly lead to a decrease of the volume available for proinsulin molecules and ΔV → 0, in this case.
and CPf represent the population density numbers of the two species, which obey the following equation, CPm + CPf = 1. The molecular volume of a native P molecule is VPf and that corresponding to a misfolded one is VPm. We assume that the volume of the secretory compartment can be written as V ≅ NPVPmCPm + NpVPf + ΔV. ΔV represents the space available for adding new proinsulin molecules in the local environment which, in normal β-cells, is much larger than VPf, ΔV ≫ VPf. In contrast, β-cells under critical conditions accumulate toxic residues, which will quickly lead to a decrease of the volume available for proinsulin molecules and ΔV → 0, in this case.
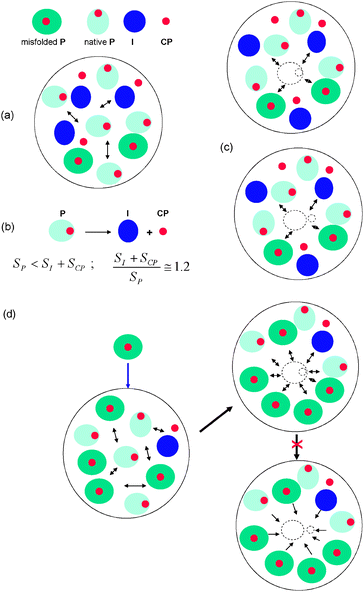 | ||
| Fig. 3 Pictorial representation of the molecular crowding effect on the proinsulin to insulin conversion. (a) In a crowded environment, molecules undergo steric repulsions. (b) Following the dissociation of P in I and CP, the total molecular surface area exposed to the surrounding environment increases; the surface area of I and CP is larger than that of single P. (c) Within circumstances assumed by the molecular crowding theory r/r0 < 1, which is a consequence of the volume exclusion effects within the finite space of the secretory compartment (i.e.vesicles). (d) Accumulation of misfolded proinsulin increases the level of molecular crowding leading to enhanced steric repulsions between molecules and volume exclusion. The activity coefficient of proinsulin is decreased which, in turn, leads to a decrease of proinsulin to insulin conversion. | ||
Let fPm be the volume fraction occupied by the misfolded proinsulin in the volume V, which can be written as
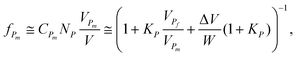 | (2) |
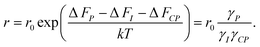 | (3) |
Eqn (1)–(3) provide the mathematical correlation between the molecular crowding effects associated with an accumulation of misfolded proinsulin in the secretory pathway and the efficacy of β-cells to convert the remaining, native proinsulin to insulin.
3.2 Mechanisms underlying altered proinsulin to insulin conversions by molecular crowding forces
Based on eqn (1), it is not too difficult to understand that, in an overcrowded environment, folding pathways that involve transition states49 having small volumes will prevail over those requiring larger volumes. Thus, if the transition state leading to native proinsulin has a larger volume than that of the misfolded proinsulin, the proper precursor of the folded proinsulin (i.e. the unfolded proinsulin) tends to misfold under crowding conditions. Under such circumstances, the presence in the ER of toxic residues (misfolded proinsulins) that increase the local molecular crowding will be a source of an accelerated denaturation of proinsulin. Such crowding effects can be associated with a sustained functional stimulation of β-cells by glucose that favors proinsulin misfolding.8,9Similar molecular crowding considerations apply to the dissociation of native proinsulin into insulin molecule and C peptide (P → I + CP). In this chemical reaction (Fig. 3b and c), the total macromolecular surface area exposed to the local environment (SI+SCP) corresponding to the two product molecules (I and CP) is larger than the surface area (SP) exposed by the precursor molecule (P), SI+SCP > SP. Therefore, in accordance with eqn (1), we obtain γP < γIγCP, which means that an increase of the molecular crowding (f→1) will favor preservation of the intact form of proinsulin rather than splitting it in two parts (Fig. 3d).
Moreover, an increased crowding can obstruct targeting P molecules by the endoproteases PC2 and PC3 which detach the Cpeptide within the secretory vesicles. Misfolded proinsulin, which cannot be clipped to become active insulin, may act as a crowding agent interfering with the conversion of native proinsulin to insulin.
4. Conclusion
Starting from basic molecular principles, we derived simple mathematical correlations between the increase of the molecular crowding in beta cells and decrease of the kinetic coefficient of the conversion of native proinsulin to insulin. Our studies8–10 suggest that steric effects and volume exclusion generated by an accumulation of misfolded proinsulin in secretory compartments can reduce the activity coefficient of the native proinsulin and preclude the transformation in insulin. Moreover, the proper precursor of the natively folded proinsulin (i.e. the unfolded proinsulin) may also tend to misfold under crowding conditions. Additional crowding agents, such as preamyloid IAPP oligomers and other intracellular toxic residues, are likely to contribute to this effect as well. Therefore, effects of cellular crowding on the efficiency of beta cells to produce insulin represent a general feature in the pathology of diabetes.According to the present results, the immediate physiological consequence of an increased crowding in β-cells would be a low kinetic coefficient of the proinsulin to insulin conversion and formation of immature insulin vesicles. Stimulation of β-cells containing immature insulin vesicles and subsequent secretion of these vesicles increases the level of intact proinsulin in the blood. Elevated levels of proinsulin have been found both at the onset of type 1 diabetes50–54 and in type 2 diabetes,54–71 as well. Two hypotheses have been suggested to explain the increased amount of proinsulin in the blood:72,73 (i) a dysfunction of the enzymatic proinsulin processing mechanism and (ii) lack of maturation of insulin vesicles due to an intense hyperglycemic stimulation. Predictions derived from our computer simulations8–10 support these hypotheses and indicate the occurrence of crowding effects as the main source of the altered proinsulin to insulin conversion. The theory predicts that the secretion of intact proinsulin from β-cells increases progressively with the intracellular accumulation of toxic residues, i.e. with the development of the disease, which is an agreement with general clinic observations.74–76
Apparently, β-cells containing misfolded proinsulin are predisposed to ER stress.22ER stress usually enhances the propensity of proteins to misfold and aggregate (reviewed by Hayden et al.),23 thereby accelerating the induction of crowding effects in secretory compartments. Numerous studies have indicated the stress on ER as the main cause of β-cell dysfunction leading to the development of diabetes.23,28–32 It was demonstrated23,29–32 that β-cells respond to an ER stress by activating the unfolding protein response (UPR). This is part of a complex mechanism by which cells limit or repair the molecular damage. The mechanism involves an increased synthesis of molecular chaperones which can protect unfolded proteins to aggregate or target misfolded proteins for degradation. If the ER stress is prolonged, or the adaptive response fails, apoptosis is triggered.32,39,40 The presence of considerable amounts of protein aggregates in apoptotic cells39,40 suggest that the decrease of β-cell mass is a consequence of the failure of the protein regulation mechanism.77 Our results8–10 suggest that an increase of the quantity of misfolded proinsulin to about 50% coincides with a sharp decrease of the conversion of the remaining, native proinsulin to insulin (<30% of synthesized proinsulin). It is known that 50% denatured proteins in a local environment leads to the onset of massive protein aggregation,42,43 which triggers cell apoptosis.39,40 This implies that the induction of apoptosis in beta cells by protein aggregation mechanisms could correlate with the occurrence of a sudden drop (Δr > 0.6) of β-cell efficiency to convert proinsulin to insulin. These findings are consistent with recent experimental results showing that supranormal production of nonnative proinsulin may predispose to cell toxicity and premature loss of pancreatic β-cell.53,78–80 Establishing a correlation between the level of proinsulin to insulin conversion (r) and loss of β-cell mass is of immediate clinical relevance and may improve the diagnosis of diabetes.81 In addition, results of the present study imply that the efficiency of drugs used to stimulate the production of insulin from malfunctioning β-cells declines in time. Rather, therapeutic strategies should focus on removing toxic β-cell residues and reestablishing the insulin synthesis to normal.34,36
The present study suggests that even a temporary increase of the frequency of mutations in the insulin gene, or other transient replication errors, may have actually long term consequences on β-cell function. Thus, if the increase of proinsulin misfolding events cannot be handled efficiently by the intrinsic cellular repair mechanism11–14 then, the accumulated toxic residues in the secretory pathway can decrease the chemical activity of the native proinsulin and interfere with the conversion to insulin. In normal β-cells, where the level of proinsulin misfolding is assumed to be within the physiological limit, a sustained functional stimulation by glucose may actually exceed proinsulin misfolding due to crowding effects. Thus, under conditions in which the volume available in the ER for newly synthesized proinsulin vanishes to zero, the precursor of the folded proinsulin may have the tendency to misfold, as previously suggested.8–10 Further studies are required to elucidate whether the volume of the transition state leading to the folded structure is larger than that corresponding to the misfolded one.
Appendix 1: List of mathematical symbols and definitions
r—the rate of proinsulin to insulin conversionr 0—the rate of proinsulin to insulin conversion corresponding to ideal (crowding free) processing conditions
Δr—the relative departure of the actual rate of proinsulin to insulin conversion (r) under crowding conditions from that corresponding to proinsulin to insulin conversion under crowding free conditions (r0), 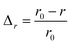
ΔV—the volume available in the ER for processing additional proinsulin molecules
N P —the amount of proinsulin molecules in a given secretory compartment
W—the volume occupied by a given amount (NP) of additional proinsulin molecules in the ER
V—the volume of given compartment of the secretory pathway
C P f —the population density number within the proinsulin normally folded states.
C P m —the population density number within the proinsulin misfolded states
V P f —the molecular volume of a native proinsulin molecule
V P m —the molecular volume of proinsulin in misfolded state
K P —the kinetic coefficient of the proinsulin transition between native and misfolded states at equilibrium
f P m —the volume fraction occupied by the misfolded proinsulin in the volume V
γ j —the apparent activity coefficient of the macromolecules of a given type j in a multicomponent system
γ P —the apparent activity coefficient of proinsulin
γ I —the apparent activity coefficient of insulin
γ CP —the apparent activity coefficient of C peptide
S P —surface area exposed by proinsulin
S I —surface area exposed by insulin
S CP —surface area exposed by C peptide
V k —the average molecular volume of the crowding agent
f—the volume fraction of the crowding agent
F j —the Helmholtz function
ΔFP—the variation of Helmholtz function due to the addition in the local environment of native proinsulin
ΔFI—the variation of Helmholtz function due to the addition in the local environment of insulin
ΔFCP—the variation of Helmholtz function due to the addition in the local environment of C peptide
kT—the thermal energy.
Acknowledgements
FD acknowledges grant support from American Heart Association and 2010 Vision Grant Award from the University of California, Davis.References
- W. Kemmler, J. D. Peterson, A. H. Rubenstein and D. F. Steiner, Diabetes, 1972, 21, 572–581 CAS.
- J. R. Ellis, Curr. Opin. Struct. Biol., 2001, 11, 114–119 CrossRef CAS.
- J. R. Ellis and A. P. Minton, Nature, 2003, 425, 27–28 CrossRef CAS.
- D. Hall and A. P. Minton, Biochim. Biophys. Acta, Proteins Proteomics, 2003, 1649, 127–139 CrossRef CAS.
- S. Cheung, D. K. Klimov and D. Thirumalai, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 4753–4758 CrossRef CAS.
- F. Despa, D. P. Orgill and R. C. Lee, Ann. Biomed. Eng., 2005, 33, 1125–1131 CrossRef.
- F. Despa, D. P. Orgill and R. C. Lee, Ann. N. Y. Acad. Sci., 2005, 1066, 54–66 CrossRef CAS.
- F. Despa, Biophys. Chem., 2009, 140, 115–121 CrossRef CAS.
- F. Despa, Biophys. J., 2010, 98, 1641–1648 CrossRef CAS.
- F. Despa and R. S. Berry, J. Diabetes Sci. Technol., 2010, 4 Search PubMed.
- C. Zuber, J. Y. Fan, B. Guhl and J. Roth, FASEB J., 2004, 18, 917–919 CAS.
- T. Yoshinaga, K. Nakatome, J. Nozaki, M. Naitoh, J. Hoseki, H. Kubota, K. Nagata and A. Koizumi, Biol. Chem., 2005, 386, 1077–1085 CrossRef CAS.
- M. Liu, I. Hodish, C. J. Rhodes and P. Arvan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15841–15846 CrossRef CAS.
- J. Støy, E. L. Edghill, S. E. Flanagan, H. Ye, V. P. Paz, A. Pluzhnikov, J. E. Below, M. G. Hayes, N. J. Cox, G. M. Lipkind, R. B. Lipton, S. A. Greeley, A. M. Patch, S. Ellard, D. F. Steiner, A. T. Hattersley, L. H. Philipson and G. I. Bell, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 15040–15044 CrossRef.
- S. Y. Park, H. Ye, D. F. Steiner and G. I. Bell, Biochem. Biophys. Res. Commun., 2010, 391, 1449–1454 CrossRef CAS.
- C. Alarcón, B. Lincoln and C. J. Rhodes, J. Biol. Chem., 1993, 268, 4276–4280 CAS.
- C. Alarćon, J. L. Leahy, G. T. Schuppin and C. J. Rhodes, J. Clin. Invest., 1995, 95, 1032–1039 CrossRef CAS.
- G. T. Schuppin and C. J. Rhodes, Biochem. J., 1996, 313, 259–268 CAS.
- X. F. Huang and P. Arvan, J. Biol. Chem., 1995, 270, 20417–20423 CrossRef CAS.
- T. Izumi, H. Yokota-Hashimoto, Z. Shengli, J. Wang, P. A. Halban and T. Takeuchi, Diabetes, 2003, 52, 409–416 CAS.
- F. Despa and R. S. Berry, Biophys. J., 2007, 92, 373–378 CAS.
- J. Nozaki, H. Kubota, H. Yoshida, M. Naitoh, T. Yoshinaga, K. Mori, A. Koizumi and K. Nagata, Genes Cells, 2004, 9, 261–270 CrossRef CAS.
- M. R. Hayden, S. C. Tyagi, M. M. Kerklo and M. R. Nicolls, JOP. J. Pancreas, 2005, 6, 287–302 Search PubMed.
- C. Y. Lin, T. Gurlo and R. Kayed, et al. , Diabetes, 2007, 56, 1324–1332 CrossRef CAS.
- P. Westermark, Z. C. Li, G. T. Westermark, A. Leckstrom and D. F. Steiner, FEBS Lett., 1996, 379, 203–206 CrossRef CAS.
- A. E. Butler, J. Janson, W. C. Soeller and P. C. Butler, Diabetes, 2003, 52, 2304–2314 CrossRef CAS.
- S. Despa, L. Chen, B. Cummings, P. J. Havel, K. B. Margulies, A. A. Knowlton, D. M. Bers and F. Despa, Circulation, 2009, 120, S457.
- H. P. Harding and D. Ron, Diabetes, 2002, 51, S455–S461 CrossRef CAS.
- S. Oyadomari, E. Araki and M. Mori, Apoptosis, 2002, 7, 335–345 Search PubMed.
- E. Araki, S. Oyadomari and M. Mori, Intern. Med., 2003, 42, 7–14 Search PubMed.
- P. Pirot, D. L. Eizirik and A. K. Cardozo, Diabetologia, 2006, 49, 1229–1236 CrossRef CAS.
- D. R. Laybutt, A. M. Preston, M. C. Akerfeldt, J. G. Kench, A. K. Busch, A. V. Biankin and T. J. Biden, Diabetologia, 2007, 50, 752–763 CrossRef CAS.
- R. W. Carrell and D. A. Lomas, Lancet, 1997, 350, 134–138 CrossRef CAS.
- V. M. Rivera, X. Wang, S. Wardwell, N. L. Courage, A. Volchuk, T. Keenan, D. A. Holt, M. Gilman, L. Orci, F. Jr. Cerasoli, J. E. Rothman and T. Clackson, Science, 2000, 287, 826–830 CrossRef CAS.
- B. Solomon, Curr. Med. Chem., 2002, 9, 1737–1749 CAS.
- E. De Lorenzi, S. Giorgetti, S. Grossi, G. Merlini, G. Caccialanza and V. Bellotti, Curr. Med. Chem., 2004, 11, 1065–1084 CrossRef CAS.
- C. J. Rhodes and P. A. Halban, J. Cell Biol., 1987, 105, 145–153 CrossRef CAS.
- S. Sizonenko, J. C. Irminger, L. Buhler, S. Deng, P. Morel and P. Halban, Diabetes, 1993, 42, 933–936 CrossRef CAS.
- J. Janson, R. H. Ashley, D. Harrison, S. McIntyre and P. C. Butler, Diabetes, 1999, 48, 491–498 CrossRef CAS.
- R. A. Ritzel and P. C. Butler, Diabetes, 2003, 52, 1701–1708 CrossRef CAS.
- A. E. Butler, J. Jang, T. Gurlo, M. D. Carty, W. C. Soeller and P. C. Butler, Diabetes, 2004, 53, 1509–1516 CrossRef CAS.
- G. I. Makhatadze and P. L. Privalov, Adv. Protein Chem., 1995, 47, 307–425 CAS.
- B. van Den Berg, C. M. Dobson and R. J. Ellis, EMBO J., 1999, 18, 6927–6933 CrossRef CAS.
- G. Rivas, J. A. Fernandez and A. P. Minton, Biochemistry, 1999, 38, 9379–9388 CrossRef CAS.
- G. Rivas, J. A. Fernandez and A. P. Minton, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3150–3155 CrossRef CAS.
- R. Engel, A. H. Westphal, D. H. E. W. Huberts, S. M. Nabuurs, S. Lindhoud, A. J. W. G. Visser and C. P. M. van Mierlo, J. Biol. Chem., 2008, 283, 27383–27394 CrossRef CAS.
- J. L. Lebowitz, E. Helfand and E. Praestgaard, J. Chem. Phys., 1965, 43, 774–779 CAS.
- T. Boublìk, Mol. Phys., 1974, 27, 1415–1427 CAS.
- F. Despa and R. S. Berry, J. Chem. Phys., 2004, 120, 5164–5168 CrossRef CAS.
- J. Ludvigsson and L. G. Heding, Acta Diabetol. Lat., 1982, 19, 351–358 Search PubMed.
- S. G. Hartling, F. Lindgren, G. Dahlqvist, B. Persson and C. Binder, Diabetes, 1989, 38, 1271–1274 CrossRef CAS.
- O. Snorgaard, S. G. Hartling and C. Binder, Diabetologia, 1990, 33, 36–42 CrossRef CAS.
- H. P. Harding, H. Zeng, Y. Zhang, R. Jungries, P. Chung, H. Plesken, D. D. Sabatini and D. Ron, Mol. Cell, 2001, 7, 1153–1163 CrossRef CAS.
- C. Ionescu-Tirgoviste, Rom. J. Intern. Med., 2007, 45, 3–15 Search PubMed.
- P. Gordon, C. M. Hendricks and J. Roth, Diabetologia, 1974, 10, 469–474 CrossRef.
- M. E. Mako, J. I. Starr and A. H. Rubenstein, Am. J. Med., 1977, 63, 865–869 CrossRef CAS.
- L. G. Heding, J. Ludvigsson and T. Kasperska-Czyzykowa, Acta Med. Scand., 1981, 5–9 Search PubMed.
- W. K. Ward, E. C. LaCava, T. L. Paquette, J. C. Beard, B. J. Wallum and D. Jr. Porte, Diabetologia, 1987, 30, 698–702 CrossRef CAS.
- N. Yoshioka, T. Kuzuya, A. Matsuda, M. Taniguchi and Y. Iwamoto, Diabetologia, 1988, 31, 355–360 CrossRef CAS.
- R. C. Temple, C. A. Carrington, S. D. Luzio, D. R. Owens, A. E. Schneider, V. J. Sobey and C. N. Hales, Lancet, 1989, 333, 293–295 CrossRef.
- M. F. Saad, S. E. Kahn, R. G. Nelson, D. J. Pettitt, W. C. Knowler, M. W. Schwartz, S. Kowalyk, P. H. Bennett and D. Porte, J. Clin. Endocrinol. Metab., 1990, 70, 1247–1253 CrossRef CAS.
- S. M. Haffner, R. R. Bowsher, L. Mykkänen, H. P. Hazuda, B. D. Mitchell, R. A. Valdez, R. Gingerich, A. Monterossa and M. P. Stern, Diabetes, 1994, 43, 1490–1493 CrossRef CAS.
- S. E. Kahn, D. L. Leonetti, R. L. Prigeon, E. J. Boyko, R. W. Bergstrom and W. Y. Fujimoto, Diabetes, 1995, 44, 173–179 CrossRef CAS.
- L. Mykkänen, S. M. Haffner, C. N. Hales, T. Ronnemaa and M. Laakso, Diabetes, 1997, 46, 1990–1995 CrossRef CAS.
- M. E. Roder, A. Vaag, S. G. Hartling, B. Dinesen, S. Lanng and H. Beck-Nielsen, J. Clin. Endocrinol. Metab., 1995, 80, 2359–2363 CrossRef.
- I. Inoue, K. Takahashi, S. Katayama, Y. Harada, K. Negishi and J. Ishii, et al. , Diabetic Med., 1996, 13, 330–336 CrossRef CAS.
- D. Porte and S. E. Kahn, Diabetes, 2001, 50, S160–S163 CrossRef CAS.
- J. L. Leahy, Diabetes Care, 1990, 13, 992–1010 CrossRef CAS.
- J. L. Leahy, S. Bonner-Weir and G. C. Weir, Diabetes Care, 1992, 15, 442–454 CrossRef CAS.
- L. Mykkänen, D. J. Zaccaro, C. N. Hales, A. Festa and S. M. Haffner, Diabetologia, 1999, 42, 1060–1066 CrossRef CAS.
- C. Ionescu-Tirgoviste, Diabetologia, 2005, 48, 203–204 CrossRef CAS.
- D. Porte and S. E. Kahn, Diabetes, 1989, 38, 1333–1336 CrossRef CAS.
- C. J. Rhodes and C. Alarćon, Diabetes, 1994, 43, 511–517 CrossRef CAS.
- A. Pfützner, A. H. Pfützner, M. Larbig and T. Forst, Diabetes Technol. Ther., 2004, 6, 405–412 CrossRef.
- A. Pfützner, T. Kunt and C. Hohberg, et al. , Diabetes Care, 2004, 27, 682–687 CrossRef.
- A. Pfützner, E. Standel and C. Holbert, et al. , Diabetes Technol. Ther., 2005, 7, 478–486 CrossRef.
- M. R. Hayden and S. C. Tyagi, JOP. J. Pancreas, 2001, 2, 124–139 Search PubMed.
- D. Ron, J. Clin. Invest., 2002, 109, 443–445 CAS.
- P. Zhang, B. McGrath, S. Li, A. Frank, F. Zambito, J. Reinert, M. Gannon, K. Ma, K. McNaughton and D. R. Cavener, Mol. Cell. Biol., 2002, 22, 3864–3874 CrossRef CAS.
- M. Liu, Y. Li, D. Cavener and P. Arvan, J. Biol. Chem., 2005, 280, 13209–13212 CrossRef CAS.
- B. Ahrén, Curr. Mol. Med., 2005, 5, 275–286 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |