A trick of the light: the optical properties of living cytoplasm which can mislead
Maurice B.
Hallett
*a and
S.
Dewitt
b
aNeutrophil Signalling Group, School of Medicine, Cardiff University Heath Park, Cardiff, CF14 4XN, UK. E-mail: hallettmb@cf.ac.uk
bNeutrophil Signalling Group, School of Dentistry, Heath Park, Cardiff, CF14 4XY, UK. E-mail: dewitt@cf.ac.uk
First published on 4th January 2011
Abstract
Our understanding of the dynamic chemical changes within living cells has increased enormously as a direct result of imaging and manipulating techniques which rely on the use of light to penetrate the cell. These optical techniques are at the intersection of the three sciences: physics, chemistry and biology. However, the understanding of the physics of illumination (e.g. lasers, confocal microscopy) and the chemistry of fluors (synthetic and protein) is way ahead of the understanding of the biological interface posed by the cell itself. In this critical review we will show that ignoring the optical properties of living cells can lead to serious errors of interpretation and that even seemingly compelling images can result from a “trick of the light”.
Insight, innovation, integrationDynamic chemical changes within living cells control their activity and decision making processes. We are increasingly able to probe and monitor these changes using imaging and photo-manipulating techniques. As these methodologies rely on the penetration of light into the cell, they require an understanding of the way that illumination (e.g. lasers, confocal microscopy) interacts with the living cell. Yet, although the chemistry of fluors (synthetic and protein) and caged compounds is increasingly understood, the biological interface is rarely discussed. Here, we show that the optical properties of living cells cannot be ignored without the risk of serious misinterpretation of the data and warn that even seemingly compelling “pictorial” evidence can result from a ‘‘trick of the light’’. |
Introduction
The use of light to interrogate the dynamic chemistry within living cells underlies a powerful set of techniques. These techniques use fluorescent probes such as those for ions such as cytosolic free Ca2+ and cytosolic pH; an array of fluorescent proteins which can act as markers of protein location within the cell, such as chimeric GFP, YFP and RFP; and FRET (fluorescent resonant energy transfer) probes for the proximity of binding pairs within the cell.1 In addition to passively visualising the subcellular intensity of these probes, light is also used to change a property of the probes. For example, the diffusion of fluorescent probes within the cytosol or membrane can be visualised and measured by localised bleaching of the probe or by its photoactivation. Finally, light is also used to manipulate the chemistry of the cell, by the use of caged compounds which can be introduced into the cell in a biologically inert (caged) form. The caging moiety is cleaved by photolysis, releasing the biological active molecule when and where required. In all these techniques, and many other examples, it is the penetration of light into the cell which gives the technique its power. Although there has been a lot of effort in pushing the chemistry of the probes and developing greater and greater precision from the fluorescent signal, it could be argued that little attention has been paid to the optical environment within the cell itself. We point out in this short critical review that far from being a simple environment, the cytosol of living cells is optically complex and changing. While the optical complexity of the living cytoplasm can be often be ignored without significant damage to the veracity of the conclusion reached, this is not always the case and it is worth considering what properties of the cytoplasm can influence the recorded fluorescent signals or efficiency of photolytic uncaging or photoactivation.Optically-confounding constituents of cytoplasm
We will separate out three properties of the living cytoplasm which impinge on the interpretation of fluorescent signals, namely soluble components of the cytosol, cytoplasmic organelles and the dynamic changes in cell shape and microanatomy.Firstly, if there are organelles which exclude the molecule of interest e.g. a fluor or caged compound, the concentration of the molecule is effectively “diluted” by the presence of excluding organelles. For example, as the volume occupancy of granules in neutrophils in the nonnuclear cytoplasm is ∼20%,5 the effective gross concentration of cytosolic fluors is 20% lower in the granular cytoplasm than in granule-free cytoplasm and the intensity of fluorescence at these granule-free sites is increased by a factor of 1.25.6 This effect can also be thought of as a change in effective excitation path length, and is thus both a spatial, and an optical effect. This effect can be easily seen in cells with obvious (i.e. large) granules such as human eosinophils (Fig. 1). At a resolution (or magnification) too low to resolve individual granules, it appears as if the effective concentration of probe in the cytosol is locally reduced and the intensity of the fluorescent signal appears non-uniform within the cell (Fig. 1a). However, at higher resolution, the fluorescent signal from the dimmer region of cytoplasm is seen to be contaminated by fluor-excluding granules (Fig. 1b). The cytosol between granules has a uniform intensity and the apparent non-uniformity reflects the non-uniformity of distribution of granules (Fig. 1). A similar but reverse effect can be seen in the nucleus. Since the cytosol has inclusions, even if sub-light microscopic, but the nucleoplasm has not, the effective concentration of fluor within the nucleus is higher. This nuclear effect has been recognised as contributing to misinterpretation of nuclear Ca2+ signals, where the Ca2+ indicator, e.g. fluo3 is higher in the nucleus.7 In polarised cells having granules in only one part of the cell, a similar disparity in the apparent concentrations in the probe would exist. For example, in pancreatic cells, the granular cytoplasm is restricted to the apical part. Changes in fluorescence intensity in the two regions within the cell therefore differ as a result of differences in the effective concentration of probe at the two sites. This effect may account for early reports that Ca2+ gradients exist in polarised (granular) cells or that there is locally high cytosolic free Ca2+ in pseudopodia.8,9 If performed correctly, these problems are minimised with ratio imaging, and the gradients in Ca2+ are not seen.10,11
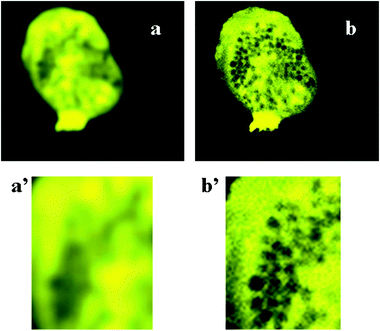 | ||
| Fig. 1 The effect of fluor exclusion by cytoplamsic organelles . Both images show a confocal plane of a human eosinophil loaded with fluo4 pseudocolured in yellow. This dye is excluded from the granules which occupy the region of the cell between the nucleus and the leading edge. (a) A low resolution image of the cell taken confocally, in which the fluorescence signal appears to be non-uniform across the cell. (b) The same cell as shown in (a) but at a higher resolution so that the individual granules are seen. (a’,b’) Higher magnifications of the granular zones of the images (a) and (b) to show how the region of lowered fluorescence is the result of the presence of fluor-excluding granules in that region. | ||
A similar argument, of course, applies to other molecules within the cytoplasm such as caged compounds. For example, the effect of uncaging IP3 in the granular region of the cell would generate less IP3 than in the clear (granule-free) cytoplasm simply because the amount of caged compound exposed to UV light would be less in the granular region. Care must therefore be taken when drawing conclusions about differences in responses to uncaging of biological active compounds if based on regional differences in the micro-anatomy of the cell.
The second way in which small inclusions, whose dimensions are near the wavelength of the incident light, can influence fluor excitation is via the light scattering effect. If the incident light is scattered, the excitation and uncaging efficiencies are consequently reduced. This effect can be profound, especially when optical confocal sections are taken “deeper” into the cell.6 Mie scattering theory provides a mathematical basis for predicting the light-scattering effect of particles suspended within a medium12 and so approximately to the situation in the cytoplasm. This calculation shows that particles whose size is near the wavelength of the incident light, and have a significant refractive index13,14 generate significant light scattering. Care must therefore be taken when comparing effects between granular cells or in regions of the cells which contain granules. For example, in granular cytoplasm containing granules with diameters 0.2–0.3 μm and physiological numbers of granules/volume of cytoplasm, the attenuation coefficient could be as high as 700 mm−1 (ref. 15). This effect can be seen in orthogonal (z plane) images,6 where the z-plane image of fluorescein in free solution and immobilized on a latex sphere are compared. The object with high light-scattering properties has significant attenuation of fluorescent signal (Fig. 2a). Similarly the efficiency of uncaging of compounds within the granular (light scattering) cytoplasm will be reduced deeper into the cell. This effect can generate false impressions in cells which change shape dynamically.
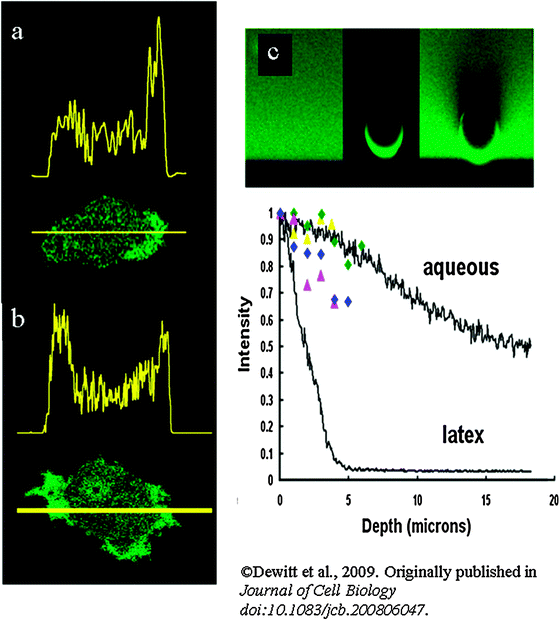 | ||
Fig. 2
Asymmetrical fluorescent signals inside and outside
cells
caused by
light scattering
. (a) The typical distribution of fluorescence intensity of cytoplasmic fluorescein along the axis of a polarized human neutrophil is shown. The dynamic nature of the fluorescence asymmetry of fluorescein can be seen in Video 1 at http://www.jcb.org/cgi/content/full/jcb.200806047/DC1. (b) The typical distribution of fluorescence intensity of cytoplasmic GFP in human cord blood-derived neutrophils along the axis shown of a polarizing cell. Bars in (a) and (b) 5 μm. (c) The fluorescent signals in the xz plane through fluorescein in solution (100 μM), and fluorescein conjugated to a latex sphere (10-μm-diam), and the latex sphere in the fluorescein solution. The attenuation of fluorescent signal is apparent in the latex bead and is plotted in the graph for comparison with free solution and fluorescein in cell-types with differing granularities ( fluorescein-loaded HECV cell, fluorescein-loaded HECV cell,  fluorescein-loaded PC3 cell, fluorescein-loaded PC3 cell,  GFP-expressing dictyostelium, and GFP-expressing dictyostelium, and  fluorescein-loaded human neutrophil. (For full details see ref. 6). fluorescein-loaded human neutrophil. (For full details see ref. 6). | ||
Dynamic changes in living cells
Measurements made in a fluorimeter are usually made in a cuvette whose dimensions are fixed and whose contents are defined. In cells which undergo dynamic changes in shape, there are obvious changes in the thickness of the cells as it stretches out or contracts. Even if the concentration of fluor is uniform within the cell, the amount of fluor will be lower in the thinner part of the cell. Without confocal imaging, the fluorescence signal from a thinner part of the cell will be lower than from a thicker part of the cell. In principle, confocal optical sections of defined size should avoid this problem because the amount of fluor in the section would be uniform in both the thinner and thicker parts of the cell. However, if the confocal section is placed at any plane where it includes non-cellular regions where the cell is thinner, a very significant apparent attenuation of signal will be seen (Fig. 3). As living cells can continually change their shape during imaging, selection of an appropriate confocal plane can be difficult.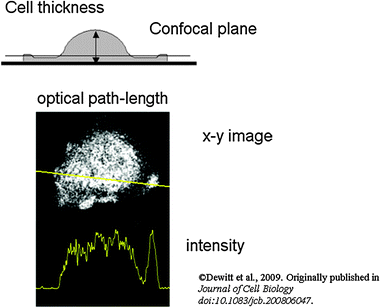 | ||
| Fig. 3 Excitation path-length changes caused by difference in cell thickness. The top diagram shows how cell thickness varies across the cell, which has a thin skirt around it and a thicker terminal region. The position of a confocal optical section is shown which would include the thicker terminal region but not the skirt. An example of this artefact is shown below (spreading human neutrophil), together with its intensity profile. For this and other confocal images shown in this paper, the resonant scanning head of the Leica SP2 confocal microscope with a 63× oil immersion objective NA 1.32 (HCX-PL- APO) was used. | ||
Perhaps a lesser recognised problem, but one which gives significant cause for concern arises in cells which polarise and form leading edges or pseudopodia since these involve a change in the local optical properties of the leading edge or pseudopodia.6 The leading fronts of motile cells and pseudopodia contain an actin mesh which provides the force for deforming the plasma membrane, and which also excludes organelles.16 There is usually an obvious organelle-free cytoplasmic zone at the front of these cells and pseudopodia are usually recognised by phase contrast microscopy by being optically “clear”, as opposed to the light-scattering and absorbing cytoplasm in the bulk of the cell. This difference in optical properties of the cytoplasm at the leading edge will mean that the efficiency for excitation will be significantly higher for fluors located here.
