Insights on the permeability of wide protein channels: measurement and interpretation of ion selectivity
Vicente M.
Aguilella
*,
María
Queralt-Martín
,
Marcel
Aguilella-Arzo
and
Antonio
Alcaraz
Dept. Physics, Lab. Molecular Biophysics, Universitat Jaume I, 12080 Castellón, Spain. E-mail: aguilell@uji.es
First published on 6th December 2010
Abstract
Ion channels are hollow proteins that have evolved to exhibit discrimination between charged solutes. This property, known as ion selectivity is critical for several biological functions. By using the bacterial porin OmpF as a model system of wide protein channels, we demonstrate that significant insights can be gained when selectivity measurements are combined with electrodiffusion continuum models and simulations based on the atomic structure. A correct interpretation of the mechanisms ruling the many sources of channel discrimination is a first, indispensable step for the understanding of the controlled movement of ions into or out of cells characteristic of many physiological processes. We conclude that the scattered information gathered from several independent approaches should be appropriately merged to provide a unified and coherent picture of the channel selectivity.
Insight, innovation, integrationStudying wide ion channels like bacterial porins, toxins and mitochondrial channels gives new insights into the general problem of characterizing membrane permeabilization by proteins and offers some innovative clues for the exploration of antibiotic resistance in bacteria, toxin inhibition and mitochondrial nucleotide exchange. This review shows that protein microscopic (atomic) information can be successfully integrated with in vitro electrophysiological measurements, in silico simulations and macroscopic electrodiffusion models to provide an original and attractive vision of protein channel function. Scattered, apparently divergent, selectivity values found in the literature are harmonized in a single coherent picture after a careful analysis of the experimental conditions and assessment of the models used for the elucidation of membrane selective permeability. |
1. Introduction
Ion channels are a large family of specialized pore-forming proteins present in all living organisms that play a decisive role in the communications between and within cells. Proper functioning of ion channels is crucial for neural transmission as well as for key physiological processes that involve cardiac, pulmonary and muscle systems.1–3 Investigations of the function of biologically critical proteins bridge basic science with clinical medicine. Such connections have been particularly evident in the field of ion channels since the discovery of channelopathies: human diseases caused by the disturbed function of ion channel subunits or the proteins that regulate them.4 As might be expected considering the variety of functions performed by ion channels, channelopathies range from inherited cardiac arrhythmias to muscle disorders and forms of diabetes.4,5 The mechanisms by which ion channels regulate the transport of molecules and electric signal transduction are often complex and even highly sophisticated. However, the simplest picture of an ion channel may be a pore of nanometre dimensions that acts either as a passive filter or as an externally activated valve within a biological membrane. Due to the hydrophobicity and low polarizability of the lipid constituents of biological membranes, for ions and many metabolites the channel route is by far more energetically favourable than crossing the membrane.6 Thus, ion channels act as gatekeepers for charged solutes that enter or exit the cells and their organelles.1,2,7 The manipulation of biological membranes and ion channels to take advantage of their ‘sensing’ properties has been successful in a variety of biotechnological and analytical applications.8–10 Similar efforts have been directed to develop synthetic pores that mimic several relevant functions of ion channels.11,12Electric signalling by way of ion channels is associated with the movement of charges across a membrane.13 This task of carrying electrical current is performed by the ions present in all living systems (typically, but not exclusively, by Na+, K+, Cl−, Ca2+). Besides, a polarized interface is quite often a requirement of some physiological function. Any electric potential difference needs charge separation and this is accomplished in cells by keeping the concentration of positive and negative ions unbalanced between both sides of the membrane. In the movement of ions through channels down their electrochemical gradients the net ionic transport reflects the combined effect of the activity gradient and the electric potential gradient. The vast majority of ion channels are able to display a preference for certain kind of ions and this property is generally known as ion selectivity.2 Such a selection may be done simply by the ion charge (valence selectivity) or may be specific for particular ions or molecules. On the one hand, the valence selectivity allows a broad classification of channels into cation selective and anion selective, depending on whether cation or anion transport is favoured by the protein channel. On the other hand, the specific selectivity serves very often to name the channel, e.g.chloride ion channel, potassium channel family, etc.14–17
Ion channels must discriminate efficiently between different charged species, which is most easily achieved by a narrow pore. However, ion channels should also transport ions as fast as possible, which is easier through a wider pore. The compromise between this two contrasting features configures the specific function of each channel. This review is focused on the measurement and interpretation of ion selectivity in multiionic channels, collectively known as wide or mesoscopic channels because their pore dimensions: their size is too large to be specific but the channel walls still do have an influence on the permeability. Understanding the mechanisms by which wide channels discriminate between different species of cations and anions is a first, necessary step for an adequate interpretation of the specific selectivity in narrow channels.15 Besides, as will be shown below, charge effects at the nanoscale are not trivial at all and need to be correctly interpreted before attributing particular selective properties to a channel.18
We propose here to use the ion-channel analogue of the well-known concept of “model system” in biology. Models are those organisms with a wealth of biological data that make them attractive to study as examples for other species that are more difficult to analyze directly. One of the first model systems for molecular biology was the bacterium Escherichia coli, present in the human digestive system. Several bacterial viruses (bacteriophage) that infect E. coli have been also very useful for the study of gene structure and gene regulation (e.g.phages Lambda and T4). The search for a model system representative of wide channels must tackle a crucial issue. When the channel structure is known in detail, the combination of several approaches—molecular dynamics (MD), Brownian dynamics (BD) and continuum theories—provides a sound interpretation of certain function-related magnitudes (conductance, selectivity, etc.) measured in electrophysiology experiments.17 But, unfortunately, such methodologies are only possible in the small group of channel-forming proteins whose structure has been solved at atomic resolution. In the rest of proteins, the lack of structural details is a breeding ground of speculation about the folding path, the actual location of the residues and their ionization state and the validity of the available theoretical approaches.
The first structures (from the 1990s) of ion channels solved at atomic resolution, i.e. below 3 Å, were those of the bacterial porins, a family of homo-trimeric channel proteins. Therefore, one of them, the OmpF porin, a protein that forms wide channels just in the outer membrane of E. coli seems the ideal candidate to become a “model system” revealing the fine details of the structure/function correlation. The atomic structure of OmpF porin was first obtained from X-ray analysis in 1992 with 2.4 Å resolution.19 Since then, subsequent structures of OmpF have been reported in the presence of salts of monovalent cations20 and divalent ones.21
Most bacterial porins fall into the category of multiionic channels. Each subunit contains 16 to 18 transmembrane, anti-parallel β-strands forming a β-barrel structure. The β-strands are amphipathic: they contain alternating polar and non-polar residues and the inter-strand interaction is fully saturated with H-bonding. This creates a hydrophilic environment providing a water filled channel weakly selective for small ions. The OmpF porin has an upper exclusion size limit corresponding to solute molecular weights ca. 600 Da. Because most metabolites have molecular weights lower than 600 Da and have been shown to pass through porin channels, porins are called general diffusion pores.22,23 The study of their selectivity is essential for many reasons, of which one of the most important is the design of molecules with antibiotic properties and good permeability across the outer membrane of Gram-negative bacteria. As is known, porins provide a path through the outer membrane to small hydrophilic antibiotics.24 Other interesting protein channels that exhibit multiionic transport are some bacterial toxins. The most widespread bacterial toxin found in the human organism is α-hemolysin secreted by Staphylococcus aureus. This channel is a good example of how the interest of a particular toxin may go beyond basic research up to novel biotechnology applications like single molecule detection, chemical sensing, drug screening, RNA/DNA sequence readout, etc.8,10,25
In the following sections we will first introduce the different sources of selectivity in our OmpF model system. Then, several ways of quantifying selectivity through experiments (in channels reconstituted on planar lipid bilayers) will be reviewed as well as the corresponding approaches to selectivity used in MD and BD simulations. Finally, we will discuss what is needed for an adequate interpretation of those measurements and calculations that may give a unitary view of channel selectivity.
2. The origin of ionic selectivity of multiionic channels
2.1 Narrow channels vs. multiionic wide channels
Channel selectivity is routinely measured in the laboratory as an element of the process of characterization of any transmembrane protein forming channels.2,26 One might think that a charged pore should allow passage of charged solutes basically depending on their size and charge. Although this is essentially true, the physicochemical mechanisms controlling ion selectivity are largely dependent on each particular type of channel.16 For instance, such size and charge controlled permeability would not be enough to understand how aquaporins selectively conduct water molecules in and out of the cell, while they are completely impermeable to charged species, even to protons.27,28 Other factors like the characteristic aqueous solvation of each ionic species may be determinant in channel selectivity.29 In potassium channels, to mention another example, the coordination of the protein-channel carbonyl groups with the unsolvated permeating ion is essential to achieve a thousand-fold K+/Na+ selectivity.30–32Leaving apart important channel features like their activation mechanism, their voltage-dependent function and the transition between states of different conductance, a broad classification of ion channels can be made between multiionic channels and ion-specific channels. The first ones are often known as mesoscopic channels and have pore dimensions that allow simultaneous passage of water molecules, positive and negative ions of several types that may enter the pore without losing their hydration layer and even metabolites like ATP33 or antibiotic molecules.34 The second type selects a particular ion so that permeability to other ions is usually very low (e.g.potassium channels have a permeability ratio for K+ over Na+ > 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and calcium channels select for Ca2+ over Na+ with a ratio > 1000:1). To achieve this high ion specificity, a close interaction between the permeating ion and the protein residues is needed.2 For this reason, channels displaying specific selectivity have pore dimensions comparable to the size of the permeating ion or at least there is a narrow region called selectivity filter—a term coined by Hille in 197135—that discriminates between ionic species. Ion transport in such narrow channels is often termed single-file because only one ion at a time can cross the selectivity filter.
:1 and calcium channels select for Ca2+ over Na+ with a ratio > 1000:1). To achieve this high ion specificity, a close interaction between the permeating ion and the protein residues is needed.2 For this reason, channels displaying specific selectivity have pore dimensions comparable to the size of the permeating ion or at least there is a narrow region called selectivity filter—a term coined by Hille in 197135—that discriminates between ionic species. Ion transport in such narrow channels is often termed single-file because only one ion at a time can cross the selectivity filter.
Fig. 1 illustrates the different occupancy of selective narrow and large ion channels as well as the solvated or unsolvated state of the permeating ions. The widechannel on the left represents a typical bacterial porin, whose slight cationic or anionic selectivity is low enough not to exclude cations or anions. The narrowchannel on the right represents a potassium channel specifically designed to let only bare K+ ions in and out. The division into narrow and wide channels, simple as it may appear, is very useful for the purpose of analyzing channel selectivity. However, such categories are not totally exclusive. In a large variety of channels the discrimination between ions can be partially done in the short section called selectivity filter while other wider regions of the pore aid rapid diffusion.15
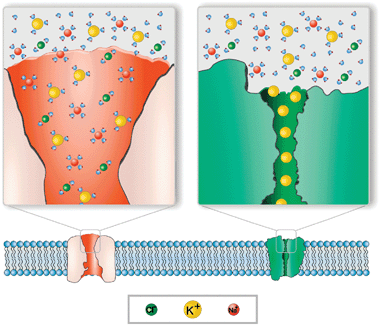 | ||
| Fig. 1 Cartoon illustrating the difference between single-file transport and multiionic transport. Left: OmpF channel. Cations and anions may enter the pore without losing their hydration layer. The channel displays a slight preference for cations at neutral pH. Right: potassium channel. Only unsolvated K+ ions are allowed to enter the pore. K+ transport takes place in a single-file fashion. | ||
2.2 The various sources of ion selectivity
The discrimination between ions depends not only on the ion intrinsic properties (size, solvation, diffusivity in water, shielding effects, etc.) but also on the particular interaction of permeating ions with the channel residues,2 as shown in Fig. 2. This general statement implies that ion selectivity is not an intrinsic property of the channel but necessarily includes both the protein channel and the electrolyte flowing through it.36 When this is particularized to the selectivity of large channels towards small inorganic ions, the two leading contributions to channel selectivity are the differences between cation and anion mobilities and the electrostatic exclusion due to the interaction between permeating ions and channel ionizable residues.26,37 Very often, a high selectivity is exclusively associated with the latter, thus overlooking not only diffusional effects, but also additional factors such as entropic effects related to the preferential rejection of ions because of their size, short range non-electrostatic interactions and osmotic effects.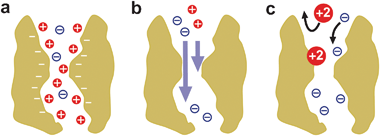 | ||
| Fig. 2 Several contributions to large channel selectivity under a salt concentration gradient. (a) A channel with net negative fixed charge accumulates cations and partially excludes anions. (b) When cations and anions have different mobilities, there appears a net electric current across the channel. (c) Short-range, non-Coulombic, interactions between permeating ions and protein residues may alter and even reverse the partitioning of cations and anions between solution and the channel pore and their respective mobilities. | ||
In principle, diffusional effects can be identified and minimized. For instance, in salts where the cation and the anion have almost equal bulk diffusivities (e.g.KCl or CsCl), the diffusional contribution is negligible. This explains why so many measurements of channel selectivity have been performed in KCl solutions. Under such conditions, the channel selectivity can be correlated rather well with the net fixed charge of the channel coming from the ionizable residues of the protein.36–38 This fact, combined with the technique of site directed mutagenesis, which allows altering the charge of the channel by substituting charged residues with neutral ones, lies on the basis of numerous studies of channel selectivity in OmpF39–41 and other wide channels like VDAC.42 However, it is not always reliable as will be explained later in the paper.
The role of diffusion can be studied separately as well. In channels with very small net charge this is straightforward, since the accumulation of counterions and the exclusion of coions are insignificant and do not contribute to channel discrimination between cations and anions. But in channels with non-negligible charge an alternative strategy may give useful information on the actual relative mobilities of ions inside the channel: the measurement of selectivity under conditions where the partitioning of ions inside the channel reaches a limit (for instance, when bulk salt concentration is much higher than the effective fixed charge concentration of the channel).38
3. Measurement and modelling of channel selectivity
As commented before, the selectivity of large channels includes two components: partitioning, an equilibrium measure of the channel preference for a particular ionic species, and diffusion, a non-equilibrium average measure of the relative mobility of ions in the channel.2,26,38 This fact becomes crucial when one realizes that all the protocols used to estimate the ion channel selectivity are based on electrokinetic measurements that operate under non-equilibrium conditions (concentration gradients of neutral and/or charged solutes, applied voltage, applied pressure, etc.). Thus, each particular way of quantify selectivity will involve a different balance between ion partitioning and electrodiffusion, so that disparate outcomes could be obtained even for the same channel.43 Here, we summarize the most common approaches to measure channel selectivity in channels reconstituted on planar lipid bilayers.3.1 Reversal potential
The reversal potential (RP) is defined as the applied transmembrane voltage that yields zero electric current when there is an activity gradient across the channel. RP is the method of choice to quantify selectivity for the sake of simplicity: the sign of the RP provides a quick estimation of the channel selectivity and its preference for cations or anions. Furthermore, by using the Goldman–Hodgkin–Katz (GHK) equation the measured RP can be converted into a single parameter for each permeant ion: the permeability. Unfortunately, the GHK equation allows one to calculate permeability ratios, but not absolute permeabilities.2 For a monovalent salt the cation/anion permeability ratio derived from the GHK equation reads: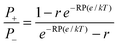 | (1) |
A typical series of experiments consists of measuring RP keeping a fixed salt concentration on one side of the channel and varying the concentration on the other side.36,44,45 However, in many situations the reversal potential measured depends not only on the concentration ratio but on the absolute value of concentrations, the pH of the solutions and even the orientation of the channel with respect to the salt gradient. The lipid composition of the membrane where the channel is embedded may have also some influence since it can modulate the local ion concentrations near the channel openings.36
3.2 Bi-ionic potential
The bi-ionic potential (BP) is the zero current potential measured in symmetric bi-ionic conditions: two different salts with a common ion on each side of the channel but both solutions with the same concentration. This approach is not suitable to measure cation/anion selectivity but to measure the relative selectivity of the channel to different cations (in salts of a common anion) or to different anions (in salts of a common cation). The BP measurements, like the RP ones, are also translated into permeability ratios. According to GHK equation,46 for a pair of solutions of NaCl|KCl, we get the following relationship between BP and the ionic permeabilities Pi: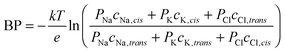 | (2) |
| PNa/PK = eBP(e/kT) | (3) |
3.3 Conductance ratio
Single channelI–V curves are measured by applying a voltage across the system in symmetric conditions (the same solution in both baths). The current–voltage ratio G = I/V yields a measurement of its conductance. A way of quantify the relative preference of the channel by two different, say, cations is to measure the channel conductance in solutions of salts with a common anion. For instance, the ratio of the channel conductances measured in two different experiments with NaCl and KCl salt solutions with the same concentration provides a measurement of the selectivity of the channel. Thus, GNaCl/GKCl is regarded as a measure of the relative channel selectivity to Na+ and K+ cations. This is especially appropriate for highly selective narrow channels where the whole electric current is carried almost exclusively by one ionic species. However, measuring conductance in wide channels in order to elucidate channel selectivity could lead to manifest contradictions when compared to other protocols.45 As commented in the case of Bi-ionic Potential, the current flow across wide weakly selective channels involves a large amount of correlated ions48,49 (both cations and anions) and cannot be understood under the assumption of the total exclusion of coions.3.4 Modelling channel selectivity
There is no unique theoretical model that may account for all the transport properties of such a complex structure like a biological ion channel.17 For this reason, several approaches have been used, each one addressed to a particular aspect. The most powerful method to model channel selectivity consists in a full all-atom non-equilibrium MD simulation whenever the atomic 3D structure of the channel is available.50–53 In large channels MD could be extremely challenging because it involves the explicit consideration of protein atoms, water molecules and ions and, most important, long enough simulation runs to get statistically meaningful quantities. In such cases, coarse grained models where the number of particles is considerably reduced are a good alternative. In spite of this, MD simulations have provided precious details about the permeation pathways of ions along a variety of channels54–58 and thanks to recent advances, it is now possible to compute explicitly channel conductance using all-atom MD simulations and analyze the behaviour of a single protein residue.52,59,60 In addition to computing power, the accuracy of the various MD methods relies on a good representation of the interaction between the mobile ions themselves and the protein atoms. Different sets of parameters commonly referred to as force fields have been developed to this end.61,62 There is also a small range of computing packages.63–65There are other methods that avoid a full atomistic representation in exchange for computational efficiency. BD, dynamic lattice Monte Carlo and grand canonical Monte Carlo BD have in common the implicit representation of the solvent and the membrane while ions are modelled explicitly.48,66–68 The trajectory of each ion in the system can be obtained by solving a particular formulation of Newton's second law: the so-called Langevin equation. A major advantage of the BD approach is that it allows a direct simulation of ion-ion interactions. Since the dynamics of the water molecules and the protein atoms are no longer included and time steps can be longer, BD is many orders of magnitude faster and much less computationally costly than MD.69,70
At the next stage in the microscopic level of representation of protein atoms, solvent molecules and ions, lies a continuum electro-diffusion model in which the mobile ions are treated as concentration profiles.71–73 Their distribution and motion are determined by random diffusion and by electrostatic forces (a mean field approximation). The atomic 3D structure of the protein channel is used to get the spatial charge distribution. In practice, Poisson equation of electrostatics and Nernst–Planck drift-diffusion equations are combined in a set of coupled partial differential equations. In the context of ion permeation through channel proteins, this approach is known as Poisson–Nernst–Planck (PNP-3D) theory.74–76 If the 3D channel structure is not available, the 3D concentration function can be replaced by a 1D concentration (with the meaning of a cross-section averaged ion concentration) and the whole problem becomes one-dimensional.77 Several numerical procedures have been developed in recent years to solve the PNP system of equations as well as refinements of the theory to get a more accurate representation of the protein-solution boundary.75,78
For several decades, the macroscopic electrodiffusion model based on 1D Nernst–Planck and Poisson equations has been used to give a quantitative description of the ion transport properties of porous membranes, single microscopic or nanoscopic pores and biological ion channels.2 Since PNP equations are a system of coupled non-linear differential equations that cannot be solved in closed form (even in the simplest one-dimensional case), two main approximations have been used to obtain analytical solutions for the RP. One of them is based on the assumption of charge neutrality all over the pore and the other, initially proposed by Goldman by assuming a constant electric field along the pore,79 led to the well known GHK equation (eqn (2) in the text) widely used today to express channel relative selectivity in a single parameter, the permeability ratio. The literature about the conditions under which GHK equation can be safely used is abundant.2,26,47,80–83 To put it briefly, constant electric field and independence of ion fluxes are the basic requirements to ensure GHK equation validity. Strictly speaking, those conditions are only rarely met. Nevertheless, there are exceptions to the rule which show that constancy of the field must not necessarily be fulfilled.47,48 Indeed, even in wide channels like OmpF, VDAC or α-hemolysin displaying intricate amphoteric surfaces where the constant field assumption along the channel seems totally unrealistic, there are situations where GHK equation fails to describe properly the channel cationic selectivity36,37 as well as cases where it works fine apparently.48 Im & Roux have used the calculated free energy profile along the OmpF channel as a paradigmatic example to discuss this matter. According to them, the number of free energy barriers and their relative position in the channel could explain the surprising validity of GHK equations in some specific cases.48
In any case, it should be borne in mind that the real question is whether the ionic permeabilities Pi involved in GHK equation are well-defined, physically-sound parameters for every ionic species in a given channel or whether they are empirically defined and allowed to be arbitrary functions of voltage and concentration. In the latter case, they become ineffective to rationalize selectivity data in a wide range of ionic species, salt concentrations and applied voltages.36,43
4. Structural insights from selectivity experiments and continuum models
The number of protein channels whose atomic structure has been resolved by X-ray or NMR methods is still limited. Therefore, additional methods providing information about channel tertiary structure are well received. Selectivity can be helpful in this task because, under certain conditions, the amount of channel charged residues and their space distribution can be probed by experiments. As mentioned in previous sections, when diffusional effects can be reasonably excluded, channel selectivity correlates with the channel net charge.36 Because channels accumulate ions with sign opposite to the net fixed charge of the ionizable residues, a channel with positive net charge (i.e. more ionized basic residues than acidic residues) will exhibit anionic selectivity. On the contrary, a channel with negative net charge will favour transport of cations. This fact, combined with measurements over a wide range of pH and a reasonable estimate of the pKa of the suspected ionizable residues may provide useful information.4.1 Selectivity measurements to explore residue titration
The elucidation of the structure/function correlation should start on a proper understanding of the channel structure.16,36 The electrostatic interaction between the channel charges and the permeating ions crucially regulates the transport through the channel,26 so that the first issue to investigate is the charge state of the ionizable residues of the protein. This is not a trivial task because of the interplay of several factors. Each titratable residue of the channel is in an environment of low electric permittivity as is the protein. It interacts with the protein permanent fixed partial charges (due to the different electronegativities of the atoms in the molecule) and with the rest of titratable residues.84,77 In addition, these interactions are influenced by the shielding effect of the ions in solution. This implies that the proton dissociation constant Ka of the residue (or its equivalent in the logarithmic scale, the pKa) inside the protein may differ from that in free solution.85 Therefore, one needs to calculate the apparent pKa of each titratable residue inside the protein. The protocol followed to perform this calculation, based on the procedure described by Antosiewicz et al.,86,87 has been described elsewhere.36 A finite difference Poisson–Boltzmann solver can be used to compute the apparent pKa from the 3D atomic crystal structure of OmpF.88The pKa calculations performed in a very acidic environment (pH = 2) in one of the three identical OmpF monomers indicate that 41 residues are charged. 31 of them have positive charge whereas 10 have a negative one (Fig. 3a). This positive net charge correlates qualitatively well with the reported anionic selectivity of the channel under such conditions. At neutral pH (Fig. 3b), the balance between positive and negative residues is inverted. There are 30 positives and 41 negatives, yielding a net negative charge. However, the situation is quite different under basic conditions (pH = 12). According to the calculations 58 residues are charged, 14 of them are positive and 44 are negative (Fig. 3c). This is consistent with the cation selectivity of the channel found in experiments at pH 7 and 12. Therefore, considering all charged residues with their properly calculated pKa, the channel selectivity exhibits a good qualitative correlation with the total monomer charge. This conclusion does not hold if one assumes that the residues that face the channel lumen or are in the vicinity of the aqueous pore are presumably most relevant to the channel selectivity. Taking a reasonably cut-off distance of 3 Å to the pore solvent accessible surface, there are only 21 residues among the overall 102 ionizable residues of each monomer that match such condition. The result (for this region “accessible” to permeant ions) is somewhat counterintuitive. At neutral pH, 13 residues are positively charged and 8 residues negatively charged. The net 5 positive charges per monomer are clearly irreconcilable with the measured cationic selectivity of the channel at neutral pH.
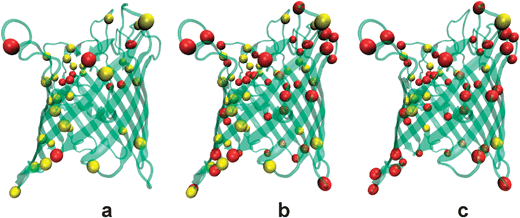 | ||
| Fig. 3 Cartoon that illustrates the amount of charged OmpF residues in three environments at different pH. A ribbon representation of the tertiary structure of the OmpF porin is combined with colored balls representing the positively charged (yellow) and negatively charged (red) ionizable residues. (a) At pH 2 the positive residues exceed by 21 the negative ones, which is consistent with the anionic selectivity of the channel. (b) At pH 7 the net balance switches to negative (−11). (c) At pH 12 negative residues are more than double the number of positive ones, what agrees with the large cationic selectivity measured at this pH. | ||
Although the computed total net charge correlates qualitatively well with the reported ionic selectivity, this is not the case quantitatively. Electric charges of such magnitude (∼40 elementary charges) would create colossal electrical fields (1010–1011 V m−1) around the protein that seem totally unfeasible.54,89 This indicates that understanding ion permeation through the channel involves something else than counting charges in the protein. The dielectric environment, the mutual interaction between residues and the distance from each particular residue to the permeation path of the ions becomes essential to explain possible screening effects. A detailed theoretical model connecting the structural data to the experimental observations seems mandatory.
Despite their low resolution, mean field theories, such as PNP or the Teorell–Meyer–Sievers (TMS) theory, have been used to calculate electrodiffusion quantities in many ionic systems like synthetic nanopores, ion exchange membranes, polyelectrolyte multilayers and ion channels. In their simplest versions, those approaches do not incorporate structural information but use effective parameters which are fitted to the experimental data.36 A slightly different, more elaborated, continuum approach consists in combining a 1D PNP model with cross-section averaged structural information extracted from the 3D atomic structure of the channel. With a minimum computational effort, a good quantitative correlation is obtained between the measured selectivity and the effective fixed charge of the channel.43,77 This effective charge is around 1–2 elementary charges, what originates realistic electric fields in the protein.77,89Fig. 4 shows measurements of RP (dots) over a wide range of pH together with the calculated RP (solid line).
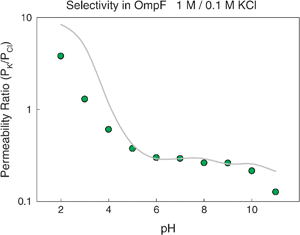 | ||
| Fig. 4 Permeability ratio in OmpF channel as a function of pH. Dots correspond to measurements in 1 M/0.1 M KCl solutions. Solid line represents the calculated permeability ratio. Details of the experiments are given elsewhere.36 | ||
These results show that average electrostatic properties obtained directly from the microscopic structure of the protein regulate the ionic transport under a variety of conditions. This conclusion holds also for some engineered OmpF mutants with very different electrostatic properties but similar atomic structure to that of wild type OmpF.37,90 The model outlined above constitutes a macroscopic electrodiffusion theory of the ion transport across the pore, but it is based on microscopic structural information. One could wonder whether a rough 1D PNP approach using effective magnitudes can accurately describe the transport properties of a system in the nanoscale as is the OmpF porin. Recent studies in both synthetic and biological nanopores support this idea. One dimensional approaches based on Smoluchowski equation, Fick–Jacobs approximation and Poisson–Nernst–Planck equations provide interesting clues for planning more efficient synthetic biosensors, nanopumps and nanodiodes.91–93 In addition, satisfactory comparisons between mean field theories and BD simulations are found for wide biochannels.94 In fact, several PNP approaches describing satisfactorily the ion transport across different biological ion channels have already been reported: The acetylcholine receptor,95 the L-type calcium channel,71 the calcium release channel,72 the gramicidin A channel,74 the mitochondrial channel VDAC76,96,97 or the OmpF porin38,43,77 among others.76,75,36
4.2 Role of the selectivity filter in wide channels
The traditional way to explore the channel distribution of ionizable residues is site directed mutagenesis. If the measured channel selectivity is not affected by the mutation, i.e. replacement of a given charged residue by a similar neutral one, this is regarded as a proof that such residue is not exposed to the ion stream.39,42,98 This is based on the assumption that only the ionizable residues facing the aqueous pore or located at the selectivity filter contribute to the channel selectivity. This may be true in channels like VDAC where practically there are no residues buried99,100,101 but it seems inaccurate or even wrong in other channels like OmpF where there are ionizable residues buried in a low polarizability environment that greatly contribute to channel selectivity,84,36 as discussed in the above section. Actually, the atomic structure of this channel shows that the overall net charge of ionizable residues facing the aqueous pore is positive while the channel is cation selective. Obviously this means that the channel selectivity is the result of the concerted action of many residues, not simply of those lining the pore or placed at the channel narrow constriction and it implies that the concept of selectivity filter,35 crucial in highly selective (narrow) channels is less meaningful in wide multiionic channels (see section 4.1).The OmpF channel has an hour-glass shape with a narrow constriction located about half of the channel total length.19,21,20Fig. 5a shows a schematic view of the constriction zone, showing two acidic residues (D113, E117) that face a cluster of three positive arginines (R42, R82, R132). At neutral pH, all these five residues are ionized and the net charge at the channel constriction is +1e, what seems difficult to reconcile with the observed cationic selectivity of the channel under such conditions.77 The fact that channel selectivity is not only regulated by the channel constriction is even more evident when site-directed mutations of the acidic residues in the selectivity filter are performed. As can be observed in Fig. 5b, the substitution of two carboxylates D113 and E117 by neutral cysteines does not eliminate the cation selectivity of the channel observed around neutral pH. The qualitative trend in the D113C/E117C mutant seems the same as that observed in the wild-type (WT) OmpF: the channel preference for cations turns into a predilection for anions as pH decreases and acidic residues are successively protonated.
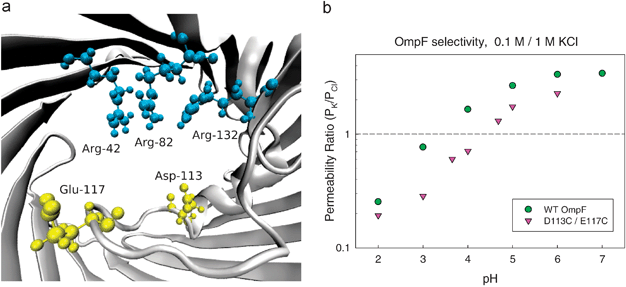 | ||
| Fig. 5 (a) View of OmpF selectivity filter with two acidic residues (D113, E117) facing a cluster of three positive arginines (R42, R82, R132). (b) Permeability ratio of WT OmpF and D113C/E117C mutant as a function of solution pH (0.1 M KCl cis|1 M KCltrans). | ||
It is also true that some quantitative differences between WT OmpF and the D113C/E117C mutant are manifest. At pH = 4 the former is selective to cations and the latter is selective to anions. This indicates that in the OmpF channel the selectivity filter plays an important role, but it is not the sole determinant of the measured selectivity. A large number of residues besides the five located in the selectivity filter seem to be involved.40,41,77,102
In addition, the selectivity filter, being the narrowest region of the pore, is a key factor both regulating the channel conductance to small ions and limiting the size of the solutes that cross the channel. Detailed MD and BD simulations suggest that the charge asymmetry in the constriction zone, clearly visible in Fig. 5a, originates a strong electric field transverse to the pore axis.54,103,98,48,49 This channel feature could be vital for the transport of dipolar molecules like some antibiotics34,104 and deserves a closer analysis.
Fig. 6 shows contour maps of the electric potential (in kT/e units) in the cross-sectional plane (at axial coordinate z = 3.42 nm) of the central constriction of the WT-OmpF channel (Fig. 6a) and the D113C/E117C mutant (Fig. 6b) where the two carboxylates D113 and E117 have been replaced by neutral cysteines.41,102 In both panels the blue dot line separates the aqueous pore (enclosed region) and the surrounding protein domain, non accessible to solvent. Fig. 6a shows the strong negative electric potential due to the channel acid groups and also a region of positive potential located near the cluster of arginines. This is in good agreement with MD simulations reporting that cations and anions follow well-separated permeation pathways along the OmpF porin in screw-like way.48,49Fig. 6b shows that the region of negative potential vanishes when two neutral residues replace the key acids D113 and E117. Such substitution clearly distorts the subtle permeation mechanism described in Fig. 6a and hinders the transport of cations through the constriction. This conjecture is supported by the fact that in experiments performed in 2 M KCl the conductance of the D113C/E117C mutant (∼2 nS) is less than one third of the conductance measured in WT OmpF (∼7 nS).53
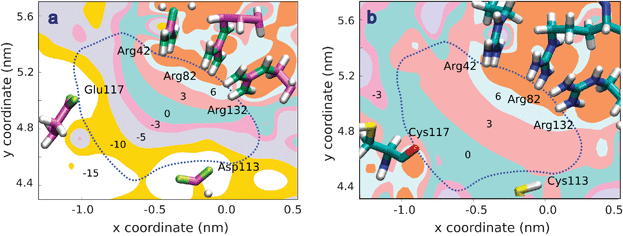 | ||
| Fig. 6 Contour plot of the electric potential in a cross-sectional plane of the OmpF channel central constriction (at z = 3.42 nm). The coordinates x, y, z are those of the first OmpF atomic structure.19 Numbers denote the maximum value of the potential on each region in kT/e (∼25.7 mV) units. The dot line depicts the limit between the aqueous pore (inner region) and the surrounding protein domain, non accessible to solvent. (a) Constriction of WT-OmpF. Even though the channel is cation selective, calculations show that there are two regions of positive and negative potential which may be the path for anions and cations when crossing the selectivity filter. This assumption has been confirmed by MD simulations.48,49 (b) Constriction of the D113C/E117C mutant. The region of negative potential is lost and cation permeation is hindered. | ||
In addition, for a proper interpretation of the role of selectivity filter in wide channels, it must be borne in mind that any electrostatic interaction between ions and protein charges is enhanced in this region.89Water in confined geometries exhibits properties very different from those in bulk. In such a small region and surrounded by charged residues, water molecules reduce their translational and rotational mobility.105 The decreased polarizability of water is reflected in a reduction of the dielectric constant known as dielectric saturation of water. Some studies addressed this problem by assigning to the effective dielectric constant of water in the channel a single value lower than the well known ε ∼ 80.106,107 For the specific case of OmpF, continuum electrostatic calculations combined with electric field dependent dielectric constants have suggested that the water dielectric constant at the constriction may be reduced down to 50% of its usual value in bulk water.89 This estimation changes considerably the prediction of the free energy well that cations encounter as they go through the channel eyelet.
4.3 Selectivity measurements to explore channel asymmetry
The distribution of ionizable residues in many protein channels is not homogeneous but asymmetric. This means that the part of the channel facing the extracellular side may exclude/accumulate ions in a different way than the part of the channel that faces the cytosol or the intermembrane space.2 If the orientation of the channel when reconstituted in the lipid membrane is mostly unidirectional, as happens with the OmpF porin,36 this asymmetry can be probed by selectivity measurements.36,108The RP measured in OmpF depends on the direction of the concentration gradient, i.e. the absolute value of RP for a given ratio Ccis/Ctrans = r and its inverse Ccis/Ctrans = 1/r is different. For a totally symmetric channel the two experiments should give the same RP, but opposite in sign.36,108 The difference between RP in the two cases may be used to probe channel orientation if the channel structure is known. Alternatively, it can be used to explore channel asymmetry when the channel orientation in experiments is known. Fig. 7 shows the results of RP measurements for a wide range of concentration ratio values (For making easier the comparison, absolute values of RP are plotted). Model calculations can be used to rationalize the asymmetry of the channel selectivity in terms of the distribution of channel ionizable residues in the axial direction. This asymmetrical channel charge distribution cannot be inferred from the usual current–voltage measurements unless its effect is high enough to produce current rectification. As Fig. 8 shows, in OmpF almost no rectification is observed.109 Interestingly, charge asymmetry has been reported in conical synthetic nanopores, showing that in such system it is possible to use it as a basis to construct ionic nanofilters.93
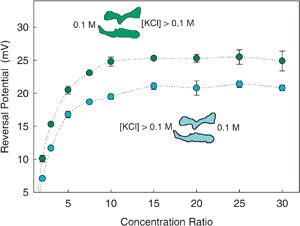 | ||
| Fig. 7 Change of OmpF reversal potential (absolute value) with salt concentration ratio in KCl solutions at pH 6. The lower electrolyte concentration was kept at 0.1 M while the higher concentration was changed to obtain the desired concentration ratio. Details of the experiments are given elsewhere.36 | ||
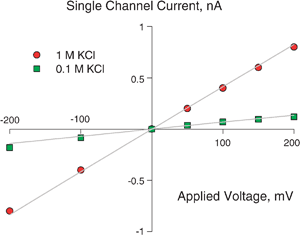 | ||
| Fig. 8 Experimental (symbols) and theoretical (solid lines) current–voltage curves in OmpF channel for two KCl electrolyte solutions. The model correctly predicts the change in the slope of the curves. | ||
4.4 Selectivity inversion by multivalent cations
The close relationship between channel charge and selectivity mentioned in previous sections is lost when another type of short-range interaction between permeant ions and protein groups outweighs the Coulombic attraction or repulsion.37,41 Any short-range interaction between ions and protein sites is likely to take place in the narrow selectivity filter where the distances are much smaller. In this context, it becomes relevant a recent publication of a 1.6 Å OmpF structure in 1 M MgCl2, which showed that one Mg2+ ion is bound in the selectivity filter between D113 and E117 of loop 3.21 This feature is probably behind the reported electrophysiological response of the channel: the well-known moderate cationic selectivity of the bacterial porin OmpF in salts of monovalent ions36,38,44,45,110 turns into anionic selectivity in salts of divalent40,41,37 and trivalent cations.102Fig. 9a shows the permeability ratios obtained viaGHK equation from RP measurements performed under a 10-fold concentration gradient of KCl and MgCl2. Also, sample current traces of the corresponding RP measurements are displayed in Fig. 9b and 9c.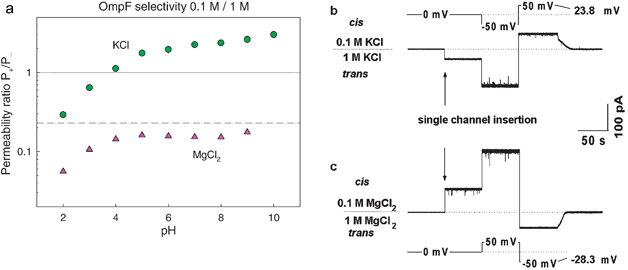 | ||
| Fig. 9 Selectivity inversion by divalent cations. (a) Permeability ratio of OmpF channel in salts of KCl and MgCl2. The lines represent the diffusion potential obtained under a concentration gradient a 0.1 M/1 M for KCl (solid line) and MgCl2 (dashed-dotted line). (b) and (c) Current traces of spontaneous channel insertion at 10-fold gradients of KCl (b) and MgCl2 (c). The potentials that should be applied in order to zero currents in the two cases are of opposite signs. | ||
In the literature of ion channel biophysics, the selectivity experiments are customary interpreted in terms of an effective channel charge. This concept refers to the charge that gives rise to the electric field actually felt by the ions permeating through the channel.2,26 Following an intuitive reasoning one could speculate that the anion selectivity (P+/P− ≈ 0.2) found for MgCl2 around neutral pH could come from a “charge inversion” phenomenon, i.e. from an effective positive charge in the channel.102 As is known, charge inversion occurs when interfacial charges attract counterions in excess of their own nominal charge111 and this phenomenon has been reported in several charged biomolecules.112 However, the connection between charge and selectivity is not so straightforward. Recall that the large difference between cation and anion diffusivities in salts of multivalent cations originates a significant diffusion potential. The diffusion potential shown in Fig. 9 (dashed-dotted line) would be the measured selectivity in a neutral ideal channel devoid of any electrostatic interaction and just filled with a MgCl2 solution. Furthermore, in a channel with negative net charge the diffusional contribution and the electrostatic partitioning contribution have opposite signs.38 This means that the selectivity inversion may be simply a consequence of the counterbalance between the diffusional contribution and the channel electrostatic preference for cations.102 This stresses the point already made in section 3.4: the GHK equation provides a poor characterization of the system because it merges different sources of selectivity in a single parameter that cannot be unambiguously rationalized.
5. Merging information from selectivity measurements and simulations
5.1 Different experimental protocols may yield dispersion of selectivity values
Channel selectivity can be quantified in several ways and each particular method involves a different balance between ion partitioning and electrodiffusion. In section 3 we have shown that it is not possible to choose a model-independent selectivity “value”, at least one should select the method most suitable for determining the selectivity in a particular situation.26 For determining the cation over anion selectivity the method of choice must be measuring RP, given that neither BP nor conductance ratios could provide such information. However, previous results suggest that a single experiment could be insufficient to assess channel selectivity. A combination of several experiments under a variety of conditions is mandatory to probe the interaction of ions with the channel. For instance, since electrostatic partitioning in the channel and the difference in cation/anion diffusivities may contribute with opposite sign to the measured zero current potential, the dependence of RP with concentration ratio may be biphasic.38 Thus, a single RP measurement for a typical ten-fold concentration ratio could be not representative of the channel selectivity because the same RP value is measured for a higher concentration ratio.The measurement of BP is more suitable for determining channel relative permeabilities of cations (in a cation selective channel) or anions (in an anion selective channel). However, the usual conversion of BP into relative permeabilities through the GHK equation can only be done if the anion channel permeability is negligible (see section 3.2) and in multiionic channels this seldom happens unless the salt concentrations of the solutions are very low in comparison with the channel effective fixed charge concentration.
Generally speaking, conductance and RP experiments give similar information but differ in some particular details. For instance, the selectivity inversion induced by divalent cations is not revealed by conductance measurements. In addition, RP measurements have another practical advantage over conductance ratio: conductance measurements require a single channel while RP can be measured no matter the number of protein channels inserted in the lipid membrane.
5.2 Conflicting data in RP experiments
Even for the same OmpF channel and under identical conditions of salt concentration, pH, lipid composition of the membrane, etc., the reported selectivity “numbers” exhibit certain dispersion.43 It is generally admitted that upon reconstitution in a lipid bilayer, this channel is weakly cation selective in salts of monovalent cations (KCl, NaCl, CsCl, LiCl)36,44,45,98,39 and becomes anion selective in salts of divalent cations (CaCl2, MgCl2, BaCl2, NiCl2).37,102,41,90,40 However, by analyzing in detail such wealth of selectivity measurements one cannot get from them a clear picture of the molecular basis of selectivity. In particular, the study of the cation-anion permeabilities in chlorides of alkali metals does not correlate well with the hydrated cation size. According to some studies OmpF prefers smaller cations98,45,39 while others report the opposite.36,38,40 The conflict does not only come from the use of different techniques (patch clamp, planar lipid membranes) or different protocols (reversal potential, channel conductance, bi-ionic potential or other experiments) to measure selectivity.26 Discrepancies are found even between experiments performed following the same technique and the same protocol under identical conditions. Thus, from RP measurements (1 M|0.1 M) in planar lipid membranes, several laboratories infer that OmpF channel is more permeable to K+ than to Na+36,38,44,110,40 and some others reach the opposite conclusion.98,39,45,103 Some of the disagreements found have been traditionally ascribed to the protein preparation and to the fine details of the channel reconstitution technique.22 Having in mind the number of factors contributing to the final result (chamber geometry, “painted-bilayer” versus “raised bilayer” technique, electrode assemblies, detergents used to solubilise the protein, etc.) it is clear that a side-by-side comparison of the results obtained is not always possible. However, when exhaustive information about the whole experimental procedure is available, many of these apparent conflicts vanish. The direction of the salt gradient with respect to the channel orientation in the membrane and the way liquid junction potentials are accounted for are two examples of causes of misinterpretation of RP measurements.37In this sense, we would like to make a technical point about the contribution of liquid junction potentials at electrode assemblies. Electrophysiological measurements are mostly done with reversible Ag/AgCl electrodes, which in many cases are immersed in salt bridges. When measuring electric potential differences between two solutions of different concentration (as is the case of RP and BP), one cannot ignore that each bridge is in contact with a different solution. The potential difference generated across each bridge/solution interface, known as liquid junction potential (LJP), is different, so that there is a non-zero contribution of both LJP to the total measured potential. Seminal studies by Barry and Neher113,114 indicated that LJP is small (∼1 mV) in RP experiments done with KCl that involve KCl bridges. That is the reason why these corrections are usually bypassed when measurements are performed at physiological conditions (moderate gradients of KCl solutions at decimolar concentrations buffered at neutral pH, occasionally containing also salts of divalent cations in micromolar or millimolar concentrations). However, in experiments with other salts (NaCl, LiCl, CaCl2, MgCl2, etc.) the LJP contribution becomes significant and may be comparable to the actual RP. The situation aggravates when electrode salt bridges are diluted, sometimes with the purpose to prevent contamination of the measuring solutions. LJP can be then several times larger than the actual RP or BP. This fact has long been pointed out115 but repeatedly ignored, thus leading to some inconsistencies in the selectivity data. A detailed study of LJP calculations and their contribution to the measured selectivity can be found elsewhere.37
5.3 Comparing measurements with computed selectivity from simulations
Simulation of ion transport by using any of the MD or BD approaches mentioned above has become increasingly common thanks to the availability of many protein atomic structures obtained from crystal X-ray diffraction or NMR.54–58 Simulations have provided useful information on ion diffusivities, ion partitioning within the channel, specific ion affinity of some channel binding sites, role of particular ionizable residues, etc.48,17 However, there remain some challenges like predicting channel selectivity in a way directly comparable with measurements of RP and BP. The main technical difficulty lies in the MD or BD simulation of an ion concentration gradient across the channel as the one imposed in RP and BP experiments. This means that, in practice, simulations are customary built with the same number of ions on both solutions.50–53 Then, it is obvious that simulations can hardly reproduce the same experimental conditions met in RP and BP measurements. The non-equilibrium component of channel selectivity that in RP and BP experiments arises from a concentration gradient is obtained in MD and BD simulations from the application of an external electric field. Consequently, the outcomes of experiments and simulations are not directly comparable.Simulations quantify channel selectivity as the ratio between simulated currents of cations and anions, I+/I−. From all the trajectories of the ions, a certain fraction β will pass through the channel (sometimes referred as “transfer efficiency”66,98,103). If the fraction of cation trajectories β+ in the simulation is greater than that of anions β−, then it is interpreted that the channel is selective to cations, otherwise it is anion selective. The determination of the fraction β can be done simply by counting the number of paths for a given ionic species that cross the channel in relation to the total number of paths. There is also a more sophisticated method to estimate this magnitude. This consists in calculating the electric current carried across the simulation box by all the ions.59,52,53 This is done at every time step of the simulation and it is repeated for all ionic species, so that one can calculate their ratio and hence obtain a measure of selectivity at each step in the simulation. This type of approach has the advantage of reducing the fluctuations of the derived magnitudes (by working on a larger number of atoms) and that it shows if the steady state is reached (i.e., if the system is equilibrated and therefore if simulation time is long enough).
MD and BD simulations can also answer the question of how ions partition inside the channel. In fact this is probably a problem well suited to these two techniques because we only need equilibrium simulations to obtain the relevant data. From the trajectories one can obtain the relative residence time of ions of each kind inside the channel, that is, the ratio between the time spent inside the channel to the total time duration of the trajectory.59,53 The average of these ratios for all the available trajectories is regarded as a measure of the selectivity. In MD simulations an equivalent method consists on summing up the number of ions of each kind inside the channel at each time step, and dividing by the total number of steps to get the average number of ions of each kind inside the channel.59,53 Then, a direct measure of the partitioning of ions inside the channel in this case is given by their ratio n+/n−. Since this ratio only reflects the equilibrium component of channel selectivity (mostly electrostatic interaction between mobile ions and protein charges in wide channels), it is evident that it cannot be generally regarded as a measure of selectivity.
Despite the above considerations, it is tempting to compare the permeability ratio P+/P− derived from RP measurements (viaGHK equation) with the ratio of simulated currents I+/I− or the occupancy ratio n+/n−. As an example, we show here a particular case where this may be acceptable. We compare the RP measured in the OmpF channel under a small concentration gradient (1.5 M|1 M) of KCl (a salt where cations and anions have almost equal diffusivities) with a MD simulation of OmpF bathed by a 1 M KCl solution. Since the diffusion potential of KCl is small and even barely measurable under such a small concentration gradient, channel selectivity arises mainly from the accumulation of cations and exclusion of anions in the channel. Then, it is expected that the permeability ratio P+/P− be close to the current ratio and the occupancy ratio.
Table 1 shows the results obtained from a MD simulation (∼25 ns) performed in OmpF53 together with cation/anion permeability ratio extracted from RP measurements performed under a small KCl concentration gradient.36,117 The agreement is fairly good for these particular conditions. Fig. 10 displays a snapshot of that simulation, one of the longest MD simulations performed on a wide protein channel up to date.
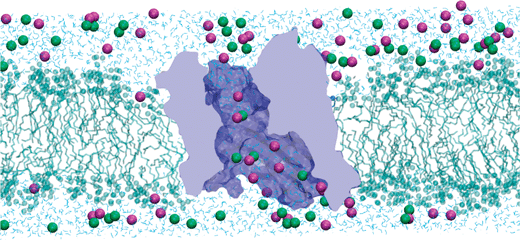 | ||
| Fig. 10 Snapshot of a MD simulation of KCl (1 M) transport across the OmpF channel embedded in a lipid bilayer. K+ ions are represented by magenta spheres and Cl− ions by green spheres. The structure of the protein has been constructed from the X-ray crystallographic data.19 For clarity of the representation, many water molecules, many lipids and the other two monomers are not shown. The picture has been produced using VMD.116 | ||
| 1.5 M|1 M KCl | 1 M KCl | |
|---|---|---|
| P K/PCl | β K/βCl | n K/nCl |
| 1.86 | 1.89 | 1.7 |
Conclusions
We have reviewed here several ways of measuring the selectivity of large multiionic channels, with particular emphasis on results obtained from the bacterial porin OmpF. Understanding the various sources of channel selectivity is shown to be a first, necessary step for a correct interpretation of selectivity measurements as well as for linking channel function with structural properties. Even though the differences in ion diffusivities and the different electrostatic partitioning of ions inside the channel are interconnected, some measurements of selectivity can be performed to analyze separately each one of these two contributions.Selectivity measurements can also be helpful in providing information about the tertiary structure of protein channels whose atomic structure has not been resolved by X-ray or NMR methods. In particular, RP measurements can be used to explore the titration of ionizable residues. In cases where the crystal structure is known—as happens in OmpF, VDAC, α-hemolysin, and other channels—RP measurements give insights on the pKa of the residues involved in channel selectivity. Interestingly, by confronting OmpF selectivity measurements with a detailed analysis of the distribution of the protein charged residues, it is found that the channel selectivity is not completely ruled by the charges at the channel constriction (what is usually known as selectivity filter in many channels) but is the result of the concerted action of many other residues. Experiments with OmpF mutants in which key acidic residues of the constriction were replaced by neutral ones support this conclusion.
None of the common experimental methods used to quantify channel selectivity is fully comprehensive of the channel preference for a particular type of ions. The translation of selectivity into ion permeability ratio viaGHK equation should be made with caution because it is valid only under some limitations. Because each particular method involves a different balance between partitioning and diffusion, it is necessary to merge the information gathered in several experiments (changing pH, salt concentration, etc.). It should be also taken into account that in some particular cases short-range interactions between multivalent cations and protein residues can reverse the channel selectivity and alter significantly the balance between the two contributions of partitioning and diffusion to selectivity.
By combining measurements (RP, BP, conductance ratio) with model calculations based on the 3D structure of the OmpF channel as well as MD simulations a consistent, unitary picture of channel selectivity, role of ionizable residues, and ion pathways across the channel is achieved. Finally, the challenges of comparing selectivity experiments with molecular simulations are discussed.
Acknowledgements
Support from the Spanish Ministry of Science and Innovation, (MICINN Projects No. FIS2007-60205 and FIS2010-19810) and Fundació Caixa Castelló-Bancaixa (Project No. P1-1A2009-13) is acknowledged.References
- D. J. Aidley and P. R. Stanfield, Ion Channels: Molecules in Action, Cambridge University Press, New York, 1996 Search PubMed.
- B. Hille, Ion Channels of Excitable Membranes, Sinauer Associates, Sunderland, MA, 3rd edn, 2001 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, M. Raff, K. Roberts and P. Walter, Molecular Biology of the Cell, Garland Science, New York, 4th edn, 2002 Search PubMed.
- R. S. Kass, J. Clin. Invest., 2005, 115, 1986 CrossRef CAS.
- B. Dworakowska and K. Dolowy, Acta Biochim. Pol., 2000, 47, 685 CAS.
- V. A. Parsegian, Nature, 1969, 221, 844 CAS.
- F. Ashcroft, D. Benos, F. Bezanilla, K. Chien, S. Choe, D. Clapham, D. Dougherty, M. Lazdunski, I. Levitan and R. Lewis, et al. , Nat. Rev. Drug Discovery, 2004, 3, 239 CrossRef.
- H. Bayley and P. S. Cremer, Nature, 2001, 413, 226 CrossRef CAS.
- V. M. Aguilella and A. Alcaraz, Nat. Nanotechnol., 2009, 4, 403 CrossRef.
- G. Maglia, A. J. Heron, W. L. Hwang, M. A. Holden, E. Mikhailova, Q. Li, S. Cheley and H. Bayley, Nat. Nanotechnol., 2009, 4, 437 CrossRef CAS.
- C. C. Harrell, Z. S. Siwy and C. R. Martin, Small, 2006, 2, 194 CrossRef CAS.
- C. Dekker, Nat. Nanotechnol., 2007, 2, 209 CrossRef CAS.
- A. L. Hodgkin, A. F. Huxley and B. Katz, J. Physiol., 1952, 116, 69–77.
- S. Laughlin, R. de Ruyter van Steveninck and J. Anderson, Nat. Neurosci., 1998, 1, 36 CrossRef CAS.
- B. Corry, Mol. BioSyst., 2006, 2, 527 RSC.
- P. Tieleman, P. C. Biggin, G. R. Smith and M. S. P. Sansom, Q. Rev. Biophys., 2001, 4, 473.
- B. Roux, T. Allen, S. Bernèche and W. Im, Q. Rev. Biophys., 2004, 37, 15 CrossRef CAS.
- Y. Luo, B. Egwolf, D. E. Walters and B. Roux, J. Phys. Chem. B, 2010, 114, 952 CrossRef CAS.
- S. W. Cowan, T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R. A. Pauptit, J. N. Jansonius and J. P. Rosenbusch, Nature, 1992, 358, 727 CrossRef CAS.
- B. Dhakshnamoorthy, S. Raychaudhury, L. Blachowicz and B. Roux, J. Mol. Biol., 2010, 396, 293 CrossRef CAS.
- E. Yamashita, M. V. Zhalnina, S. D. Zakharov, O. Sharma and W. A. Cramer, EMBO J., 2008, 27, 2171 CrossRef CAS.
- A. H. Delcour, Front. Biosci., 2003, 8, d1055–d1071 CrossRef CAS.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS.
- A. H. Delcour, Biochim. Biophys. Acta, Proteins Proteomics, 2009, 1794, 808 CrossRef CAS.
- H. Bayley, B. Cronin, A. Heron, M. A. Holden, W. L. Hwang, R. Syeda, J. Thompson and M. Wallace, Mol. BioSyst., 2008, 4, 1191 RSC.
- D. Gillespie and R. S. Eisenberg, Eur. Biophys. J., 2002, 31, 454 CrossRef CAS.
- P. Agre, G. M. Preston, B. L. Smith, J. S. Jung, S. Raina, C. Moon, W. B. Guggino and S. Nielsen, Am. J. Physiol., 1993, 265, F463 CAS.
- P. Agre, Proc. Am. Thorac. Soc., 2006, 3, 5 CrossRef CAS.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2009, 131, 8092 CrossRef CAS.
- S. Y. Noskov, S. Berneche and B. Roux, Nature, 2004, 431, 830 CrossRef CAS.
- D. A. Doyle, J. M. Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait and R. MacKinnon, Science, 1998, 280, 69–77 CrossRef CAS.
- S. Y. Noskov and B. Roux, Biophys. Chem., 2006, 124, 279–291 CrossRef CAS.
- M. Colombini, Mol. Cell. Biochem., 2004, 256, 107 CrossRef.
- E. M. Nestorovich, C. Danelon, M. Winterhalter and S. M. Bezrukov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9789 CrossRef CAS.
- B. Hille, J. Gen. Physiol., 1971, 58, 599 CrossRef CAS.
- A. Alcaraz, E. M. Nestorovich, M. Aguilella-Arzo, V. M. Aguilella and S. M. Bezrukov, Biophys. J., 2004, 87, 943 CrossRef CAS.
- A. Alcaraz, E. M. Nestorovich, M. L. López, E. García-Giménez, S. M. Bezrukov and V. M. Aguilella, Biophys. J., 2009, 96, 56 CrossRef CAS.
- M. L. López, M. Aguilella-Arzo, V. M. Aguilella and A. Alcaraz, J. Phys. Chem. B, 2009, 113, 8745 CrossRef CAS.
- N. Saint, K. L. Lou, C. Widmer, M. Luckey, T. Schirmer and J. P. Rosenbusch, J. Biol. Chem., 1996, 271, 20676 CrossRef CAS.
- H. Miedema, A. Meter-Arkema, J. Wierenga, J. Tang, B. Eisenberg, W. Nonner, H. Hektor, D. Gillespie and W. Meijberg, Biophys. J., 2004, 87, 3137 CrossRef CAS.
- M. Vrouenraets, J. Wierenga, W. Meijberg and H. Miedema, Biophys. J., 2006, 90, 1202 CrossRef CAS.
- E. Blachly-Dyson, S. Peng, M. Colombini and M. Forte, Science, 1990, 247, 1233 CrossRef CAS.
- M. L. López, E. García-Giménez, V. M. Aguilella and A. Alcaraz, J. Phys. Condens. Mat., 2010, 22, 454106 Search PubMed.
- R. Benz, A. Schmid and R. E. W. Hancock, J. Bacteriol., 1985, 162, 722 CAS.
- C. Danelon, A. Suenaga, M. Winterhalter and I. Yamato, Biophys. Chem., 2003, 104, 591 CrossRef CAS.
- A. Hodgkin and B. Katz, J. Physiol., 1949, 108, 37 CAS.
- J. P. Sandblom and G. Eisenman, Biophys. J., 1967, 7, 217 CrossRef CAS.
- W. Im and B. Roux, J. Mol. Biol., 2002, 322, 851 CrossRef CAS.
- W. Im and B. Roux, J. Mol. Biol., 2002, 319, 1177 CrossRef CAS.
- C. Chimerel, L. Movileanu, S. Pezeshki, M. Winterhalter and U. Kleinekathöfer, Eur. Biophys. J., 2008, 38, 121 CrossRef CAS.
- M. Ceccarelli and P. Ruggerone, Curr. Drug Targets, 2008, 9, 779 Search PubMed.
- S. Pezeshki, C. Chimerel, A. N. Bessonov, M. Winterhalter and U. Kleinekathöfer, Biophys. J., 2009, 97, 1898 CrossRef CAS.
- J. Faraudo, C. Calero and M. Aguilella-Arzo, Biophys. J., 2010, 99, 2107 CrossRef CAS.
- D. P. Tieleman and H. J. C. Berendsen, Biophys. J., 1998, 74, 2786 CrossRef CAS.
- T. W. Allen, S. Kuyucak and S. H. Chung, Biophys. J., 1999, 77, 2502–2516 CrossRef CAS.
- I. H. Shrivastava and M. S. P. Sansom, Biophys. J., 2000, 78, 557–570 CrossRef CAS.
- S. Bernèche and B. Roux, Biophys. J., 2000, 78, 2900–2917 CrossRef CAS.
- T. W. Allen, O. S. Andersen and B. Roux, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 117–122 CAS.
- A. Aksimentiev and K. Schulten, Biophys. J., 2005, 88, 3745–3761 CrossRef CAS.
- I. Biró, S. Pezeshki, H. Weingart, M. Winterhalter and U. Kleinekathöfer, Biophys. J., 2010, 98, 1830 CrossRef CAS.
- A. D. MacKerell, B. Brooks, C. L. Brooks, L. Nilsson, B. Roux, Y. Won and M. Karplus, The Encyclopedia of Computational Chemistry, John Wiley & Sons, Chichester, 1998, vol. 1 Search PubMed.
- J. W. Ponder and D. A. Case, Adv. Protein Chem., 2003, 66, 27 CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435 CrossRef CAS.
- J. E. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale and K. Schulten, J. Comput. Chem., 2005, 26, 1781 CrossRef CAS.
- D. A. Case, T. E. Cheatham, III, T. Darden, H. Gohlke, R. Luo, K. M. Merz, Jr., A. Onufriev, C. Simmerling, B. Wang and R. Woods, J. Comput. Chem., 2005, 26, 1668 CrossRef CAS.
- W. Im, S. Seefeld and B. Roux, Biophys. J., 2000, 79, 788 CrossRef CAS.
- S. Y. Noskov, W. Im and B. Roux, Biophys. J., 2004, 87, 2299 CrossRef CAS.
- D. Marreiro, M. Saraniti and S. Aboud, J. Phys.: Condens. Matter, 2007, 19, 215203 CrossRef.
- G. Moy, B. Corry, S. Kuyucak and S. H. Chung, Biophys. J., 2000, 78, 2349–2363 CrossRef CAS.
- W. Im and B. Roux, J. Chem. Phys., 2001, 115, 4850 CrossRef CAS.
- W. Nonner, D. P. Chen and B. Eisenberg, Biophys. J., 1998, 74, 2327 CrossRef CAS.
- D. Chen, L. Xu, A. Tripathy, G. Meissner and B. Eisenberg, Biophys. J., 1999, 76, 1346 CrossRef CAS.
- A. E. Cardenas, R. D. Coalson, P. Graf and A. Nitzan, Biophys. J., 2000, 79, 80–93 CrossRef CAS.
- M. G. Kurnikova, R. D. Coalson, P. Graf and A. Nitzan, Biophys. J., 1999, 76, 642–656 CrossRef CAS.
- P. Graf, M. Kurnikova, R. Coalson and A. Nitzan, J. Phys. Chem. B, 2004, 108, 2006–2015 CrossRef CAS.
- O. P. Choudhary, R. Ujwal, W. Kowallis, R. Coalson, J. Abramson and M. Grabe, J. Mol. Biol., 2010, 396, 580 CrossRef CAS.
- M. Aguilella-Arzo, J. J. García-Celma, J. Cervera, A. Alcaraz and V. M. Aguilella, Bioelectrochemistry, 2007, 70, 320 CrossRef CAS.
- D. S. Bolintineanu, A. Sayyed-Ahmad, H. T. Davis and Y. N. Kaznessis, PLoS Comput. Biol., 2009, 5, e1000277 CrossRef.
- D. Goldman, J. Gen. Physiol., 1943, 27, 37 CrossRef CAS.
- S. Mafé, J. Pellicer and V. M. Aguilella, Ber. Bunsen-Ges., 1986, 90, 476 CAS.
- J. Pellicer, S. Mafé and V. M. Aguilella, Ber. Bunsen-Ges., 1986, 90, 867 CAS.
- A. Syganow and E. von Kitzing, Biophys. J., 1999, 76, 768 CrossRef CAS.
- P. H. Barry, Cell Biochem. Biophys., 2006, 46, 143 CrossRef CAS.
- S. Varma and E. Jakobsson, Biophys. J., 2004, 86, 690–704 CrossRef CAS.
- C. N. Schutz and A. Warshel, Proteins: Struct., Funct., Genet., 2001, 44, 400 CrossRef CAS.
- J. Antosiewicz, J. A. McCammon and M. K. Gilson, J. Mol. Biol., 1994, 238, 415 CrossRef CAS.
- J. Antosiewicz, J. A. McCammon and M. K. Gilson, Biochemistry, 1996, 35, 7819 CrossRef CAS.
- J. D. Madura, J. M. Briggs, R. C. Wade, M. E. Davis, B. A. Luty, A. Ilin, J. Antosiewicz, M. K. Gilson, B. Bagheri, L. R. Scott and J. A. McCammon, Comput. Phys. Commun., 1995, 91, 57 CrossRef CAS.
- M. Aguilella-Arzo, A. Andrio, V. M. Aguilella and A. Alcaraz, Phys. Chem. Chem. Phys., 2009, 11, 358 RSC.
- H. Miedema, M. Vrouenraets, J. Wierenga, D. Gillespie, B. Eisenberg, W. Meijberg and W. Nonner, Biophys. J., 2006, 91, 4392 CrossRef CAS.
- A. Fulinski, I. D. Kosinska and Z. Siwy, New J. Phys., 2005, 7, 132 CrossRef.
- J. Cervera, B. Schiedt and P. Ramírez, Europhys. Lett., 2005, 71, 35 CrossRef CAS.
- I. D. Kosinka and A. Fulinski, Phys. Rev. E, 2005, 72, 011201–1 CrossRef.
- B. Corry, S. Kuyucak and S. H. Chung, Chem. Phys. Lett., 2000, 320, 35 CrossRef CAS.
- D. G. Levitt, Biophys. J., 1991, 59, 278 CrossRef CAS.
- E. B. Zambrowicz and M. Colombini, Biophys. J., 1993, 65, 1093 CrossRef CAS.
- V. Levadny and V. M. Aguilella, J. Phys. Chem. B, 2001, 105, 9902 CrossRef CAS.
- P. S. Phale, A. Philippsen, C. Widmer, V. P. Phale, J. P. Rosenbusch and T. Schirmer, Biochemistry, 2001, 40, 6319 CrossRef CAS.
- S. Hiller, R. G. Garces, T. J. Malia, V. Y. Orekhov, M. Colombini and G. Wagner, Science, 2008, 321, 1206 CrossRef CAS.
- M. Bayrhuber, T. Meins, M. Habeck, S. Becker, K. Giller, S. Villinger, C. Vonrhein, C. Griesinger, M. Zweckstetter and K. Zeth, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15370 CrossRef CAS.
- R. Ujwal, D. Cascio, J. Colletier, S. Fahama, J. Zhanga, L. Toro, P. Pinga and J. Abramson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17742 CrossRef CAS.
- E. García-Giménez, A. Alcaraz and V. M. Aguilella, Phys. Rev. E, 2010, 81, 021912 CrossRef.
- T. Schirmer and P. S. Phale, J. Mol. Biol., 1999, 294, 1159 CrossRef CAS.
- V. Levadny, V. M. Aguilella, M. Aguilella-Arzo and M. Belaya, Phys. Rev. E, 2004, 70, 041912 CrossRef CAS.
- M. S. P. Sansom, G. R. Smith, C. Adcock and P. C. Biggin, Biophys. J., 1997, 73, 2404 CrossRef CAS.
- P. C. Jordan, in Ion Channel Reconstitution, ed. C. Miller, Springer, New York, 1986 Search PubMed.
- W. Cheng, C. X. Wang, W. Z. Chen, Y. W. Xu and Y. Y. Shi, Eur. Biophys. J., 1998, 27, 105 CrossRef CAS.
- E. M. Nestorovich, V. A. Karginov and S. M. Bezrukov, Biophys. J., 2010, 99, 782 CrossRef CAS.
- A. Alcaraz, P. Ramírez, E. García-Giménez, M. L. López, A. Andrio and V. M. Aguilella, J. Phys. Chem. B, 2006, 110, 21205 CrossRef CAS.
- E. M. Nestorovich, T. K. Rostovtseva and S. M. Bezrukov, Biophys. J., 2003, 85, 3718 CrossRef CAS.
- J. Lyklema, Colloids Surf., A, 2006, 291, 3 CrossRef CAS.
- W. M. Gelbart, R. F. Bruinsma, P. A. Pincus and V. A. Parsegian, Phys. Today, 2000, 53, 38 CrossRef CAS.
- P. H. Barry and J. M. Diamond, J. Membr. Biol., 1970, 3, 93 CrossRef CAS.
- E. Neher, Methods Enzymol., 1992, 207, 123 CAS.
- P. H. Barry and J. W. Lynch, J. Membr. Biol., 1991, 121, 101 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef.
- P. Ramírez, M. Aguilella-Arzo, A. Alcaraz, J. Cervera and V. M. Aguilella, Cell Biochem. Biophys., 2006, 44, 287 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |