Solvent-free selective epoxidation of cyclooctene using supported gold catalysts: an investigation of catalyst re-use
Salem
Bawaked
,
Nicholas F.
Dummer
,
Donald
Bethell
,
David W.
Knight
and
Graham J.
Hutchings
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: hutch@cardiff.ac.uk
First published on 12th November 2010
Abstract
Selective oxidation is of immense importance in the synthesis of chemical intermediates and the epoxidation of alkenes by the electrophilic addition of oxygen to a carbon–carbon double bond remains one of the most significant challenges in oxidation catalysis. Although molecular oxygen is the most environmentally benign oxidant in many cases, far more reactive forms of oxygen are required to achieve reaction, and this can lead to by-products with a heavy environmental burden with respect to their disposal. We show that gold supported on graphite is a very effective catalyst for the epoxidation of cis-cyclooctene as long as catalytic amounts of a hydroperoxy species are present at the start of the reaction. Using mild solvent-free conditions the hydroperoxy initiator persists in solution for only a few minutes, being initially adsorbed on the catalyst surface. Subsequently, it decomposes to establish a reactive species that can propagate the selective oxidation process we observe. The observation of an induction period may in part be due to the adsorption of the radical initiator blocking surface sites as well as the establishment of the reactive species. We confirm that graphite is the best support and that tert-butyl hydroperoxide is the preferred initiator. We report extensive studies concerning the reusability of the gold/graphite catalyst as catalyst reusability is a key feature of green chemistry. The catalyst is found to be inhibited by the epoxide product but we demonstrate the effect of this is negligible for reused catalysts over a long reaction time.
Introduction
Oxidation, particularly epoxidation, lies at the heart of the modern chemical industry. It is an essential part of many chemical syntheses and is often used to functionalize less reactive starting materials to ensure they can be used in downstream processes. In terms of green chemistry molecular oxygen is the preferred oxidant. However, many molecules and catalysts at present are unreactive with molecular oxygen and consequently more reactive forms of oxygen are employed. Often these include chromium and manganese compounds and these are clearly environmentally non-benign and their use leads to significant waste.1 There is therefore a pressing need to identify environmentally friendly approaches to designing new oxidation catalysts that can operate with molecular oxygen. Alkene epoxidation has attracted significant attention, ethene is oxidized with molecular oxygen to ethylene oxide commercially using a supported silver catalyst.2 Unfortunately, to achieve high selectivities non-green additives, such as chloro-hydrocarbons and nitrogen oxides, have to be added to prevent non-selective side reactions. Ethene appears to be a special case and attempts to oxidize higher alkenes with molecular oxygen have proved more problematic. Propene can be epoxidised with hydrogen peroxide using the titanium silicalite TS-13 and this process is being commercialized. Detailed studies using gold catalysts initiated by Haruta and co-workers4,5 have shown that propene can be epoxidised using gold supported on titanium oxide supports using oxygen in the presence of hydrogen. It is considered that the O2/H2 forms a surface hydroperoxy species that is responsible for the selective oxidation. In this respect it has similarities to the reaction using TS-1 where hydrogen peroxide is the oxidant.3 The use of gold catalysts for propene epoxidation has attracted significant attention.6–20 Initially, only low selectivities based on propene were observed, but by using mesoporous titanium silicate supports high selectivities have been achieved. However, the utilization of H2 remains very low, and is significantly lower than that achievable in the commercial synthesis of hydrogen peroxide.21 Hence, at present this route cannot compete with the oxidation of propene using hydrogen peroxide rather than O2/H2 mixtures.We have shown that gold nanoparticles supported on graphite can epoxidise cyclic alkenes using molecular oxygen as long as a radical initiator is present in catalytic amounts.22 In these initial studies we showed that cis-cyclooctene could be epoxidised with a selectivity of >80% under mild solvent-free conditions. In the absence of the catalyst, some oxidation was observed but this was minor and non-selective giving very low selectivities to the epoxide. We also showed that it was possible to achieve epoxidation in the absence of the radical initiator, but typically low selectivities ensued. Hence, we concluded that for high selectivities an appropriate combination of molecular oxygen and a radical initiator was required. More recently,23 we explored the reaction in more detail. Caps and co-workers24–27 have also investigated this catalyst, and Lambert and co-workers28 have shown that very small Au55 nanocrystals supported on carbon are active catalysts for styrene oxidation with oxygen but only minor selectivity to the epoxide was observed and the major product was benzaldehyde. This result confirms our earlier observation that the radical initiator was required to ensure that high epoxide selectivity was attained.22 In this paper we extend our earlier studies21,22 to explore whether alternative supports or radical initiators can improve the performance, and in particular we examine the reusability of the catalyst as this is a key aspect of green chemistry that has not been explored previously with these catalysts.
Experimental
Catalyst preparation
Catalysts (1 wt% Au/support) were prepared using the following standard deposition precipitation method (denoted DP). A solution of HAuCl4·3H2O (5 ml, 2 g in 100 ml distilled water) was diluted with water (45 ml). Aqueous sodium carbonate was added with stirring until pH = 10 was attained. This solution was then added, with continuous stirring, to a slurry of the support in water (4.95 g in water 50 ml). The mixture was stirred for 1 h at 20 °C, maintaining the pH at 10. The mixture was heated to 70 °C and formaldehyde was added as a reducing agent. The solid was recovered by filtration and washed with water (1.2 L) until the washings were found to be chloride free. The catalyst was dried (110 °C, 16 h) prior to use. Three supports were used in this study namely, graphite (Johnson Matthey), Al2O3 (Aldrich) and SiC (Aldrich).Catalyst testing
Results and discussion
Investigation of alternative radical initiators
As noted in our earlier studies for the epoxidation of cis-cylooctene, there are key components of the reaction system that are required for high epoxide selectivity, namely a suitable supported gold catalyst and a radical initiator, as without the latter low epoxide selectivities are observed.22,28 With an appropriate supported gold catalyst and a radical initiator epoxide selectivities are typically in the region of 80% and the other oxidation products are mainly cis-cyclo-oct-2-enol and cis-cyclo-oct-2-enone formed with 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio. In our studies we have found that tert-butyl hydroperoxide (TBHP) was the most effective radical initiator of those tested. We have extended the range of radical initiators and have now investigated dibenzoyl peroxide (DBP) and azobisisobutyronitrile (AIBN). Since we are seeking to oxidise cyclooctene with molecular oxygen, it is important we determine the reactions that occur in the absence of catalyst since it is possible that molecular oxygen, a di-radical in its ground state, together with the radical initiator could effect oxidation of the cyclooctene. These non-catalysed reactions can often lead to lower selectivities being achieved and can, in the extreme cases, mask any catalytic reaction that may be occurring. Hence, as we noted in our earlier detailed study,23 it is important to determine the reaction conditions where no reaction occurs in the absence of the catalyst. In the absence both of a peroxy initiator and a catalyst, no reaction was observed over the temperature range we have studied in this paper. The three radical initiators (DBP, AIBN, TBHP) were investigated at 60–80 °C with alkene/initiator mol ratios between 74.6 and 748, and the results are shown in Table 1. For the reaction in the presence of the 1 wt% Au/graphite catalyst it is apparent that all three radical initiators give excellent selectivity to the epoxide. However, both DBP and AIBN give similar or higher conversions in the absence of the catalyst, a result that contrasts sharply with reports by Corma and co-workers,29–30 who observed enhancement of conversions of cyclohexene and of 1-octene into the epoxides in oxidation by molecular oxygen catalysed by nanoparticulate Au supported on ceria or MCM-41 when AIBN was present at the start of the reaction. Corma has presented convincing evidence that radicals from AIBN decomposition can give rise to organogold compounds and has developed mechanistic interpretations of the AIBN-promoted epoxidations in the light of this.31
:1 molar ratio. In our studies we have found that tert-butyl hydroperoxide (TBHP) was the most effective radical initiator of those tested. We have extended the range of radical initiators and have now investigated dibenzoyl peroxide (DBP) and azobisisobutyronitrile (AIBN). Since we are seeking to oxidise cyclooctene with molecular oxygen, it is important we determine the reactions that occur in the absence of catalyst since it is possible that molecular oxygen, a di-radical in its ground state, together with the radical initiator could effect oxidation of the cyclooctene. These non-catalysed reactions can often lead to lower selectivities being achieved and can, in the extreme cases, mask any catalytic reaction that may be occurring. Hence, as we noted in our earlier detailed study,23 it is important to determine the reaction conditions where no reaction occurs in the absence of the catalyst. In the absence both of a peroxy initiator and a catalyst, no reaction was observed over the temperature range we have studied in this paper. The three radical initiators (DBP, AIBN, TBHP) were investigated at 60–80 °C with alkene/initiator mol ratios between 74.6 and 748, and the results are shown in Table 1. For the reaction in the presence of the 1 wt% Au/graphite catalyst it is apparent that all three radical initiators give excellent selectivity to the epoxide. However, both DBP and AIBN give similar or higher conversions in the absence of the catalyst, a result that contrasts sharply with reports by Corma and co-workers,29–30 who observed enhancement of conversions of cyclohexene and of 1-octene into the epoxides in oxidation by molecular oxygen catalysed by nanoparticulate Au supported on ceria or MCM-41 when AIBN was present at the start of the reaction. Corma has presented convincing evidence that radicals from AIBN decomposition can give rise to organogold compounds and has developed mechanistic interpretations of the AIBN-promoted epoxidations in the light of this.31
| Radical initiator | Initiator (mol × 10−2) | Temperature (°C) | Blank a | 1%Au/graphiteb | ||
|---|---|---|---|---|---|---|
| Conversion | Selectivity (%) | Conversion (%) | Selectivity (%) | |||
| a Reaction without catalyst: cyclooctene (10 ml, 0.077 mol), 24 h, atmospheric pressure. b Reaction using 1% Au/graphite: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), 24 h, atmospheric pressure. c Dibenzoyl peroxide. d Azobisisobutyronitrile. e Tert-butyl hydroperoxide. | ||||||
| DBP c | 0.1032 | 60 | 2.3 | 77.2 | 2.4 | 77.6 |
| 0.0516 | 1.8 | 77.0 | 1.3 | 78.1 | ||
| 0.0103 | 0.36 | 80.4 | 0.22 | 80.9 | ||
| 0.1032 | 70 | 8.2 | 84.4 | 2.2 | 83.5 | |
| 0.0516 | 5.9 | 79.7 | 2.2 | 78.7 | ||
| 0.0103 | 2.7 | 76.0 | 1.5 | 75.5 | ||
| 0.1032 | 80 | 8.4 | 77.1 | 5.6 | 79.5 | |
| 0.0516 | 8.9 | 78.1 | 5.1 | 79.7 | ||
| 0.0103 | 5.6 | 83.5 | 3.3 | 79.4 | ||
| AIBN d | 0.1032 | 60 | 1.4 | 70.3 | 1.2 | 69.5 |
| 0.0516 | 0.85 | 73.5 | 0.85 | 73.5 | ||
| 0.0103 | 0.28 | 74.7 | 0.08 | 94.9 | ||
| 0.1032 | 70 | 3.6 | 79.8 | 3.1 | 84.5 | |
| 0.0516 | 2.6 | 77.7 | 2.9 | 81.1 | ||
| 0.0103 | 1.2 | 74.5 | 1.2 | 77.7 | ||
| 0.1032 | 80 | 7.8 | 81.1 | 7.6 | 75.9 | |
| 0.0516 | 6.3 | 85.4 | 5.8 | 80.5 | ||
| 0.0103 | 4.1 | 82.4 | 4.1 | 80.7 | ||
| TBHP e | 0.1032 | 60 | 0.4 | 58.4 | 1.9 | 68.3 |
| 0.0516 | 0.16 | 30.7 | 1.6 | 64.2 | ||
| 0.0103 | 0.01 | 0.01 | 0.6 | 63.9 | ||
| 0.1032 | 70 | 0.90 | 63.2 | 3.9 | 71.4 | |
| 0.0516 | 0.36 | 64.5 | 2.6 | 72.3 | ||
| 0.0103 | 0.04 | 86.4 | 1.9 | 72.3 | ||
| 0.1032 | 80 | 4.46 | 74.2 | 8.5 | 78.7 | |
| 0.0516 | 1.79 | 62.6 | 6.6 | 78.7 | ||
| 0.0103 | 0.1 | 42.6 | 4.0 | 78.2 | ||
The greater efficacy in stimulating catalytic oxidation of TBHP over the other initiators used in our experiments could be due to the lower stabilities of DBP and AIBN compared with TBHP (temperatures for 10 h half lives of radical initiators: AIBN 65 °C, BBP 70 °C, TBHP 170 °C). It is possible that gold is acting as a radical scavenger, as Corma has suggested, with these reaction systems but the completely different observations in the presence of AIBN seems to suggest that the support may play an important role in promoting the catalytic mechanism when radical initiators are present. It is possible that AIBN and DBP could be used in place of a catalyst if the radical reaction can be maintained using a relatively simple homogeneous process. However, it is apparent that, for the gold-based heterogeneous catalyst, TBHP remains the preferred choice of radical initiator for this reaction; the heterogeneously catalysed reaction shows clear improvement over the non-catalysed radical pathway. It is interesting that both DBP and AIBN give high selectivities to the epoxide in the absence of the gold catalyst when this is not observed with TBHP, cumene hydroperoxide, DBP2 and hydrogen peroxide22 This shows the importance of the radicals formed on decomposition since the hydroperoxides and H2O2 all generate R-O˙, whereas DBP gives PhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O·O˙ (and subsequently Ph˙) and AIBN decomposes to (CH3)2C˙(CN), the latter two being much more nucleophilic radical species.
O·O˙ (and subsequently Ph˙) and AIBN decomposes to (CH3)2C˙(CN), the latter two being much more nucleophilic radical species.
In view of these results TBHP was selected as the radical initiator for use with the gold catalysts. However, it was essential that low concentrations were used and consequently a concentration of 1.03 × 10−4 mol l−1TBHP was selected as the standard concentration for the subsequent studies, as only a trace of reaction was observed under our reaction conditions (Table 1). It is noteworthy that pretreatment of the catalyst with the radical initiator in toluene or DMF at 80 °C for 36 h, followed by transfer to cyclo-octene under standard epoxidation conditions, led to no reaction. Clearly the role of TBHP is not merely to activate the catalyst surface by removing certain species but rather to interact also with other components of the reaction system.
Oxidation of cyclooctene using Al2O3 and SiC as supports
In our previous study23 we showed that carbon supports were preferred for the epoxidation of cyclooctene, and that graphite gave the best performance as it showed no blank reaction in the presence of TBHP. We have extended the range of supports investigated and now include data on Al2O3 and SiC (Tables 2 and 3). The use of SiC required an initial wash with water prior to gold loading, without this step the activity of the support was found to be higher. With the unwashed material the conversion of cyclooctene with 0.0103 × 10−2 mol of TBHP at 80 °C was found to be ca. 1.5%, this decreased to ca. 0.1% upon washing. At the lowest concentrations of TBHP Al2O3 does not show any activity in the absence of gold; however 1 wt% Au/Al2O3 (Table 2) is not as active as 1 wt% Au/graphite (Table 1) under comparable conditions. SiC (Table 3) does show activity in the absence of gold with all concentrations of TBHP used. However, the addition of gold enhances the activity significantly although the selectivity to the epoxide is slightly lower than that observed with 1 wt% Au/graphite. We do not observe CO2 as a product with 1 wt% Au/graphite and the by-product formed was the allylic alcohol. Using results from our previous studies we can conclude that based on activity the supports have the following order: graphite ≈ SiC > Al2O3 ≈ SiO2 > TiO2, and for selectivity to the epoxide the order is graphite ≈ SiO2 ≈ TiO2 > SiC > Al2O3. In view of this we selected the 1 wt% Au/graphite catalyst for detailed studies on catalyst re-use.| TBHP (mol × 10−2) | Temperature (°C) | Al2O3a | 1%Au/Al2O3b | ||
|---|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) | ||
| a Reaction using Al2O3: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), 24 h, atmospheric pressure. b Reaction using 1%Au/Al2O3: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), 24 h, atmospheric pressure. | |||||
| 0.1032 | 60 | 0.25 | 53.6 | 0.55 | 52.1 |
| 0.0516 | 0.09 | 71.8 | 0.48 | 49.7 | |
| 0.0103 | 0 | 0 | 0.13 | 54.2 | |
| 0.1032 | 70 | 1.2 | 61.7 | 1.8 | 62.4 |
| 0.0516 | 0.43 | 48.7 | 1.3 | 63.6 | |
| 0.0103 | 0.01 | 72.5 | 0.38 | 63.3 | |
| 0.1032 | 80 | 4.4 | 71.4 | 6.3 | 72.6 |
| 0.0516 | 0.2 | 46.7 | 4.6 | 74.0 | |
| 0.0103 | 0 | 0 | 2.6 | 71.8 | |
| TBHP (mol × 10−2) | Temperature (°C) | SiC a | 1%Au/SiCb | ||
|---|---|---|---|---|---|
| Conversion (%) | Selectivity (%) | Conversion (%) | Selectivity (%) | ||
| a Reaction using SiC: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), 24 h, atmospheric pressure. b Reaction using 1%Au/SiC: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), 24 h, atmospheric pressure. | |||||
| 0.1032 | 60 | 0.64 | 36.2 | 0.9 | 67.6 |
| 0.0516 | 0.2 | 44.5 | 0.63 | 66.4 | |
| 0.0103 | 0 | 0 | 0.13 | 63.5 | |
| 0.1032 | 70 | 2.1 | 72.7 | 3.2 | 72.1 |
| 0.0516 | 0.8 | 71.0 | 1.4 | 69.7 | |
| 0.0103 | 0 | 0 | 0.74 | 64.4 | |
| 0.1032 | 80 | 6.9 | 76.1 | 11 | 73.4 |
| 0.0516 | 4.4 | 78.3 | 7.7 | 71.4 | |
| 0.0103 | 0.32 | 59.4 | 4.8 | 75.2 | |
Catalyst reusability
One of the key advantages of heterogeneous catalysts is that when operated in a batch mode the catalyst can be readily recovered by filtration for re-use. The successful recovery and re-use of heterogeneous catalysts is therefore an essential aspect of green chemistry. To date the reusability of the Au/graphite catalysts for alkene epoxidation has not been considered and we now address this.Following reaction of cis-cyclooctene under standard conditions for 24 h the catalyst was recovered by centrifugation. We investigated several treatments for the drying of the catalyst prior to reuse. In all cases we used a larger batch of catalyst in the first use experiment so that following recovery and drying the correct amount of catalyst could be added to the reactor in the re-use experiments. This ensures that any loss in activity is not due to any discrepancy in the catalyst mass used. We investigated the way in which the recovered catalyst was dried after an initial washing treatment with acetone (Table 4). Drying at 20 °C for 16 h did not recover the activity or selectivity, nor did drying at a higher temperature for 1 h. Drying at temperatures of 110–170 °C for 16 h recovered the selectivity of the catalyst but the activity of the catalyst was at best ca. 60% compared with the activity of the fresh catalyst for a 24 h reaction. However, a catalyst that had been re-used once could be successfully re-used retaining this lower activity on subsequent reactions (Table 4). The effect of the drying atmosphere (Table 5) was also found to have a marked effect. Drying with static air gave the best results and use of flowing air or H2 gave a slightly decreased activity on re-use. However, using flowing O2 all samples gave a much poorer performance for the re-used catalyst. The solvent used in the catalyst treatment prior to drying was also found to be important. If no solvent extraction was used then the activity was only marginally lower than if an acetone washing step was employed, and the selectivity was retained at a high level. Washing with water, diethyl ether, ethanol, aqueous sodium hydroxide or dilute nitric acid all led to no activity being observed in the recovered catalyst. These results demonstrate the sensitivity of the catalyst to treatment with most solvents and the best results are attained either using no solvent treatment or using acetone.
| Drying temperature | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction conditions: cyclooctene 10 ml, catalyst 0.12 g, TBHP 0.01 ml, temperature 80 °C, glass reactor, 24 h, atmospheric pressure; after use the catalyst was recovered by centrifugation, washed with acetone (30 ml), and dried at the temperature and time specified in the table. b Conversion on first reuse. c Second reuse. d Third reuse. e Fourth reuse. | ||
| Fresh catalyst | 4 | 78.2 |
| 20 °C, 16 h | 0.44b | 67.1 |
| 110 °C, 1 h | 1.1b | 66.9 |
| 110 °C, 16 h | 2.4b | 75.0 |
| 170 °C, 16 h | 2.4b | 78.6 |
| 150 °C, 16 h | 2.4b | 79.0 |
| 150 °C, 16 h | 2.4c | 78.4 |
| 150 °C, 16 h | 1.9d | 78.0 |
| 150 °C, 16 h | 1.1e | 78.3 |
| Drying conditions | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction conditions: cyclooctene 10 ml, catalyst 0.12 g, TBHP 0.01 ml, temperature 80 °C, glass reactor, 24 h, atmospheric pressure; after use the catalyst was recovered by centrifugation, washed with acetone (30 ml), and dried at the temperature and time specified in the table. b Conversion on first reuse. | ||
| Fresh catalyst | 4 | 78.2 |
| Static air, 150 °C, 16 h | 2.4b | 78.6 |
| Flowing air, 150 °C, 16 h | 2.1b | 78.3 |
| Flowing 5% H2/Ar, 150 °C, 16 h | 2.2b | 76.8 |
| Flowing O2 at 150 °C, 16 h | 1.1b | 71.6 |
We also investigated the reaction of the cyclooctene recovered after it had been used for an initial reaction after removal of the catalyst (Table 6). In this case fresh (not recovered) catalyst and TBHP were added and the reaction was allowed to proceed for either an additional 2 or 24 h. The conversions we observe are similar to those observed with increasing reaction time for the conversion of cyclooctene (Fig. 4) and hence we conclude that reused cyclooctene behaves normally, giving conversions that are indistinguishable from standard reactions carried out for the same time.
| Catalyst/conditions | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Standard reaction conditions: cyclooctene 10 ml, catalyst 0.12 g, TBHP 0.01 ml, temperature 80 °C, glass reactor, atmospheric pressure. After the standard reaction the mixture was filtered and the recovered cyclooctene was reused for the stated time. The conversion is reported after the total reaction time. | ||
| Standard reaction for 24 h | 4.0 | 78.2 |
| Standard reaction for 24 h, fresh catalyst and TBHP for 2 h | 5.2 | 79.9 |
| Standard reaction for 24 h, fresh catalyst and TBHP for 24 h | 13.3 | 81.9 |
| Standard reaction for 6 h then fresh catalyst and TBHP for 24 h | 4.5 | 79.2 |
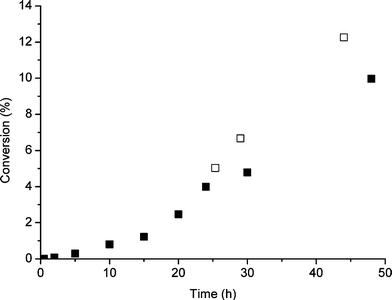 | ||
| Fig. 1 Effect of addition of TBHP after 24 h standard reaction on the conversion of cyclooctene oxidation; standard reaction (■) and after second TBHP addition (□). | ||
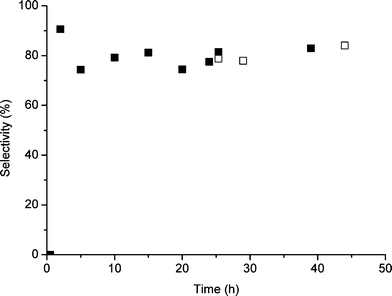 | ||
| Fig. 2 Effect of addition of TBHP after 24 h standard reaction on the selectivity of cyclooctene oxidation; standard reaction (■) and after second TBHP addition (□). | ||
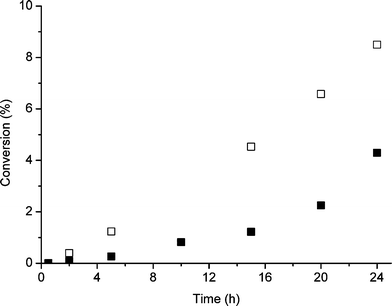 | ||
| Fig. 3 Effect of initial concentration of TBHP on the rate of cyclooctene oxidation under standard reaction conditions. Key: ■ 0.01 × 10−2 mol, □ 0.1 × 10−2 mol. | ||
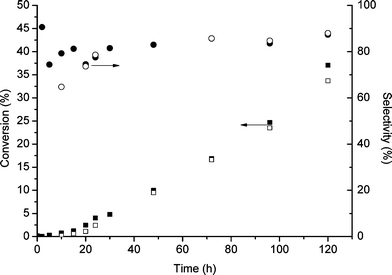 | ||
| Fig. 4 Time on line study for cyclooctene oxidation: closed symbols fresh catalyst, open symbols reused catalyst. Reaction conditions: cyclooctene 10 ml, catalyst 0.12 g TBHP 0.01 ml, temperature 80 °C, glass reactor, atmospheric pressure. | ||
In further investigations we followed the fate of TBHP in the reaction using analysis by gas chromatography and NMR spectroscopy and we observed that the TBHP was no longer detected in solution after the first 15 min of reaction. We consider this to be due to adsorption of TBHP onto the catalyst surface as the half life of homogeneous TBHP decomposition under these conditions is of the order of 10–15 h. It is possible that slow decomposition of the surface-bound TBHP leads to two effects. First it could clean the active sites so that effective catalytic species are established and secondly it participates directly in establishing the intermediates that propagate the reaction. In both cases this would lead to an induction period which is observed. We therefore investigated the addition of TBHP during the course of the reaction (Table 7, Fig. 1 and 2). Addition of a second aliquot of TBHP at 2, 6 or 24 h had a dramatic effect by increasing the activity and also enhanced the selectivity when added in the earlier period. Indeed, the addition of the second aliquot of TBHP had immediate effects and did not involve a further induction period. This shows that the TBHP acts as a source of active (radical) intermediates immediately. This is consistent with the first aliquot aiding the cleaning of the catalyst surface as we suggested. These results show that by carefully manipulating the experimental conditions enhanced activity and selectivity can be achieved.
| Reaction conditions | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Standard reaction conditions: cyclooctene 10 ml, catalyst 0.12 g, TBHP 0.01 ml, temperature 80 °C, glass reactor, 24 h, atmospheric pressure, TBHP (0.01 ml) was added to the reaction mixture without stopping the reaction. | ||
| Standard reaction, 24 h | 4 | 78.2 |
| Second aliquot of TBHP added at 2 h, 24 h total reaction | 4.6 | 80.6 |
| Second aliquot of TBHP added at 6 h, 24 h total reaction | 5.8 | 81.8 |
A further experiment was conducted in which a higher concentration of TBHP was added at the start of the reaction (Fig. 3). In this case we observe a dramatic shortening of the slow induction period at the start of the reaction. Indeed, at the higher TBHP amount the induction period almost disappears. While this is consistent with a radical chain mechanism we also note that the rate of conversion at the end of the period studied is roughly the same for both levels of TBHP. This could be interpreted as indicating that the function of the initiator is merely to activate the catalyst surface and this is achieved more quickly with the higher amount of TBHP. Since the amount of catalyst was the same in both experiments, the same rate is eventually reached in both experiments. The mechanism of activation by TBHP could possibly involve O-centred radical species reacting either directly with the metal surface or with surface contaminants. We consequently investigated the addition of two potential radical scavengers at the start of the reaction and these quenched the reaction (Table 8). These observations are consistent with the possibilities discussed above and indicate that free radicals are involved at some stage in the catalytic cycle.
| Radical scavenger | Conversion (%) | Selectivity (%) |
|---|---|---|
| a Reaction using 1% Au/graphite: cyclooctene (10 ml, 0.077 mol), catalyst (0.12 g), TBHP (0.01 × 10−2 mol), 80 °C, atmospheric pressure. b BHT = 2,6-di-tert-butyl-4-methylphenol. c PS-Tempo = 2,2,6,6-tetramethylpiperidine-1-oxyl supported on polystyrene. | ||
| None | 4 | 78.2 |
| BHT a | 0.36 | 0 |
| PS-TEMPO b | 0.21 | Trace |
A tentative mechanism that is consistent with most of our observations is set out in Scheme 1. Although largely modelled on the behaviour of free radicals in autooxidation chains, the dramatic increase in epoxide selectivity in the presence of the gold catalyst suggests that the chemistry takes place on or near the metal surface. Accordingly, we have not speculated about the possible existence and nature of chain-terminating processes.
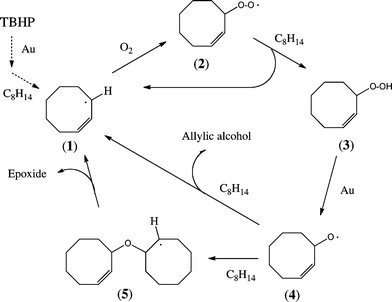 | ||
| Scheme 1 Proposed mechanism for cyclooctene oxidation using TBHP as radical initiator over supported gold catalysts. | ||
In the first step, TBHP decomposes to produce t-BuOO˙ or, more probably (see below), a t-BuO˙ radical attached to the surface of the gold catalyst. The radical so formed abstracts a hydrogen atom from cyclooctene and produces the allylic radical (1) which, via the oxygen adduct (2), produces cyclooctene hydroperoxide (3), evidence for the presence of which was presented earlier;23 this step could account for the induction period, after which the concentration of cyclooctene hydroperoxide is sufficient to sustain the oxidation of cyclooctene on the gold surface. This, we suggest, proceeds by transformation of (3) into the cyclooctenyloxy radical (4) because it has been demonstrated by DFT calculations32 that, on a ten atom gold cluster, hydrogen peroxide dissociates into two HO radicals attached to gold atoms on the surface. It is the masked radical (4) that we believe to be a key intermediate in the catalytic cycle.
As it stands, the selectivity to epoxide predicted by the catalytic cycle in Scheme 1 depends critically on the partitioning of the cyclooctenyloxy radical (4) so as to complete the catalytic cycle between (i) allylic hydrogen abstraction from a further molecule of cyclooctene or (ii) attachment to the double bond of cyclooctene, thereby forming radical (5). Cleavage of (5) affords a molecule of the epoxide and the allylic radical (1) to complete the cycle; only epoxide is produced in this way. Hydrogen atom abstraction from cyclooctene by (4) would on the other hand produce a molecule of the allylic alcohol together with (1). Our observation of epoxide selectivities in the range 70–80% would require that both routes are available. At the moment we do not have independent, direct supporting evidence for the mechanism of catalysis shown in Scheme 1. Such evidence might be sought from isotopic labelling of both of the sp2carbon atoms of cyclo-octene (or their attached hydrogens), since Scheme 1 requires that the epoxide ring incorporates only one of the labels, while an alternative direct O-transfer to the double bond would incorporate both.
As the cyclooctene could be re-used effectively (Table 6) we investigated the cause of the deactivation of the catalyst after the initial use. We studied the addition at the start of the reaction of the epoxide at a range of concentrations up to the concentration expected for 4% conversion and 80% selectivity after a 24 h reaction (Table 9). This epoxide was added together with the fresh catalyst and TBHP. We observed that the conversion at the end of the 24 h reaction period was 2.4%; i.e. identical to that of the reused catalysts that had been recovered, washed with acetone and dried in static air (Tables 4–5). Hence it seems that, after the recovery process, epoxide is retained on the surface of the catalyst and we are observing weak product inhibition.
| Entry | Catalyst | Conversion (%) | Selectivity (%) |
|---|---|---|---|
| a Reaction conditions: substrate 10 ml, catalyst 0.12 g, TBHP 0.01 ml, temperature 80 °C, glass reactor, 24 h, atmospheric pressure. | |||
| 1 | 1%Au/graphite + epoxide 0.01 g | 3.7 | 78.0 |
| 2 | 1%Au/graphite + epoxide 0.15 g | 3.4 | 78.4 |
| 3 | 1%Au/graphite + epoxide 0.3 g | 2.4 | 75.0 |
| 4 | Reuse of catalyst from entry 2 | 2.2 | 77.6 |
| 5 | Reuse of catalyst from entry 3 | 2.6 | 78.9 |
With these findings we investigated the reaction of the recovered catalyst for longer periods (Fig. 4). It is apparent that at such longer reaction times the activity of the initially fresh catalyst and the reused catalyst are indistinguishable; inhibition is only evident by comparison of fresh and recovered catalysts at short times and low epoxide conversions. Consequently the catalyst can be re-used efficiently over longer reaction times, an important facet of a green chemical process. We cannot, on the basis of the present evidence, say how the epoxide brings about the reduction in the catalyst activity. Although the inhibition is only detectable at the commencement of epoxidation, the presence of the epoxide affects the rate of conversion throughout the reaction. Further, more detailed investigation of the effect is warranted.
Conclusions
We have confirmed that gold supported on graphite is a very effective catalyst for the epoxidation of cis-cyclooctene using molecular oxygen as long as small amounts of a hydroperoxy initiator are present at the start of the reaction. The catalyst is effective under mild solvent-free conditions and selectivities to the epoxide of 75–80% can readily be achieved. TBHP is found to be the most effective initiator but, under the reaction conditions, it does not persist long, setting up the reactive species, including trappable radicals that propagate the catalytic oxidation. The catalyst is found to be inhibited by the epoxide product but we demonstrate the effect of this is negligible for reused catalysts over a long reaction time.Acknowledgements
This work formed part of an EPSRC funded project and we thank them for funding this research. We also thank King Abdul Aziz University (Saudi Arabia Government) for financial support.References
- R. A. Sheldon, Stud. Surf. Sci. Catal., 1991, 66, 33.
- R. M. Lambert, F. J. Williams, R. L. Cropley and A. Palermo, J. Mol. Catal. A: Chem., 2005, 228, 27 CrossRef CAS.
- E. Klemm, E. Dietzsch, T. Schwarz, T. Kruppa, A. Lange, de Oliveira, F. Becker, G. Markowz, S. Schirrmeister, R. Schütte, K. J. Caspary, F. Schüth and D. Hönicke, Ind. Eng. Chem. Res., 2008, 47, 2086 CrossRef CAS.
- T. Hayashi, K. Tanaka and M. Haruta, J. Catal., 1998, 178, 566 CrossRef CAS.
- M. Haruta and M. Date, Appl. Catal., A, 2001, 222, 427 CrossRef CAS.
- T. A. Nijhuis, T. Visser and B. M. Weckhuysen, J. Phys. Chem. B, 2005, 109, 19309 CrossRef CAS.
- B. S. Uphade, S. Tsubota, T. Hayashi and M. Haruta, Chem. Lett., 1998, 1277 CrossRef CAS.
- T. A. Nijhuis, B. J. Huizinga, M. Makkee and J. A. Moulijn, Ind. Eng. Chem. Res., 1999, 38, 884 CrossRef CAS.
- T. A. Nijhuis, T. Q. Gardner and B. M. Weckhuysen, J. Catal., 2005, 236, 153 CrossRef CAS.
- T. A. Nijhuis, T. Visser and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2005, 44, 1115 CrossRef CAS.
- T. A. Nijhuis and B. M. Weckhuysen, Chem. Commun., 2005, 6002 RSC.
- N. Yap, R. P. Andres and W. N. Delgass, J. Catal., 2004, 226, 156 CrossRef CAS.
- A. Zwijnenburg, M. Makkee and J. A. Moulijn, Appl. Catal., A, 2004, 270, 49 CrossRef CAS.
- C. Qi, T. Akita, M. Okumura, K. Kuraoka and M. Haruta, Appl. Catal., A, 2003, 253, 75 CrossRef CAS.
- B. Taylor, J. Lauterbach and W. N. Delgass, Appl. Catal., A, 2005, 291, 188 CrossRef CAS.
- A. K. Sinha, S. Seelan, M. Okumura, T. Akita, S. Tsubota and M. Haruta, J. Phys. Chem. B, 2005, 109, 3956 CrossRef CAS.
- B. Chowdhury, J. J. Bravo-Suarez, M. Date, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2006, 45, 412 CrossRef.
- E. Sacaliuc, A. M. Beale, B. M. Weckhuysen and T. A. Nijhuis, J. Catal., 2007, 248, 235 CrossRef CAS.
- J. Lu, X. Zhang, J. J. Bravo-Suárez, K. K. Bando, T. Fujitani and S. T. Oyama, J. Catal., 2007, 250, 350 CrossRef CAS.
- A. K. Sinha, S. Seelan, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 1546 CrossRef CAS.
- J. K. Edwards and G. J. Hutchings, Angew. Chem., Int. Ed., 2008, 47, 9192–9198 CrossRef CAS.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132 CrossRef CAS.
- S. Bawaked, N. F. Dummer, N. Dimitratos, D. Bethell, Q. He, C. J. Kiely and G. J. Hutchings, Green Chem., 2009, 11, 1037 RSC.
- P. Lignier, F. Morfin, S. Mangematin, L. Massin, J.-L. Rousset and V. Caps, Chem. Commun., 2007, 186 RSC.
- P. Lignier, S. Mangematin, F. Morfin, J.-L. Rousset and V. Caps, Catal. Today, 2008, 138, 50 CrossRef CAS.
- P. Lignier, F. Morfin, Laurent Piccolo, J.-L. Rousset and V. Caps, Catal. Today, 2007, 122, 284 CrossRef CAS.
- D. Gajan, K. Guillois, P. Delichère, J.-M. Basset, J.-P. Candy, V. Caps, C. Copéret, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2009, 131, 14667 CrossRef CAS.
- M. Turner, V. B. Golovko, O. P. H. Vaughan, P. Abdulkin, A. Berenguer-,Murcia, M. S. Tikhov, B. F. G. Johnson and R. M. Lambert, Nature, 2008, 454, 981 CrossRef CAS.
- M. Alvaro, C. Aprile, A. Corma, B. Ferrer and H. Garcia, J. Catal., 2007, 245, 249 CrossRef CAS.
- C. Aprile, A. Corma, M. E. Domine, H. Garcia and C. Mitchell, J. Catal., 2009, 264, 44 CrossRef CAS.
- C. Aprile, M. Boronat, B. Ferrer, A. Corma and H. Garcia, J. Am. Chem. Soc., 2006, 128, 8388 CrossRef CAS.
- A. Thetford, G. J. Hutchings, S. H. Taylor and D. J. Willock, Proc. R. Soc. A Search PubMed , submitted.
| This journal is © The Royal Society of Chemistry 2011 |