False sub-cellular protein translocation
The effect of granular cytoplasm is especially important when considering the translocation of fluorescent proteins to the front (leading edge) of motile or polarised cells. Because optically “clear” cytoplasm at the leading edge has an increased transmission of light, fluors located here will be excited with greater efficiency. Not only is the light scattering effect absent but the “dilution” of fluor by fluor-excluding organelles is also absent. These effects lead to a “false localization” of fluorescence signals at the front of the cell or in pseudopodia even when the concentration of fluor is uniform throughout the cell.6 For example, in human neutrophils loaded with fluorescein or GFP, the front of the cell has an increased fluorescence signal (Fig. 2b,c). This changes dynamically as the cell sends out new pseudopodia or leading fronts (see movie 1 of ref. 6). Clearly fluorescein and GFP are not reporting specific changes and the concentration of either fluor is uniform, yet the optical effects discussed earlier give rise to striking non-uniform effects. If the probe were designed to report a chemical event in the cell by translocating and binding to its partner, these fluorescent asymmetric signals could easily be misinterpreted as translocation of the probe to the front edge. However, this optical difference in pseudopodia at the front of motile cells or during phagocytosis will give an asymmetry in fluorescent signal independently of protein translocation.Synopsis
Although the use of light both for the detection of fluorescent signals and the manipulation of chemistry within living cells has produced spectacular results and has undoubtedly added to our knowledge of cell biology greatly, we feel that it is time to add a note of caution when interpreting such experiments. Because pictures are so visually compelling, (and said to be worth a thousand words), we are in danger of losing scientific scepticism about their interpretation. We suggest that the optical properties of the cellular environment which we have highlighted here are not trivial and must not be overlooked. In fact, dynamic changes in the cell micro-anatomy create some serious imaging artefacts which could easily lead to the misinterpretation of their meaning. Fortunately, however, as we have previously reported,6 the problems are easily identified by the use of control experiments, ratio imaging of two probes and simple intensity quantitative analysis.Acknowledgements
We are grateful to the Wellcome Trust for grant funding (MBH and SD) and to ARUK for fellowship funding to SD.References
- R. Y. Tsien, Fluorescent probes of cell signalling, Annual Review of Neuroscience, 1989, 12, 227–253 Search PubMed.
- S. Voronina, T. Sukhomlin, P. R. Johnson, G. Erdemli and O. H. Petersen, Correlation of NADH and Ca2+ signals in mouse pancreatic acinar cells, The Journal of Physiology, 2002, 539, 41–52 Search PubMed.
- J. C. Brasen, D. Dewitt and M. B. Hallett, A reporter of UV intensity delivered to the cytosol during photolytic uncaging, Biophys. J., 2010, 98, L25–L27 CrossRef CAS.
- H. T. Zhao, J. Joseph, H. M. Fales, E. A. Sokoloski and R. L. Levine, Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 5727–5732 CrossRef CAS.
- G. W. Schmid-Schonbein, Y. Y. Shih and S. Chien, Morphometry of human leukocytes, Blood, 1980, 56, 866–875 CAS.
- S. Dewitt, R. Darley and M. B. Hallett, Translocation or just location? psuedopodia affect fluorescent signals, J. Cell Biol., 2009, 184, 197–203 CrossRef CAS.
- F. A. Al-Mohanna, K. W. T. Caddy and S. R. Bolsover, The nucleus is insulated from large cytosolic-ion changes, Nature, 1994, 367, 745–750 CrossRef CAS.
- D. W. Sawyer, J. A. Sullivan and G. L. Mandell, Intracellular free calcium localization in neutrophils during phagocytosis, Science, 1985, 230, 663–666 CrossRef CAS.
- R. A. Brundage, K. E. Fogarty, R. A. Tuft and F. S. Fay, Ca2+ gradients underlying polarization and chemotaxis of eosinophils, Science, 1991, 254, 703–705 CrossRef CAS.
- I. Laffafian and M. B. Hallett, Does cytosolic free Ca2+ signal neutrophil chemotaxis?, J. Cell Sci., 1995, 108, 3199–3205 CAS.
- S. Dewitt and M. B. Hallett, Cytosolic free Ca2+ changes and calpain activation are required for β2 integrin-accelerated phagocytosis by human neutrophils, J. Cell Biol., 2002, 159, 181–189 CrossRef CAS.
- G. Mie, Contributions to the optics of turbid media particularly of colloidal metal solutions, Ann. Phys., 1908, 330, 377–445 CrossRef (English translationhttp://diogenes.iwt.uni-bremen.de/vt/laser/papers/RAE-LT1873-1976-Mie-1908-translation.pdf).
- R. A. Meyer, Light scattering from biological cells: dependence of backscatter radiation on membrane thickness and refractive index, Appl. Opt., 1979, 18 Search PubMed.
- G. I. Ruban, S. M. Kosmacheva, N. V. Goncharova, D. Van Bockstaele and V. A. Loiko, Investigation of morphometric parameters for granulocytes and lymphocytes as applied to a solution of direct and inverse light-scattering problems, J. Biomed. Opt., 2007, 12, 044017 CrossRef.
- S. Prahl, Mie Scattering calculator, 2007, http://omlc.ogi.edu/calc/mie_calc.html.
- K. Luby-Phelps, D. L. Taylor and F. Lanni, Probing the structure of cytoplasm, J. Cell Biol., 1986, 102, 2015–2022 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |