Selective hydrogenolysis of biomass-derived xylitol to ethylene glycol and propylene glycol on supported Ru catalysts
Jiying
Sun
and
Haichao
Liu
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Stable and Unstable Species, College of Chemistry and Molecular Engineering, Green Chemistry Center, Peking University, Beijing, 100871, China. E-mail: hcliu@pku.edu.cn; Fax: +86-10-6275-4031; Tel: +86-10-6275-4031
First published on 1st December 2010
Abstract
The selective hydrogenolysis of biomass-derived xylitol to ethylene glycol and propylene glycol was carried out on different catalysts in the presence of Ca(OH)2. The catalysts included Ru supported on activated carbon (C) and, for comparison, on metal oxides, Al2O3, TiO2, ZrO2 and Mg2AlOx as well as C-supported other noble metals, Rh, Pd and Pt, with similar particle sizes (1.6–2.0 nm). The kinetic effects of H2 pressures (0–10 MPa), temperatures (433–513 K) and solid bases including Ca(OH)2, Mg(OH)2 and CaCO3 were examined on Ru/C. Ru/C exhibited superior activities and glycol selectivities than Ru on TiO2, ZrO2, Al2O3 and Mg2AlOx, and Pt was found to be the most active metal. Such effects of the metals and supports are attributed apparently to their different dehydrogenation/hydrogenation activities and surface acid-basicities, which consequently influenced the xylitol reaction pathways. The large dependencies of the activities and selectivities on the H2 pressures, reaction temperatures, and pH values showed their effects on the relative rates for the hydrogenation and base-catalyzed reactions involved in xylitol hydrogenolysis, reflecting the bifunctional nature of the xylitol reaction pathways. These results led to the proposition that xylitol hydrogenolysis to ethylene glycol and propylene glycol apparently involves kinetically relevant dehydrogenation of xylitol to xylose on the metal surfaces, and subsequent base-catalyzed retro-aldol condensation of xylose to form glycolaldehyde and glyceraldehyde, followed by direct glycolaldehyde hydrogenation to ethylene glycol and by sequential glyceraldehyde dehydration and hydrogenation to propylene glycol. Clearly, the relative rates between the hydrogenation of the aldehyde intermediates and their competitive reactions with the bases dictate the selectivities to the two glycols. This study provides directions towards efficient synthesis of the two glycols from not only xylitol, but also other lignocellulose-derived polyols, which can be achieved, for example, by optimizing the reaction parameters, as already shown by the observed effects of the catalysts, pH values, and H2 pressures.
1. Introduction
The use of renewable biomass provides a viable route to both alleviate the shortage of fossil fuels and reduce the CO2 emissions.1–3 In this context, biomass-derived polyols emerge as promising building blocks for liquid fuels and value-added chemicals from biomass.4–10 Among them, xylitol and sorbitol are especially noticeable because they are readily available from abundant lignocellulose, and can be converted to different products such as ethylene glycol, propylene glycol and glycerol.11,12Ethylene glycol and propylene glycol are important commodity chemicals. They are widely used as functional fluids such as antifreezes and coolants, and as monomers in the synthesis of polyester fibers and resins, etc. Industrial production of ethylene glycol and propylene glycol is currently based on the multi-step transformation of petroleum-derived ethylene and propylenevia their epoxide intermediates. In comparison with ethylene and propylene, xylitol and sorbitol are not only renewable, but also structurally analogous to ethylene glycol and propylene glycol with adjacent hydroxyl groups. These features render xylitol and sorbitol to be favorable feedstocks, in term of sustainability and energy efficiency, for the synthesis of the two glycols by catalytic hydrogenolysis.
Hydrogenolysis of xylitol and sorbitol to ethylene glycol and propylene glycol requires cleavage of specific C–C and C–O bonds, which generally involves the use of Ni or Ru-based catalysts and basic promoters.13–21 Clark et al.13 reported that sorbitol can be hydrogenolyzed to ethylene glycol, propylene glycol and glycerol in yields of 16%, 17% and 40%, respectively, at 488 K and 14 MPa H2 in water on kieselguhr supported Ni catalysts with Ca(OH)2 as a promoter. Using more basic NaOCH3, Tanikella et al.14 achieved high yields of ethylene glycol (45%) and propylene glycol (33%) from xylitol in methanol on Ni/Al2O3 and Ni/SiO2 at 548 K and 27.6 MPa H2. Dubeck et al.15 found that on sulfur-modified Ru/C catalysts, sorbitol hydrogenloysis forms the three products of ethylene glycol, propylene glycol and glycerol with a combined selectivity of as high as 96% in the presence of Ca(OH)2 at 513 K and 17 MPa H2. Such a high selectivity has been attributed tentatively to the electron transfer from Ru to S, which possibly leads concurrently to the deactivation of direct C–C and C–O hydrogenolysis and the formation of hydroxyl groups on Ru as the active sites for C–C bond cleavage via retro-aldol condensation, etc.16 The modification of Ni catalysts was also explored in the patents, as exemplified by Ni–Re/C17catalysts, exhibiting improved performances in hydrogenolysis of sorbitol and xylitol. For instance, on 2.5% Ni–2.5% Re/C, xylitol hydrogenolysis is 89% selective to the three products at a ∼50% conversion in the presence of KOH under relatively mild conditions (473 K and 8.5 MPa H2). Structural effects of the C supports have also been examined.18,19 Recently, Zhou et al.20,21 reported that mesoporous carbon nanofibers are superior as the Ru catalyst supports to the commercial activated carbons, which can facilitate the mass transfer and electronic modification of the Ru particles, and thus lead to a significant increase in the selectivity to ethylene glycol, propylene glycol and glycerol from 26.1% to 51.3% at similar sorbitol conversions (70–85%) under the identical conditions (493 K and 8 MPa H2).
These previous efforts have demonstrated the feasibility of the hydrogenolysis route in the synthesis of ethylene glycol and propylene glycol. However, these studies apparently encounter low activities or selectivities for the target glycols, or harsh reaction conditions (e.g. high temperatures or H2 pressures). Addressing these problems requires the development of improved catalysts, which is dependant unequivocally on the understanding of the key fundamental issues including the effect of catalyst structures and functions and the reaction mechanism. However, these issues remain still unclear, likely as a consequence that most of the studies to date have been reported in patents with focus mainly on optimization of the catalyst compositions and reaction parameters.
Here, we report a detailed study of xylitol hydrogenolysis on supported Ru catalysts. Our choice of xylitol is based on the following consideration. Relative to more extensively studied sorbitol, xylitol possesses a shorter chain and is more structurally advantageous in understanding the hydrogenolysis mechanism in term of formation of ethylene glycol and propylene glycol, the dominant products from both xylitol and sorbitol. On the other hand, xylitol is derived from xylose that is not as efficient as glucose in production of ethanol fuelviafermentation, and thus other outlets of xylitol need to be explored including its hydrogenolysis discussed here. We examine the effects of supports and basic promoters on the activity and selectivity of the Ru catalysts. The supports include activated carbon (C), ZrO2, TiO2, Al2O3 and Mg2AlOx with a wide range of acid-base and redox surface properties, and the bases include Ca(OH)2, Mg(OH)2 and CaCO3 resulting in different pH values in water. We compare the catalytic activity and selectivity of Ru/C and other supported noble metal catalysts, Rh/C, Pd/C and Pt/C, in xylitol hydrogenolysis, and also examine the effects of reaction parameters (e.g. pH, H2 pressure and temperature). These detailed studies are aimed to probe the effects of acid-basicity and dehydrogenation/hydrogenation property of the catalysts on the reaction pathways and to elucidate the reaction mechanism for xylitol hydrogenolysis.
2. Experimental
Activated carbon (C) supported noble metal catalysts, 4 wt% Ru/C, 4 wt% Pd/C, 8 wt% Pt/C and 4 wt% Rh/C, were prepared by incipient wetness impregnation of C (AR, Beijing Dali Fine Chemical, dried at 383 K in air for 12 h) with acetone solutions of RuCl3·nH2O (GR, Sinopharm Chemical), PdCl2 (AR, Shenyang Research Institute of Nonferrous Metals) with several drops of aqueous HCl solution, H2PtCl6·6H2O (AR, Beijing Chemical) and RhCl3·nH2O (Rh 38.5–45.5%, Alfa Aesar), respectively. After evaporation of acetone, the resulting powders were dried at 383 K in air overnight, and then reduced in a flowing 20% H2/N2 at 673, 523, 473 and 673 K, respectively.Metal oxide-supported Ru catalysts were prepared using the method similar to that of Ru/C above. Monoclinic ZrO2 (m-ZrO2) was prepared hydrothermally in the way reported previously by Liet al.22Mg2AlOx (Mg/Al molar ratio of 2/1) was prepared by urea hydrolysis reaction under hydrothermal conditions. Briefly, Teflon inner vessel containing an aqueous solution of 0.16 M Al(NO3)3, 0.33 M Mg(NO3)2, and 1.6 M urea in a stainless steel jacket was heated at 393 K for 12 h. The resulting white precipitates were filtered and washed thoroughly with de-ionized water. γ-Al2O3 (Condea Chemie Gmbh), TiO2 (Alfa Aesar), m-ZrO2 and Mg2AlOx were calcined at 673 K under an air flow before impregnation of them with acetone solutions of RuCl3.
The catalysts were characterized by X-ray diffraction (XRD) and transmission electron microscopy (TEM). The XRD patterns were obtained on a Rigaku D/MAX-2400 diffractometer using Cu Kα1 radiation (λ = 1.5406 Å) operated at 40 kV and 100 mA. The TEM images were taken on a Philips Tecnai F30 FEGTEM operated at 300 kV. Samples were prepared by uniformly dispersing the catalysts in ethanol and then placing them onto carbon-coated copper grids. The average size of metal particles and their size distributions were calculated by averaging of more than 300 particles randomly distributed in the TEM images.
Xylitol hydrogenolysis reactions were carried out in a Teflon-lined stainless steel autoclave (100 ml) at a stirring speed of 800 rpm. In a typical run, 40 g of 10 wt% xylitol (99%, Alfa Aesar) aqueous solution, 0.1 g of metal catalyst and at least 0.26 g of Ca(OH)2 were introduced to the autoclave. Afterwards, the reactor was purged with H2 (>99.99%, Beijing Huayuan) three times, and pressurized with H2 to 4.0 MPa and then heated to 473 K, which was kept constant during the reaction. The reactant and liquid products, after silylation with hexamethyldisilazane (HMDS) and trimethylchlorinesilane (TMSCl) (both ≥98.0%, Sinopharm Chemical) in pyridine (AR, Shantou Xilong Chemical), were analyzed by gas chromatography (Shimadzu 2010 GC) using a capillary column OV-101 (30 m × 0.25 mm × 0.33 µm) and a flame ionization detector. The reactant and liquid products were also analyzed by high-performance liquid chromatography (Shimadzu LC-20A) using a PRP-X300 column at 304 K and a PDA detector.
3. Results and discussion
Table 1 shows the activity and selectivity of xylitol hydrogenolysis at 473 K and 4.0 MPa H2 on different supported Ru catalysts in the presence of Ca(OH)2. The supports include activated carbon (C), TiO2, m-ZrO2, Al2O3, and Mg2AlOx with a wide range of surface properties such as acid-basicity and reducibility in favor of examining the support effects. C is a known favorable support in polyol hydrogenolysis, and it is inert and stable under hydrothermal conditions even at high pressures and temperatures.23TiO2 and m-ZrO2 are also hydrothermally stable, but they are amphoteric oxides with redox functions that can catalyze alcohol dehydrogenation and dehydration.22,23Mg2AlOx and Al2O3 are unreducible oxides with basic and acid properties, respectively. The five Ru catalysts were characterized by XRD and TEM. The XRD patterns (not included here) show only the diffraction features of the respective supports. The TEM images reveal that these catalysts possessed similar mean diameters of Ru particles (ca. 1.4–1.8 nm) with narrow size distributions, as shown in Fig. 1 and 2.| Catalyst | Activitiy (h−1) | Selectivity (on a carbon basis, %) | ||||||
|---|---|---|---|---|---|---|---|---|
| Ethylene glycol | Propylene glycol | Glycerol | Threitol | Arabitol | Lactic acid | Formic acid | ||
| a Reaction conditions: 473 K, 4.0 MPa H2, 0.02–0.20 g catalysts, 0.26–0.39 g Ca(OH)2, 40 g 10 wt% xylitol aqueous solution, 1 h, ∼20% xylitol conversion. b Not detected. | ||||||||
| Ru/C | 184.4 | 32.4 | 24.9 | 9.6 | 2.3 | trace | 16.8 | 2.3 |
| Ru/ZrO2 | 87.8 | 24.2 | 20.5 | 9.6 | 3.2 | trace | 22.8 | 2.1 |
| Ru/TiO2 | 66.7 | 30.0 | 24.0 | 8.0 | 2.3 | trace | 28.3 | 3.7 |
| Ru/Al2O3 | 162.5 | 17.5 | 9.0 | 13.2 | 4.3 | trace | 25.2 | 1.6 |
| Ru/Mg2AlOx | 145.2 | 19.5 | 7.9 | 4.8 | 6.9 | 3.4 | 36.5 | 2.2 |
| Pd/C | 63.0 | 30.0 | 29.0 | 3.4 | n.d.b | n.d. | 28.4 | 0.9 |
| Rh/C | 61.2 | 26.4 | 30.0 | 7.2 | n.d. | n.d. | 23.3 | 4.6 |
| Pt/C | 754.5 | 25.0 | 23.3 | 10.5 | n.d. | 8.3 | 15.5 | 2.0 |
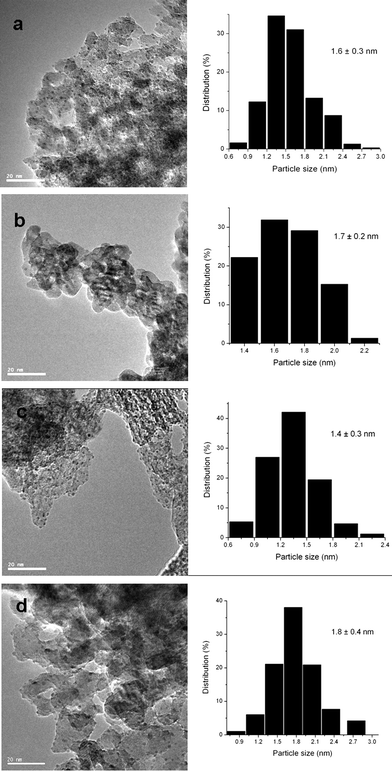 | ||
| Fig. 1 TEM micrographs and histograms of Ru particle size distribution for different Ru catalysts (scale bar = 20 nm). (a) Ru/Al2O3, 1.6 ± 0.3 nm; (b) Ru/ZrO2, 1.7 ± 0.2 nm; (c) Ru/Mg2AlOx, 1.4 ± 0.3 nm; (d) Ru/TiO2, 1.8 ± 0.4 nm. | ||
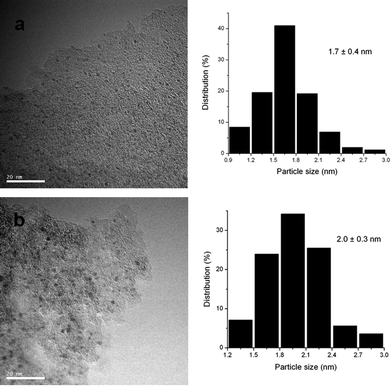 | ||
| Fig. 2 TEM micrographs and histograms of Ru particle size distribution of Ru/C before reaction and after six reaction cycles (scale bar = 20 nm). (a) Before reaction, 1.7 ± 0.4 nm; (b) after six reaction cycles, 2.0 ± 0.3 nm. | ||
On the five Ru catalysts, ethylene glycol, propylene glycol and glycerol were formed as the major products in the presence of Ca(OH)2. Lactic acid was also detected with a significant amount. Other products mainly included CH4, CO2 and some unidentified compounds possibly from condensation of xylitol or its derivatives. The carbon balance was generally better than 90%. The activities (normalized per Ru atom) and selectivities strongly depend on the identity of the supports. As shown in Table 1, the activity was 184.4 h−1 on Ru/C, which was greater than on the four metal oxide-supported Ru catalysts, especially Ru/ZrO2 (87.8 h−1) and Ru/TiO2 (66.7 h−1) at similar xylitol conversions (∼20%). Such support effect on the activity, although the underlying reason needs to be clarified, might be related to their effects on the Ru electronic property as well as to the difference in the adsorption of xylitol, its derivatives or products on these supports. As for the selectivities, Ru/C is more selective, relative to the other four catalysts, to ethylene glycol and propylene glycol with a combined selectivity of 57.3% at ca. 20% xylitol conversion (Table 1). Including glycerol, the selectivity to the sum of the three C2 and C3polyols reached 66.9% on Ru/C, which was similar to the result on Ru/TiO2 (62.0%), but much higher than those on Ru/ZrO2, Ru/Al2O3 and Ru/Mg2AlOx, being 54.3%, 39.7% and 32.2%, respectively (Table 1). In contrast, the selectivity to lactic acid increased from 16.8% on Ru/C to 36.5% on Ru/Mg2AlOx (Table 1). Such changes in these selectivities appear to be consistent with the known difference in the basicity of the five supports, i.e. C < TiO2 < ZrO2 < Al2O3 < Mg2AlOx, apparently reflecting the effects of the basicity on the xylitol reaction pathways, as discussed later.
Among the five supports examined above, C is clearly the most preferable one, leading to superior activity and selectivity to the target products for xylitol hydrogenolysis. Using C as support, three other noble metals, Pd, Rh and Pt, were also examined for comparison with Ru/C. The three catalysts were also characterized by TEM, and possessed metal particle sizes of 1.7–1.9 nm (Fig. 3), similar to the size for Ru/C (1.7 nm). In the presence of Ca(OH)2, the three catalysts were also active and selective for xylitol conversion to ethylene glycol and propylene glycol, and the combined selectivities (48.3–59.0%) were comparable with the value of Ru/C (57.3%) under the identical conditions. Pd/C and Rh/C were less active, but Pt/C offered a four times greater activity than Ru/C. The superior activity of Pt/C was also observed by Ukisu et al.24 in isopropanol dehydrogenation and by Davis et al.25 in glycerol hydrogenolysis under the basic conditions.
 | ||
| Fig. 3 TEM micrographs and histograms of metal particle size distribution for Pd/C, Pt/C and Rh/C (scale bar = 20 nm). (a) Rh/C, 1.7 ± 0.3 nm; (b) Pd/C, 1.8 ± 0.3 nm; (c) Pt/C, 1.9 ± 0.5 nm. | ||
The recyclability of these catalysts were examined in the kinetic controlled regime (at ca. 33% xylitol conversion), and the representative results on Ru/C are shown in Fig. 4. No significant decline in the xylitol activities and selectivities was observed after recycling the Ru/C catalyst over six times. This is consistent with the TEM characterization result of this catalyst (Fig. 2), demonstrating that the mean diameters of the Ru nanoparticles and their size distributions remained essentially unaltered (1.7 ± 0.4 vs. 2.0 ± 0.3 nm) after the six successive runs. These results reveal that the Ru/C catalyst is stable and recyclable under the reaction conditions in this work.
 | ||
| Fig. 4 Activities and selectivities to ethylene glycol, propylene glycol and glycerol for the six reaction cycles of xylitol hydrogenolysis on Ru/C. Reaction conditions: 473 K, 4.0 MPa H2, 0.15 g Ru/C (4 wt% Ru), 0.33 g Ca(OH)2, 40 g 10 wt% xylitol aqueous solution, 1 h. | ||
Fig. 4 displays the change of selectivities for the four major C2 and C3 products, ethylene glycol, propylene glycol, glycerol and lactic acid, as a function of the xylitol conversions on Ru/C at 473 K and 4.0 MPa H2. The selectivities to propylene glycol increased slightly from 24.0 to 28.4% with increasing the xylitol conversion from 15.0 to 98.0%, while the ethylene glycol selectivities remained essentially constant (ca. 33%). Such trend leads to the increase in the combined selectivity to 61.2% for the target glycols at nearly 100% xylitol conversion, corresponding to a 60% yield, showing the potential advantage of the Ru/C catalyst for selective hydrogenolysis of xylitol. These results reflect the stability of the two glycols under the conditions in this work, as indeed verified by their separate hydrogenolysis reactions under the identical conditions (not shown here). The non-zero selectivities for the two glycols observed as the conversions approached the zero level indicate that they are formed as primary products directly from xylitol. In contrast, the selectivities to glycerol declined dramatically from 14.6 to less than 1.0% concurrently with an increase in the lactic acid selectivity from 10.5 to 28.0%, as the conversion increased from 15.0 to 98.0%. Such concurrent changes in the lactic acid and glycerol selectivities indicate that lactic acid is formed via secondary glycerol reactions or they may be formed competitively from the same intermediates. The observed slight increase in the propylene glycol selectivities indicates that glycerol can also convert via its secondary reactions to propylene glycol. Such discussion on the glycerol conversion is consistent with the results reported previously20,25–28 and for our separate glycerol reaction. Glycerol hydrogenolysis on Ru/C in the presence of Ca(OH)2 at 473 K and 4.0 MPa H2 formed lactic acid and propylene glycol with 67.4% and 24.6% selectivities, respectively.
The reaction activities and selectivities strongly depend on the reaction parameters of H2 pressure, temperature, and pH value in the aqueous reaction solutions. Table 2 shows the effects of the pH value on xylitol hydrogenolysis on Ru/C at 473 K and 4.0 MPa H2. In the neutral solution, i.e. without addition of any bases, xylitol converted predominantly to arabitol and adonitol (totally 80%) together with small amounts of glycerol, threitol and erythritol on the Ru catalyst. Only trace amounts of ethylene glycol and propylene glycol were formed. Arabitol and adonitol are epimers of xylitol. It has been reported that polyol epimerization involves polyol dehydrogenation to the corresponding carbonyl intermediates and their subsequent hydrogenation.29 Together with the result of the control experiment showing no detectable xylitol conversion in the absence of Ru/C (with or without Ca(OH)2), the predominance of the xylitol epimers implies that xylitol hydrogenolysis on Ru/C proceeds primarily by its dehydrogenation.16,30–32 The detection of C4polyols, threitol and erythritol, is indicative that xylitol dehydrogenates at its primary hydroxyl groups to form the corresponding aldopentose (i.e.xylose) rather than ketopentoses. The reason is that hydrogenolysis of primary alcohols tends to occur by cleavage of their C–C bonds at the end of the chains via their aldehyde intermediates, while C–O bonds in secondary alcohols are more susceptible than C–C bonds to cleavage, forming dehydroxylated products on the metal catalysts.33,34
| Base | pHb | Activity (h−1) | Selectivity (on a carbon basis, %) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Ethylene glycol | Propylene glycol | Glycerol | Threitol | Erythritol | Arabitol | Adonitol | Lactic acid | |||
| a Reaction conditions: 473 K, 4.0 MPa H2, 0.02–0.20 g Ru/C (4 wt% Ru), 40 g 10 wt% xylitol aqueous solution, 1 h, ∼20% xylitol conversion. b Measured at 298 K. | ||||||||||
| — | 7.0 | 815 | trace | trace | 3.5 | 8.3 | 2.9 | 67.8 | 12.6 | n.d. |
| CaCO3 | 9.4 | 67.8 | 21.0 | 18.4 | 15.5 | 4.2 | trace | 14.9 | trace | 2.2 |
| Mg(OH)2 | 10.7 | 116.2 | 24.5 | 21.0 | 16.8 | 3.7 | trace | 11.7 | trace | 4.1 |
| Ca(OH)2 | 12.3 | 184.4 | 32.4 | 24.9 | 9.6 | 2.3 | trace | trace | n.d. | 16.8 |
Upon addition of CaCO3, Mg(OH)2 and Ca(OH)2 bases, the pH values in the reaction solutions increased to 9.4, 10.7 and 12.3, respectively (measured at 298 K), which kept constant during the reaction due to the known solubility equilibrium of these solid bases slightly soluble in the solutions. The activities decreased sharply by 4–10 fold in comparison with the activity in the absence of the bases. Such phenomenon is different from that observed by Davis and coworkers in the hydrogenolysis of glycerol that Ru/C is more active in the basic solutions.25 However, in the presence of the solid bases, the activities increased almost linearly from 67.8 to 184.4 h−1 with increasing the pH values from 9.4 to 12.3. These results show the dual effects of the bases on xylitol reaction although the underlying reason is not clear.
The addition of the solid bases and consequent increase in the pH values led to a significant change in the dominant products from the xylitol epimers to C2 and C3 products (Table 2). The selectivities to ethylene glycol, propylene glycol and lactic acid monotonically increased to 32.4, 24.9 and 16.8%, respectively, with increasing the pH values from 7.0 to 12.3; the combined selectivity to arabitol and threitol concurrently decreased to nearly zero. Glycerol was also detected with selectivities of about 16% at pH 9.4 and pH 10.7, which further decreased to 9.6% at pH 12.3. But the combined selectivities to the three C3 products were always greater at higher pH values. Such pH effects on the selectivities clearly show the direct involvement of the bases in the C–C bond cleavage of xylitol, which affects the reaction pathways of hydrogenation of the xylose intermediate to C4–C5 products and their degradation to the C2–C3 products, and ultimately the product selectivities. Obviously, appropriate pH values are required for efficient xylitol conversion to the C2–C3 products, as indeed was found in Table 2 and further confirmed by the following experiment. The use of stronger bases such as NaOH and KOH increased the pH values to 13 or above, leading to complex products due to the rapid reactions of xylitol or its derivatives with OH− in the solutions (not shown here), consistent with the finding by Zhao et al. in sorbitol hydrogenolysis.20 The result reflects the finding by Norkus et al.35 that xylitol can be fully deprotonated in the solution of pH 13, as characterized by 13C NMR.
Variation of the H2 pressure led to similar effects on the reaction pathways and selectivities. At similar xylitol conversions (∼25%), the activities increased, as expected, with increasing the H2 pressure in the range 0–10 MPa on Ru/C at 473 K and pH 12.3 (in the presence of Ca(OH)2). Under inert N2 atmosphere, the major products were lactic acid (54.9%) together with small amounts of propylene glycol and ethylene glycol. With increasing the H2 pressure, the selectivities for the two glycols increased sharply and reached the maximum values of 30.0 and 35.3%, respectively, at 2.0 MPa H2. Concurrently, the selectivities to lactic acid sharply decreased from 54.9 to 23.1% as the H2 pressures increased from 0 to 2.0 MPa, which then gradually decreased to 15.0% at 10.0 MPa. The selectivities for glycerol, arabitol and threitol increased monotonically with increasing the H2 pressure in the range 0–10 MPa. Such change in the selectivities for the C3 products indicates that they are formed via the same intermediates, and higher H2 pressures facilitate the hydrogenation reactions and the formation of propylene glycol and glycerol at the expense of lactic acid. The gradual increase in the selectivities for arabitol and threitol with the H2 pressure apparently reflects the enhanced rates at higher H2 pressures for hydrogenation of the xylose intermediate, which competes with its C–C bond cleavage to ultimately form the C2 and C3 products.
Xylitol hydrogenolysis forms C2 and C3 products by breaking its central C2–C3 bond through the xylose intermediate, for example, following the retro-aldol condensation mechanism, as proposed by Wang et al.32 This mechanism should lead to glycoaldehyde and glyceraldehyde intermediates with theoretical selectivities of 40% and 60%, respectively, as indeed was found in Fig. 6. The combined selectivities for the C3 products, lactic acid, propylene glycol and glycerol, were approximately 60% at the H2 pressures of below 2.0 MPa, near the theoretical value (60%) from xylitol. The selectivities to the C2 products (mainly ethylene glycol) were however lower than 40%, reflecting the known instability of glycoaldehyde intermediate under basic and H2-poor conditions especially at the high temperature of 473 K. Above 2.0 MPa H2, as the hydrogenation steps were accelerated, the ethylene glycol selectivities increased; the selectivity ratio of ethylene glycol to the C3 products approached the theoretical value of 2/3 (Fig. 6). This cleavage site selectivity, together with the observed pH effects, confirms the involvement of the retro-aldol condensation as the dominating C–C bond cleavage route in xylitol hydrogenolysis.
 | ||
| Fig. 5 Dependence of products selectivities on xylitol conversion for Ru/C. Reaction conditions: 473 K, 4.0 MPa H2, 0.10–0.40 g Ru/C (4 wt% Ru), 0.26–0.60 g Ca(OH)2, 40 g 10 wt% xylitol aqueous solution, 1–6 h. | ||
 | ||
| Fig. 6 Effects of H2 pressure on activities and selectivities for xylitol hydrogenolysis on Ru/C at ca. 25% conversion. Reaction conditions: 473 K, 0.10 g Ru/C (4 wt% Ru), 0.26 g Ca(OH)2, 40 g 10 wt% xylitol aqueous solution, 1 h. | ||
Fig. 7 shows the activities and selectivities for xylitol hydrogenolysis as a function of the reaction temperature on Ru/C at similar xylitol conversions (∼25%). The activities increased dramatically from 20.8 to 981.1 h−1 with increasing the temperature from 433 to 513 K. The selectivities to ethylene glycol and propylene glycol increased with increasing the temperature, and reached the maximum values between 473 and 493 K. In contrast, the selectivities to glycerol and to the sum of arabitol and threitol monotonically decreased in the range 433–513 K. Lactic acid selectivities remained almost constant (around 17%) between 433–473 K, which however sharply increased to 31.2% at 513 K, showing its favorable formation at higher temperatures, as previously found in glycerol reactions.26 The combined selectivities to the C3 products, propylene glycol, glycerol and lactic acid, continuously increased with the temperature, which indicates that the decline in the propylene glycol selectivities above 493 K is due to the favored formation of lactic acid from the same intermediates at higher temperatures. Obviously, lower temperatures favor the direct hydrogenation of the xylose intermediate while its degradation reactions prevail at higher temperatures, and lead to efficient synthesis of the target glycols at appropriate temperatures.
 | ||
| Fig. 7 Effects of temperature on activities and selectivities for xylitol hydrogenolysis on Ru/C at ca. 25% conversion. Reaction conditions: 4.0 MPa H2, 0.02–0.20 g Ru/C (4 wt% Ru), 0.26 g Ca(OH)2, 40 g 10 wt% xylitol aqueous solution, 1–4 h. | ||
Taken together, the observed effects of the catalyst and reaction parameter show the directions toward efficient xylitol hydrogenolysis by tuning of catalysts and reaction parameters, and in turn of the reaction pathways involving the formation of the xylose intermediate and its subsequent transformations. These effects have shed light on the xylitol hydrogenolysis mechanism, as discussed below.
Concerning the mechanism, although there is no clear description on xylitol hydrogenolysis, it has been proposed that polyols (e.g.sorbitol) are hydrogenolyzed generally involving two key steps, i.e.dehydrogenation of polyols to the corresponding carbonyl intermediates on metal catalysts, and their subsequent C–C bond cleavage with bases, most likely via the retro-aldol condensation. Such proposition was verified by Wang et al.,32 based on their study on hydrogenolysis of different 1,3-diol model compounds on Raney Cu and Ni catalysts in the presence of NaOH. In combination with the current mechanistic understandings, we propose the reaction pathways for xylitol hydrogenolysis under the conditions employed in this work. As depicted in Scheme 1, the first step is dehydrogenation of xylitol to the corresponding xylose intermediate on the metal catalysts as discussed by others.16,30–32 Subsequently, the xylose intermediate are either hydrogenated on the metal catalysts to the xylitol epimers (i.e.arabitol and adonitol) and C4polyols or undergo the base-involved retro-aldol condensation to yield glycoaldehyde and glyceraldeyde, which are then hydrogenated to ethylene glycol and glycerol, respectively, or in parallel react with OH− ions. The glyceraldehyde intermediate undergoes dehydration readily to pyruvaldehyde, followed by its hydrogenation to propylene glycol or by its base-catalyzed benzilic acid rearrangement to lactic acid, as found previously by Davis, Liu and their coworkers.25,26 The formation of the C3 products from the same glyceraldehyde intermediate can account for the observed effects of the H2 pressure on their selectivity trends, i.e. the concurrent increase in the propylene glycol and glycerol selectivities and decrease in the lactic acid selectivities with increasing the H2 pressure (Fig. 6). The hydrogenation of glyceraldehyde to glycerol is reversible and glycerol can convert ultimately to propylene glycol and lactic acid especially at the higher xylitol conversions, as reflected by the observed concurrent increase in the propylene glycol and lactic acid selectivities and decrease in the glycerol selectivities as xylitol conversion increases (Fig. 5). Such glycerol reaction is consistent with the previous studies on glycerol conversion,20,25–28 and it was also verified by our separate experiment in this work, as described above.
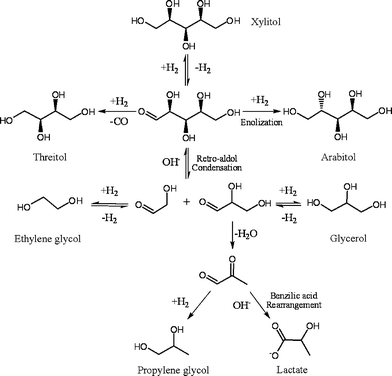 | ||
| Scheme 1 | ||
In addition, the xylitol dehydrogenation step appears to be prerequisite for its hydrogenolysis according to Scheme 1. This step may dictate the overall hydrogenolysis rate. For verification, xylose hydrogenolysis was examined under the standard conditions of 473 K and 4.0 MPa H2 as well as at lower temperatures (e.g. 373 K) in the presence of Ca(OH)2. It was found that xylose readily converted to a complex mixture of products, such as xylitol, arabitol, and various organic acids, even at 373 K or lower (e.g. 313K), reflecting the high reactivity of sugars with H2 on Ru surface and with bases in the reaction solutions.36 However, ethylene glycol, propylene glycol and glycerol were always detected with a combined selectivity of less than 30% under a wide range of reaction conditions. This finding additionally implies that xylose, although it is the intermediate for xylitol hydrogenolysis, is less effective than xylitol for direct synthesis of ethylene glycol and propylene glycol, which is consistent with the conclusion reported by Müller et al.37
Clearly, these proposed reaction pathways demonstrate that while the rate of xylitol hydrogenolysis appears to be limited by its primary dehydrogenation step, the final product distributions are controlled by the competition between the hydrogenation steps on the metal surfaces and the base-catalyzed reactions. Such understanding of these pathways enables one to improve the efficacy of xylitol hydrogenolysis into the target glycols, for example, by optimizing the reaction parameters, as revealed, at least partially, from the effects of the catalyst, pH, and H2 pressure.
4. Conclusions
Supported noble metals, Ru, Pd, Rh and Pt efficiently catalyze xylitol hydrogenolysis into ethylene glycol and propylene glycol in the presence of Ca(OH)2. The activities and selectivities strongly depend on the nature of the metals and their underlying supports. Ru supported on C exhibits superior activities and glycol selectivities than on TiO2, ZrO2, Al2O3 and Mg2AlOx, and Pt is found to be the most active metal compared at similar particle sizes, apparently as a result of the difference in their dehydrogenation/hydrogenation activities and surface acid-basicities, and the consequent effects on the reaction pathways of xylitol hydrogenolysis. The activities and selectivities also depend largely on the H2 pressures, reaction temperatures, and pH values varied by using different solid bases, CaCO3, Mg(OH)2 and Ca(OH)2, which influence the hydrogenation and base-catalyzed steps involved in xylitol hydrogenolysis. Taken together, it is apparent that xylitol hydrogenolysis to ethylene glycol and propylene glycol proceeds by its kinetically relevant dehydrogenation of xylitol to xylose on the metal surfaces, and its subsequent base-catalyzed retro-aldol condensation to form glycolaldehyde and glyceraldehyde, the intermediates for the two glycols. The involved hydrogenation of the aldehyde intermediates on the metal surfaces and their competitive reactions with the bases clearly dictate the ultimate selectivities to the two glycols.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 20825310, 20733009, 20973011 and 50821061) and National Basic Research Project of China (Grant No. 2006CB806100).References
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- P. Gallezot, Catal. Today, 2007, 121, 76–91 CrossRef.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gärtner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS.
- P. L. Dhepe and A. Fukuoka, ChemSusChem, 2008, 1, 969–975 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- M. Schlaf, Dalton Trans., 2006, 4645–4653 RSC.
- A. Behr, J. Eilting, K. Irawadi, J. Leschinski and F. Lindner, Green Chem., 2008, 10, 13–30 RSC.
- Y. Nakagawa, Y. Shinmi, S. Koso and K. Tomishige, J. Catal., 2010, 272, 191–194 CrossRef CAS.
- A. Villa, G. M. Veith and L. Prati, Angew. Chem., Int. Ed., 2010, 49, 4499–4502 CrossRef CAS.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- B. Blanc, A. Bourrel, P. Gallezot, T. Haas and P. Taylor, Green Chem., 2000, 2, 89–91 RSC.
- I. T. Clark, Ind. Eng. Chem., 1958, 50, 1125–1126 CrossRef CAS.
- M. S. S. R. Tanikella, US Pat., 4 404 411, 1983.
- M. Dubeck and G. G. Knapp, US Pat., 4 430 253, 1984.
- C. Montassier, J. C. Ménézo, L. C. Hoang, C. Renaud and J. Barbier, J. Mol. Catal., 1991, 70, 99–110 CrossRef CAS.
- T. A. Werpy, J. G. Frye, A. H. Zacher and D. J. Miller, US Pat., 7 038 094, 2006.
- S. P. Chopade, D. J. Miller, J. E. Jackson, T. A. Werpy, J. G. Frye and A. H. Zacher, US Pat., 6 291 725, 2001.
- G. Gubitosa and B. Casale, US Pat., 5 326 912, 1994.
- L. Zhao, J. H. Zhou, Z. J. Sui and X. G. Zhou, Chem. Eng. Sci., 2010, 65, 30–35 CrossRef CAS.
- J. H. Zhou, M. G. Zhang, L. Zhao, P. Li, X. G. Zhou and W. K. Yuan, Catal. Today, 2009, 147S, S225–S229 CrossRef.
- W. Z. Li, H. Huang, H. J. Li, W. Zhang and H. C. Liu, Langmuir, 2008, 24, 8358–8366 CrossRef CAS.
- W. C. Ketchie, E. P. Maris and R. J. Davis, Chem. Mater., 2007, 19, 3406–3411 CrossRef CAS.
- Y. Ukisu and T. Miyadera, React. Kinet. Catal. Lett., 2004, 81, 305–311 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
- Y. Shen, S. Zhang, H. Li, Y. Ren and H. Liu, Chem. Eur. J., 2010, 16, 7368–7371 CrossRef CAS.
- S. Wang, Y. Zhang and H. Liu, Chem. Asian J., 2010, 5, 1100–1111 CAS.
- T. Miyazawa, S. Koso, K. Kunimori and K. Tomishige, Appl. Catal., A, 2007, 329, 30–35 CrossRef CAS.
- L. Wright and L. Hartmann, J. Org. Chem., 1961, 26, 1588–1596 CrossRef CAS.
- D. K. Sohounloue, C. Montassier and J. Barbier, React. Kinet. Catal. Lett., 1983, 22, 391–397 CrossRef CAS.
- E. Tronconi, N. Ferlazzo, P. Forzatti, I. Pasquon, B. Casale and L. Marini, Chem. Eng. Sci., 1992, 47, 2451–2456 CrossRef CAS.
- K. Y. Wang, M. C. Hawley and T. D. Furney, Ind. Eng. Chem. Res., 1995, 34, 3766–3770 CrossRef CAS.
- C. A. M. Abreu, N. M. Lima and A. Zoulalian, Biomass Bioenergy, 1995, 9, 487–492 CrossRef CAS.
- V. N. Ipatieff, W. W. Thompson and H. Pines, J. Am. Chem. Soc., 1951, 73, 553–555 CrossRef CAS.
- E. Norkus, J. Vaičiūnienė, T. Vuorinen, E. Gaidamauskas, J. Reklaitis, A. S. Jääskeläinen and D. C. Crans, Carbohydr. Res., 2004, 339, 599–605 CrossRef CAS.
- J. M. de Bruijn, A. P. G. Kieboom and H. van Bekkum, Recl. Trav. Chim. Pays-Bas, 1986, 105, 176–183 CAS.
- C. Müller, P. Rimmelin, J. P. Hindermann, R. Kieffer and A. Kiennemann, in Studies in Surface Science and Catalysis, ed. M. Guisnet, J. Barrault, C. Bouchoule, D. Duprez, G. Perot, R. Maurel and C. Montessier, Elsevier, Amsterdam, 1991, Vol. 59, pp. 237–244 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |