Eco-friendly liquid chromatographic separations based on the use of cyclodextrins as mobile phase additives†
Víctor
González-Ruiz
a,
Andrés G.
León‡
a,
Ana I.
Olives
a,
M. Antonia
Martín
*a and
J. Carlos
Menéndez
b
aSección Departamental de Química Analítica, Facultad de Farmacia, Universidad Complutense, 28040, Madrid, Spain. E-mail: mantonia@farm.ucm.es; Fax: +34 91 3941754; Tel: +34 91 3941756
bDepartamento de Química Orgánica y Farmacéutica, Facultad de Farmacia, Universidad Complutense, 28040, Madrid, Spain
First published on 6th December 2010
Abstract
Acetonitrile and methanol are the most popular solvents employed in analytical HPLC, but they suffer from a number of drawbacks from the environmental point of view. Alternative, greener mobile phases employing methanol or the less toxic solvent ethanol as the sole organic solvent are proposed in this paper, and applied to the problem of the separation of β-carbolines on C18-stationary phases. The use of β-cyclodextrin (β-CD) and (2-hydroxypropyl)-β-cyclodextrin (HPβ-CD) as mobile phase additives allowed us to increase the proportion of water in the mobile phases without loss in the resolution or efficiency of the separations, leading initially to a considerable reduction of the proportion of methanol in the mobile phase (from 70% to 50%) and at a later stage, to the development of a mobile phase containing only 30% of ethanol. The β-carboline–cyclodextrin association constants were determined by HPLC, and the inclusion complexes were also characterized by 1H-NMR, 13C-NMR and 2D-ROESY experiments, and these studies were used to explain the chromatographic behaviour. The new chromatographic methodology developed was validated and applied to the quantitation of β-carboline derivatives in spiked human serum samples. For the extraction of β-carboline alkaloids from serum samples, liquid–liquid extraction (LLE) and solid-phase extraction (SPE) procedures were compared. It was concluded that the combination of a pre-treatment procedure (ionic exchange SPE) with a water-enriched chromatographic separation leads to a promising, environmentally friendly new methodology.
Introduction
Cyclodextrins (CDs) are cyclic oligosaccharides with fascinating molecular recognition properties that have been exploited for the design of other host molecules. As stated by Sezjtli,1 CDs are very attractive for the development of environmentally friendly technologies because they are semi-natural compounds produced from starch (a renewable material) by enzymatic conversion. They present a variety of applications in chemistry2 and also in pharmaceutical, food and cosmetic industries because they are non-toxic and inexpensive. Furthermore, chemically modified CDs with tailor-made cavity dimensions are easily synthesized, and they can be designed to be soluble in water or organic solvents. In analytical chemistry, they are employed in separation techniques,3 including GC, SFC, HPLC and CZE. Their use as mobile phase additives (MPAs) in HPLC is less developed, in spite of the fact that CDs have the advantage over other additives of being transparent in the UV-Vis range and that they also enhance the HPLC resolution of non-chiral compounds like indole derivatives4 or steroids.5 CDs are also very popular in HPLC and CZE as chiral selectors, and thus CDs enhance the enantiomeric HPLC resolution of chiral compounds like norgestrel6 and sertraline.7 The effects of the addition of neutral and charged CDs in CZE and to the mobile phase in HPLC have been compared to establish the stereoselective interactions that contribute to chiral discrimination.8 A favourable consequence of the presence of CDs in the mobile phases is the possibility to reduce the organic solvent/water ratio without a decrease in the selectivity or resolution. For instance, the separation of several pharmaceutically interesting compounds (citalopram, pantoprazol, nitrofurazone, trans-resveratrol) was performed in acetonitrile–buffered aqueous solution6,9 or in methanol–buffered aqueous solution4,10 in the presence of different types of CDs as MPA.β-Carbolines are natural alkaloids occurring in plants and mammalian tissues. They have a great variety of biological activities, including mutagenic behaviour due to their capability of binding to DNA,11 and various neurological effects due to their interactions with benzodiazepine and imidazoline receptors.12 Clinical studies correlated increased plasma concentrations of β-carbolines13 with neurological diseases14 and also with high consumption of alcohol,15heroin16 and tobacco.17 The structures of the β-carboline alkaloids studied in this paper are shown in Fig. 1.
 | ||
| Fig. 1 β-Carbolines studied in this paper. | ||
Different chromatographic methods18 have been employed for the determination of β-carbolines in several samples and matrices. Thus, RP-HPLC coupled to mass spectrometry, fluorimetric and UV-Vis detection has been employed to quantify β-carboline in food and foodstuffs,19 human blood20 and hair,21 and HPLC with electrochemical detection is useful for determining β-carboline in foods and beverages22 as an alternative to gas chromatographic23 or capillary electrophoretic techniques.24 Among the liquid chromatographic procedures described for quantitation of β-carbolines, isocratic or gradient elution has been found to require aqueous mixtures with a high proportion of acetonitrile25 or methanol,14,20 or alternatively, acetonitrile–methanol mixtures.22,26 Lowering the pH of the aqueous component to pH 3.0 allows the reduction of the organic solvent proportion in the mobile phase,19a,21 but these conditions increase the degradation of the stationary phases.
HPLC is a powerful analytical technique that allows the fast separation and quantification of compounds in complex matrices with high efficiency, resolution and sensitivity, and is the most widely employed separation technique for routine analysis in industrial settings. The volume of organic solvent waste originating from HPLC analysis is very high, due to the huge number of instruments in operation worldwide. Reversed-phase HPLC, the most popular separation method, traditionally involves the use of mixtures of water and acetonitrile. Due to the toxicity of acetonitrile, these mixtures have to be treated as chemical waste, and for this reason there is a growing interest in the use of non-toxic solvents, specially alcohols, as an alternative.27 Replacing acetonitrile is not an easy task because acetonitrile–water mobile phases have a number of advantages including their lower viscosity and higher transparency in the UV region, coupled with better chromatographic efficiencies. There is a growing interest in the replacement of acetonitrile by ethanol to solve some specific chromatographic problems,28 but these methodologies are still in their early stages. In the case of ionic analytes such as uric acid, efficient RP-HPLC separation can be achieved employing ammonium phosphate as the mobile phase, but this requires a guard column to avoid the easy degradation of the analytical column.29 Other alternatives to reduce the organic solvent consumption involve the use of micellar liquid chromatography (MLC), an approach that combines conventional stationary phases with mobile phases modified by the presence of surfactants and a reduced proportion of organic solvents.30 One drawback of this technique is the tendency of the surfactants to become adsorbed on the chromatographic stationary phases.
The present paper describes the potential of β-CD and HPβ-CD as MPA for greener RP-HPLC separations. β-CD and HPβ-CD were selected over other CDs taking into account their benefit/cost ratio. We present here our results on the use for this purpose of modified methanolic mobile phases, which are compared with the greener ethanolic mobile phases. This has been illustrated for the case of the separation of the biologically relevant β-carboline alkaloids norharmane, harmane and harmine in human serum samples. Taking into account that the complexation equilibrium competes with the chromatographic distribution equilibrium, and in order to achieve the maximum reduction in the organic solvent proportion we also considered relevant a detailed knowledge of the stability and structure of the inclusion complexes to obtain a more detailed picture of its influence on the chromatographic process. To this end, we undertook the chromatographic determination of the CD–carboline association constants and the structural characterization of the complexes by NMR techniques. In order to reduce the use of organic solvents also at the pre-treatment stage, our study involves the comparison of the traditional Liquid–Liquid Extraction (LLE) protocol with another one based on Solid-Phase Extraction (SPE), which requires a much lower amount of organic solvent and which had not been previously applied to the analysis of carboline derivatives in blood or serum samples.
Experimental section
Apparatus and reagents
All reagents and solvents were analytical, spectroscopic or chromatographic grade and were used without further purification. Water was doubly distilled and deionized (Milli-Q system) prior to its use. Norharmane, harmane and harmine were purchased from Sigma (Steinheim, Germany) as well as the cyclodextrins employed, namely β-CD and HP β-CD (average degree of substitution: 0.8 hydroxypropyl groups per glucose unit).NMR spectra were recorded with a Bruker Advance 250 spectrometer (Servicio de Resonancia Magnética Nuclear, UCM) using D2O or d6-DMSO as solvents.
A liquid chromatographic system (Hitachi-Merck, Tokyo, Japan) consisted of a quaternary gradient pump L-7100 coupled with a fluorescence detector L-7485, and a second was equipped with an isocratic pump L-6000 coupled to a fluorescence detector F-1050. For both instruments the temperature of the columns was controlled with a L-2300 oven and the samples were injected through a Rheodyne 7725i injector (20 μL loop). The two chromatographic systems were under computer control through the HPLC System Manager software, version 4.1. The analytical C18 columns (Spherisorb, ODS2, 5 μm, 150 × 4.6 mm) were purchased from Waters (Milford, Massachusetts) and they were utilized in combination with ethanolic and methanolic mobile phases. The mobile phases were filtered through a 0.45 μm × 47 mm membrane filter (GPH, Waters, Bedford, MA, USA and Sartorius Stedim Biotech GmbH, Goettigen, Germany) under vacuum followed by the application of ultrasound for 5 min. The flow rate was 0.9 mL min−1 (pressure of 80–120 bar). The fluorescence detection conditions used an excitation wavelength of 290 nm and an emission wavelength of 430 nm.
Procedures
Stock solutions of norharmane, harmane or harmine (3.0 × 10−3 M in ethanol) were freshly prepared. Aliquots of these stocks were taken to prepare solutions around 3.0 × 10−5 M of the alkaloids in 0.1 M HCl. These acidic solutions were spectrophotometrically measured and their molar absorptivities were considered in order to calculate accurately the real concentration of the stock solutions.31 Then, suitable volumes of the standarized stock solutions were employed to prepare the standards for validation of chromatographic procedure and also to spike the serum samples studied.The β-carbolines–CD solid inclusion complexes were prepared at stoichiometric ratios of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:2. An appropriate amount of norharmane, harmane and harmine was weighed. Then the corresponding amount of CD was added to the solid β-carboline derivative and an adequate volume of water to assure the solubilization of the cyclodextrins. The mixture was magnetically stirred in a water bath at 50 °C for 1 week. The mixture was then cooled to 8 °C and the solid complexes obtained were filtered and washed with ethanol followed by water to eliminate free β-carboline and free CDs. Finally, the complexes were freeze-dried for 48 h. Then, adequate amounts of the complexes obtained were dissolved in d6-DMSO or D2O containing a trace of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal standard, at a concentration of 0.01 M for β-CD and 0.1 M for HPβ-CD, in order to obtain the 1H-NMR and 13C-NMR spectra.
:1 and 1:2. An appropriate amount of norharmane, harmane and harmine was weighed. Then the corresponding amount of CD was added to the solid β-carboline derivative and an adequate volume of water to assure the solubilization of the cyclodextrins. The mixture was magnetically stirred in a water bath at 50 °C for 1 week. The mixture was then cooled to 8 °C and the solid complexes obtained were filtered and washed with ethanol followed by water to eliminate free β-carboline and free CDs. Finally, the complexes were freeze-dried for 48 h. Then, adequate amounts of the complexes obtained were dissolved in d6-DMSO or D2O containing a trace of 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS) as an internal standard, at a concentration of 0.01 M for β-CD and 0.1 M for HPβ-CD, in order to obtain the 1H-NMR and 13C-NMR spectra.
For the chromatographic determination of the association constants, isocratic mobile phases of methanol–buffered phosphate (pH 7.8), 50:50, (v:v) and isocratic mobile phases of ethanol–buffered phosphate (pH 7.8), 30:70, (v:v) were employed. The mobile phases were modified by the addition of appropriate amounts of CDs. The range of concentrations assayed was 0–3.0 mM (7 levels of concentration, methanolic mobile phases) and 0–11.0 mM (7 levels of concentration, ethanolic mobile phases) for β-CD and 0–17 mM for HPβ-CD (10 levels of concentration, methanolic and ethanolic mobile phases). The association constants were determined at 25, 35 and 45 °C. The columns were washed daily according to a previously described procedure,32 which allowed each column to be employed at least in 370–400 assays for the analysis of β-carbolines in serum samples.
With the aim to demonstrate the applicability of the method to the separation and quantitation of β-carbolines in real samples, human serum specimens (∼2–3 mL) belonging to 20 healthy volunteers were mixed to obtain a representative serum pool exempt from β-carboline derivatives. Two aliquots (S1 and S2) were taken from this pool and then adequate volumes of standard solutions containing norharmane, harmane and harmine were added to the serum samples, the concentration of each alkaloid (norharmane, harmane and harmine) being 0.250 μM (S1) and 0.500 μM (S2). The serum samples were spiked with the standard solutions of the alkaloids in a volume no higher than 5% of the initial serum volume. Two different procedures for sample clean-up were employed, namely the liquid–liquid extraction (LLE) procedure20a for the determination of harmane and harmine in blood samples, and the solid-phase extraction (SPE) procedure described for analysis of β-carboline derivatives in food samples.19SPE was carried out employing cartridges of 3 mL capacity packed with 200 mg propylsulfonic acid-derivatized silica gel (Isolute SCX 2, International Sorbent Technology, Mid-Glamorgan, UK) placed in a vacuum manifold. The purified samples were then injected onto the chromatographic system using methanolic and ethanolic mobile phases modified by the addition of appropriate amounts of CDs (3 mM β-CD or 15 mM HPβ-CD).33
Results and discussion
1H- and 13C-NMR characterization of the β-carboline–cyclodextrin inclusion complexes
Although different methodologies and instrumental techniques can be employed for the structural characterization of CD inclusion complexes, NMR is well-established as a powerful tool for investigating the through-space interactions of CDs and their ligands, both inside and outside the CD cavities. In the present work, we have employed for this purpose a combination of 1D 1H-NMR and 13C-NMR and 2D ROESY data. We have previously described a preliminary 1D NMR study of the harmane and harmine CD inclusion complexes, but under the conditions employed the carboline signals could not be observed, and the study was based on the changes observed in the glucose signals.34a We report now the use of more sensitive conditions that allow the observation of the alkaloid signals and perform a much more refined structural study.Several significant changes were observed in the 1H-NMR spectra of the complexes in comparison to those of the corresponding free cyclodextrin and guest. As an example, Fig. 2 shows the 1H-NMR spectrum of β-CD and its 1:1 complex with harmane. CD complexation led to decoupling of the OH groups at C-2, C-3 and C-6. This reflects a fast proton exchange, which is a consequence of alterations of the hydrogen-bonding network at the rims of the cyclodextrin. Related changes were observed for HPβ-CD (Fig. S1†). Comparison of the 1H-NMR spectra of free CDs with the ones obtained for the inclusion complexes, significant spectral changes can also be observed in the 3.40 ppm zone. In the case of free CDs, an intense peak is observed in this region due to the water molecules included inside the CD cavity,34b but for the inclusion complexes this signal disappears because water is displaced from the CD cavity by the β-carboline molecules.
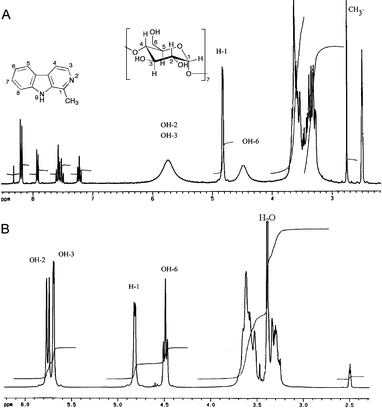 | ||
| Fig. 2
1H-NMR
spectrum of (A) the harmane–β-CD complex (1:1) and (B) reference β-CD. The spectra were obtained at 250 MHz in d6-DMSO. | ||
13C-NMR measurements showed increased displacements for harmane, chosen as a representative example for this discussion, and higher displacements for the 1:2 stoichiometry, showing a deeper penetration of the ligands inside the CD cavities. In the case of the 1:1 complexes, the signals that showed the highest displacements were those of C-5, C-6, C-7 and C-8 (Table 1), which proved that the more lipophilic benzene rings of the alkaloids enter the cavity first. The significant increase in Δδ for C-4 for the 1:2 stoichiometry is consistent with a complex containing two CD molecules per ligand. The absence of changes in the CH3 group suggests that, due to geometrical reasons, this group is outside the CD. Fig. 3 summarizes the displacements found for the carbon signals of harmane. Harmine showed a very similar behaviour to harmane in all respects. However, in the case of norharmane the signals due to the benzene ring and those of the pyridine ring showed similar displacements, and indeed the most significant one corresponded to C-1. This suggests a different mode of inclusion, where both the benzene and the pyridine rings at the ends of the molecule can enter the CD cavities. For the full data and copies of representative spectra of the complexes, see the ESI (Fig. S2–S10†).
:1 and 1:2 cyclodextrin inclusion complexes. Data are given as δcomplex − δguest molecule
| Norharmane–β-CD | Norharmane–HPβ-CD | Harmane–β-CD | Harmane–HPβ-CD | Harmine–β-CD | Harmine–HPβ-CD | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Δδ 1:1 | Δδ 1:2 | Δδ 1:1 | Δδ 1:2 | Δδ 1:1 | Δδ 1:2 | Δδ 1:1 | Δδ 1:2 | Δδ 1:1 | Δδ 1:2 | Δδ 1:1 | Δδ 1:2 | |
| C-1 | 17.8 | 14.9 | 6.1 | −1.3 | 2.9 | 8.8 | 2.5 | 6.5 | −0.5 | 4.5 | 1.7 | 5.0 |
| C-3 | −3.7 | −2.2 | 7.6 | 3.5 | 5.0 | 7.8 | 4.8 | 6.5 | 1.0 | 2.8 | 2.2 | 3.0 |
| C-4 | 3.6 | 7.8 | 4.0 | 8.5 | 6.9 | 16.7 | 6.3 | 13.3 | 2.3 | 7.6 | 4.5 | 7.8 |
| C-4a | −1.4 | −0.5 | −2.4 | 0.9 | 0.4 | 4.8 | 0.5 | 3.8 | −0.8 | 1.8 | 0.6 | 2.0 |
| C-4b | 1.7 | 6.7 | 4.2 | 8.7 | 2.3 | 13.4 | 2.2 | 10.7 | 0.4 | 6.4 | 2.6 | 7.0 |
| C-5 | 4.2 | 8.9 | 4.3 | 9.6 | 7.0 | 17.7 | 4.6 | 14.2 | 3.0 | 7.8 | 4.8 | 7.7 |
| C-6 | 3.4 | 6.6 | 3.5 | 2.1 | 6.9 | 14.9 | 6.3 | 11.8 | 3.2 | 3.4 | 4.6 | 8.5 |
| C-7 | 4.4 | 9.4 | 5.2 | 10.2 | 7.0 | 18.4 | 6.3 | 14.5 | 0.5 | 5.3 | 2.4 | 5.6 |
| C-8 | 0.8 | 4.3 | 0.8 | 30.0 | 4.2 | 12.1 | 3.8 | 10.0 | 0.3 | 4.0 | 2.0 | 4.5 |
| C-8a | 0.4 | 3.0 | 0.8 | 4.2 | 0.8 | 9.0 | 0.5 | 6.8 | 0.0 | 2.9 | 1.4 | 3.0 |
| C-9a | −2.6 | −0.2 | 3.6 | 0.2 | 0.6 | 7.0 | 0.5 | 5.2 | −0.7 | 2.7 | 0.9 | 3.1 |
| CH3 | — | — | — | — | 0.3 | 0.8 | 0.1 | 0.2 | −0.7 | −0.7 | −0.3 | −0.6 |
| OCH3 | — | — | — | — | — | — | — | — | 2.5 | 6.5 | 4.1 | 6.6 |
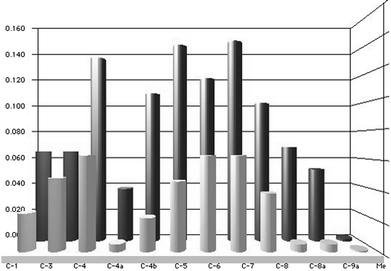 | ||
| Fig. 3 Comparison of the chemical shift changes (ppm) for the C-signals of the harmane in the inclusion complexes with harmane–CD stoichiometry of 1:1 (front) and 1:2 (back), in comparison to free harmane. | ||
A more detailed picture of the solution geometry of the complexes can be obtained by measuring NOE enhancements, which show the spatial proximity of protons. However, cyclodextrin complexes fall in the region where ωtc ≈ 1, and consequently small NOE effects are expected. Therefore, rotating-frame nuclear Overhauser effect (ROE) experiments, which give a positive response over the whole molecular weight range,35 were employed. 2D ROESY experiments for the 1:1 and 1:2 harmane–HPβ-CD complexes are shown in Fig. 4 as representative examples. Clear correlations were observed between the signals corresponding to the protons of the benzene rings H-5, H-6, H-7 and H-8 on the β-carboline ring and the group of signals corresponding to protons H-3, H-5 and H-6 of the glucose rings, but not to H-2 and H-4. Since H-3 and H-5 are oriented towards the inner side of the CD cavities and H-2 and H-4 are outside, these results prove the penetration of the alkaloids into the host molecules. Another intense correlation is observed for H-4 of the carboline system, which is sufficiently close to the rim of the CD molecule, while H-3 shows a clear correlation only for the case of the 1:2 complex. The methyl group of harmane also shows a correlation, although rather weak. Harmine shows a similar behaviour to harmane. In the case of norharmane, strong ROESY correlations were observed for H-1, H-3, H-4, H-5, H-6, H-7 and H-8, confirming that the benzene and pyridine rings can be included with similar probability (ROESY experiments not reproduced here can be found in the ESI, Fig. S11 to S15†).
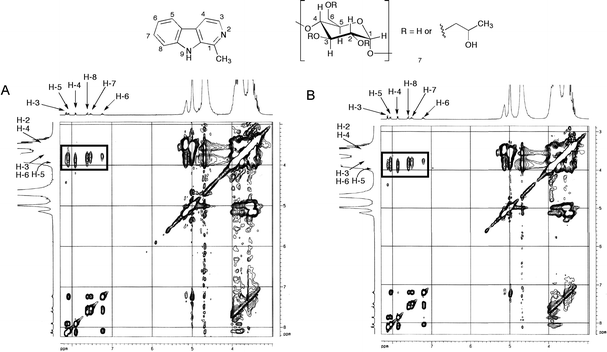 | ||
| Fig. 4 Partial contour plots of 2D-ROESY experiments (D2O, 250 MHz) corresponding to the inclusion complexes (A) harmane–HPβ-CD (1:1) and (B) harmane–HPβ-CD (1:2). | ||
The 13C-NMR studies, coupled with the ROESY correlations, lead us to propose the schematic model for the inclusion of harmane and harmine shown in Fig. 5A. On the other hand, the 1:1 norharmane complexes can be considered as mixtures of two species, where the inclusion takes place by either end of the molecule (Fig. 5B). According to these models, the more lipophilic part of the molecule, which is the one responsible for the interaction with the stationary phases, is inside the CD cavity. Therefore, diminished retention times can be expected in the presence of CDs, even in mobile phases containing high proportions of water, in comparison with the situation in the absence of cyclodextrins. For the case of norharmane, the effective interactions of both aromatic rings (benzene and pyridine) with the CDs increase the solubility of the ligand in the aqueous mobile phases and thus decrease its retention on the stationary phase, contributing to a further reduction of the retention times. This is in agreement with the higher values found for the association constants of norharmane in comparison with harmane and harmine (see Table 2).
| MeOH–buffer | EtOH–buffer | |||||||
|---|---|---|---|---|---|---|---|---|
| β-CD | HPβ-CD, Kass ± σn−1 (M−1) | β-CD, Kass ± σn−1 (M−1) | HPβ-CD, Kass ± σn−1 (M−1) | |||||
| 25 °C | 35 °C | 45 °C | 35 °C | 45 °C | 35 °C | 45 °C | ||
| a The values obtained were not proccesable because R2 < 0.80. | ||||||||
| Norharmane | —a | 19.82 ± 0.16 | 10.05 ± 0.44 | 10.63 ± 1.63 | 11.21 ± 2.07 (64.78 ± 11.95) | 6.70 ± 0.28 (38.71 ± 1.64) | 21.59 ± 2.47 | 15.99 ± 0.96 |
| Harmane | —a | 16.48 ± 0.35 | 5.81 ± 0.43 | 9.19 ± 1.78 | 8.30 ± 1.68 (47.98 ± 9.74) | 4.89 ± 0.94 (28.30 ± 5.42) | 16.46 ± 2.11 | 13.06 ± 0.34 |
| Harmine | —a | 18.01 ± 0.28 | —a | 9.47 ± 2.09 | 6.62 ± 3.03 (—a) | —a (—a) | 12.58 ± 3.23 | 11.49 ± 0.51 |
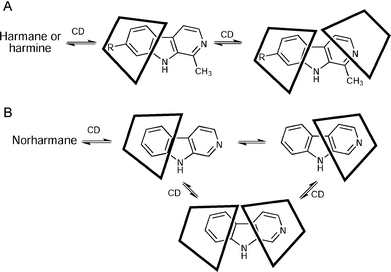 | ||
| Fig. 5 Proposed models of the inclusion of β-carboline alkaloids into the CD cavities. (A) Inclusion complexes of harmane or harmine. (B) Inclusion complexes of norharmane. | ||
Chromatographic determination of the β-carboline–cyclodextrin association constants
The presence of CDs in the mobile phases has been used to enhance the chromatographic separation of closely related compounds.3,36 When CDs are added to the mobile phases, the retention factors decrease as a consequence of the formation of inclusion complexes during the chromatographic separation. The determination of the complex stability constants and the evaluation of the strength of guest–host interactions by HPLC are of interest because the existence of competitive chromatographic and inclusion equilibria can lead to alterations not only of the retention factors but also of the elution order and the selectivity. Studying the association constants can therefore contribute to optimize the mobile phase and the chromatographic conditions for routine analysis. Mobile phases with a very high proportion of water favour the formation of CD inclusion complexes, but they cannot be employed in practice because the retention times became prohibitively long, even in the presence of CDs in the mobile phase.The apparent association constants (Kass) can be determined using eqn (1),37 where k and k0 are the retention index values in the presence and absence of cyclodextrin, respectively. The plot of 1/k vs. [CD] gives a straight line, where the value of Kass can be calculated as slope/ordinate.
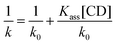 | (1) |
Table 2 shows the values obtained for the β-carboline–CD association constants (Kass), which can be seen to bear an inverse relationship with temperature. Inclusion complexes were more stable in ethanol–water mixtures than in methanol, and complexes with HPβ-CD were more stable than those with β-CD for the ethanolic mobile phases, which is an important result in terms of the use of ethanol as real alternative in the design of greener mobile phases. It is well known that increased temperatures are accompanied by better chromatographic features, and for this reason we undertook a brief study of the influence of temperature on complex stability. As shown in Table 2, it can be observed that increasing temperatures favour the dissociation of the β-carboline–CD complexes, an effect that is well documented in the literature for some types of ligands. This observation led us to exclude the use of temperatures above 35 °C to increase the chromatographic efficiency. For both mobile phases and both CDs, the magnitude of the association constants for the three β-carbolines studied varied as follows: norharmane > harmane > harmine. These values are in agreement with NMR results and with the models of inclusion proposed in Fig. 5.
The literature association constants for the methanol–β-CD and ethanol–β-CD complexes are 0.32 and 0.93 M−1, respectively.38 Considering these values, the alkaloids should be more easily displaced from the CD cavities by ethanol than by methanol, but the presence of a higher proportion of water in the ethanolic mobile phases more than makes up for this effect, as previously observed by other workers, who noticed that an increase of 5% in the proportion of water of the mobile phases produced a significant enhancement of the association constant values for terpene derivatives.39 This effect can be attributed to the predominance of hydrophobic interactions inside the CD cavity, which favours the formation of analyte inclusion complexes.40Table 2 also includes the values of Kass calculated considering the competition of the solvent and β-carboline alkaloids, where Ksolvent is 0.32 for methanol and 0.93 for ethanol, and the free CD concentration present in the mobile phase ([CD]M) can be determined as shown in eqn (2).38,41
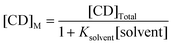 | (2) |
As expected, a notable increase in the stability constants was observed when the competition of the solvent for the β-CD was considered (Table 2). From these results can be deduced that the water/organic solvent ratio employed in our experiments is adequate to obtain a reasonable time of analysis and the combination C18 and ethanol–buffered aqueous solution with HPβ-CD as MPA is a valuable approach to the development of green chromatographic separations.
Influence of CDs on the chromatographic separation and quantitation of β-carbolines
The concentration of CDs in the mobile phases needs to be adjusted to the experimental conditions. Higher CD concentrations reduced the retention factor although, due to the limited solubility of CDs in the alcohol–water mixtures, the pressure in the column is increased and therefore the column efficiency is reduced. Based on our previous experience on the influence of the composition of the mobile and stationary phases, temperature and CDs concentration in the mobile phase on the separation of β-carboline alkaloids,33 15 mM HPβ-CD and 3 mM β-CD concentrations were chosen for the quantitation of norharmane, harmane and harmine in biological samples. We have also described the chromatographic determination of the association constants of 2,6-di-O-methyl-β-cyclodextrin and 2,3,6-tri-O-methyl-β-cyclodextrin and β-carboline alkaloids,32 but these CDs are too expensive for routine chromatographic analysis, compared to native β-CD and HPβ-CD.The addition of β-CD and HPβ-CD as MPA caused a notable decrease in the retention times and in the retention factors of the β-carboline alkaloids under study, as shown in Fig. 6 for the separation of the solutions of standards (see also Fig. 7 below for the results obtained for spiked serum samples). The chromatographic behaviour obtained was the same for the standards and for real samples. The decrease in the retention time was higher for the ethanolic than for methanolic mobile phases and the decrease observed in the retention factors was higher for HPβ-CD than for β-CD. These effects can be explained considering that the concentration of β-CD is lower than that of HPβ-CD, the proportion of water was higher in the ethanolic mixtures than in the methanolic ones and hence the formation of inclusion complexes is favoured in the ethanolic mobile phases, and also taking into account that the association constant values are higher for HPβ-CD than for β-CD. The decrease in the retention factors is a consequence of the higher solubility of the inclusion complexes in the mobile phase36 in comparison to free alkaloids present in the non-modified mobile phases.
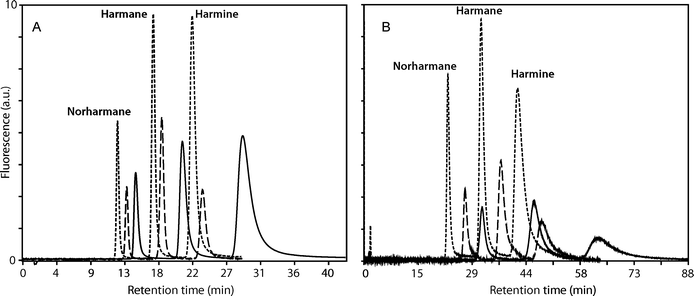 | ||
| Fig. 6 Effects of CDs as mobile phase additives on the separation of β-carboline alkaloids in the standard solutions, at 35 °C. (A) Mobile phase: methanol–buffered aqueous solution. (B) Mobile phase: ethanol–buffered aqueous solution. Without CDs (solid line), with β-CD 3 mM (dashed line) and HPβ-CD 15 mM (dotted line). | ||
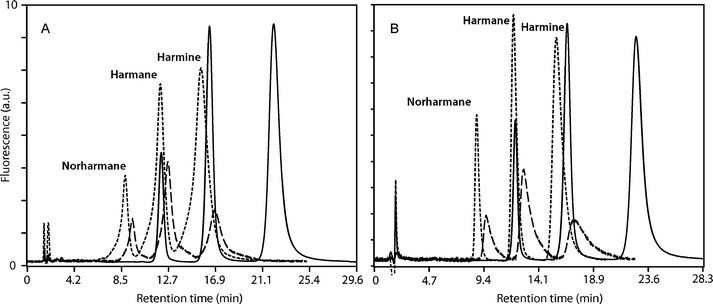 | ||
| Fig. 7 Chromatographic analysis of β-carboline alkaloids in human serum samples. (A) Chromatographic separation after liquid–liquid extraction (LLE). (B) Chromatographic separation after solid-phase extraction (SPE). Mobile phase: methanol–buffered aqueous solution, at 35 °C. Without CDs (solid line), with β-CD 3 mM (dashed line) and HPβ-CD 15 mM (dotted line). | ||
The reliability of the chromatographic method was shown by the agreement in the retention time obtained for β-carbolines in the standard solutions and those obtained for the spiked serum samples. A good reproducibility in the retention times was obtained in non-modified mobile phases and in the presence of CDs. Thus, after 12 consecutive injections of the standard solutions, the change in the retention times was in the 0.5–2% range for all the analytes using mobile phases without CDs. Similarly, no significant changes in the retention times were observed in the presence of CDs (1–3%). A percentage of reduction in the retention time of 3–7% was observed after 35–40 injections in three consecutive days with CDs. The highest reduction was observed in ethanolic mobile phases for both β-CD and HPβ-CD. The peak area reproducibility was also evaluated by injecting the standard solutions.
Validation of the chromatographic method
Linearity ranges and limits of detection were deduced using standard solutions of norharmane, harmane and harmine dissolved in the mobile phases in the 0.1–1.0 μM concentration range. Duplicate injections were made for each standard solution. The limit of detection (Table 3) was determined by injecting a solution of concentration 0.1 μM 6 times, and then performing calculations as described by Zheng.20a The calibration curves for the β-carbolines studied were linear over the 0.1–1.0 μM concentration range. The correlation coefficients (R2) were obtained from linear regression treatment, using the peak area versus concentration for each standard compound. An increase in the correlation and in the slopes of the calibration curves was observed in the presence of CDs in ethanolic or methanolic mobile phases. When HPβ-CD was employed as mobile phase additive, the slopes for the three alkaloids were twice as high in comparison with the non-modified mobile phases. The limits of detection (LODs) obtained in the presence of CDs were better than the ones obtained in non-modified mobile phases for both types of CDs and mobile phases studied (Table 3). The enhancement of LODs was most noticeable for ethanolic mobile phase and HPβ-CD. Fluorimetric detection is adequate for the quantitation of these alkaloids, whose high native fluorescence is enhanced by the formation of CD inclusion complexes, increasing the sensitivity for their detection.42 The LOD values obtained (2–6 × 10−8 M, which corresponds to 3.6–10.8 ng mL−1) were in the same range as other chromatographic procedures described for the determination of β-carbolines in blood samples.20a–c These values were better than those required for the analysis of the alkaloids17,18b in foodstuffs, cigarette smoke condensates or plant extracts.| Mobile phase | Compound | R 2 | LOD |
|---|---|---|---|
| a R2 Coefficient of determination; LOD limit of detection (n = 6). | |||
| Without CDs in the mobile phase | |||
| MeOH–buffer 50:50 | Norharmane | 0.9970 | 5.92 × 10−8 M |
| Harmane | 0.9960 | 6.10 × 10−8 M | |
| Harmine | 0.9959 | 3.77 × 10−8 M | |
| EtOH–buffer 30:70 | Norharmane | 0.9887 | 1.04 × 10−7 M |
| Harmane | 0.9903 | 1.20 × 10−7 M | |
| Harmine | 0.9521 | 1.41 × 10−7 M | |
| 3 mM β-CD in the mobile phase | |||
| MeOH–buffer 50:50 | Norharmane | 0.9993 | 2.03 × 10−8 M |
| Harmane | 0.9996 | 2.07 × 10−8 M | |
| Harmine | 0.9997 | 3.66 × 10−8 M | |
| EtOH–buffer 30:70 | Norharmane | 0.9975 | 6.33 × 10−8 M |
| Harmane | 0.9987 | 5.30 × 10−8 M | |
| Harmine | 0.9940 | 7.46 × 10−8 M | |
| 15 mM HPβ-CD in the mobile phase | |||
| MeOH–buffer 50:50 | Norharmane | 0.9987 | 5.15 × 10−8 M |
| Harmane | 0.9991 | 5.36 × 10−8 M | |
| Harmine | 0.9992 | 4.70 × 10−8 M | |
| EtOH–buffer 30:70 | Norharmane | 0.9978 | 6.14 × 10−8 M |
| Harmane | 0.9974 | 5.83 × 10−8 M | |
| Harmine | 0.9975 | 5.47 × 10−8 M | |
Precision was evaluated by the intra-day and inter-day repeatability relative standard deviation (RSD, %). For intra-day assays the standard solution containing the mixture of alkaloids was injected 6 times in the same day. For the inter-day RSD calculation the standard solutions containing the mixture of alkaloids were injected in duplicate sets for 6 consecutive days. Intra-day and inter-day precision studies were carried out at two concentrations: 0.1 μM and 1.0 μM (Table 4). The precision of the method was good, as shown by the fact that RSD values were lower than 8% in 64 cases out of 72. The RSD values were higher for the ethanolic mobile phases than for the methanolic ones for both concentration levels. These results are a consequence of the efficiency of the chromatographic conditions, since the peaks are wider in ethanol than in methanol, and this affects the quantitation based on peak areas. The intra-day precision was enhanced in the presence of β-CD as MPA for both mobile phases with regard to the results obtained without CDs (for a graphical comparison, see Fig. S16†). The intra-day precision for β-CD was better than in the case of HPβ-CD for both mobile phases. The differences between the RSD values for intra-day assay and inter-day assay are negligible, which shows a good response of the chromatographic system in the presence of CD additives in the mobile phase, owing to the adequate selection of CD concentrations and the efficiency of the daily washing procedure.32,33 Thus, comparing the same mobile phase and the same CD as MPA, the RSD values for inter-day assay are lower than the corresponding intra-day assay RSD values. These results can be ascribed to the fact that the inter-assay experiments were developed each day after the night washing procedure and hence the detrimental effect of the presence of CDs in the mobile phases for several hours is avoided. Accuracy was estimated as the percentage of the measured concentration (on the calibration curves) over the nominal concentrations. The accuracy of the chromatographic separation and of the total methodology including the pre-treatment procedures has been determined to show the utility of CDs as MPA. The accuracy of the chromatographic procedure was evaluated in terms of recovered concentrations. An exactly measured volume of the standarized solutions of the β-carbolines under study was added to the extracted serum samples containing a concentration of 0.5 μM of each β-carboline alkaloids. Then the samples were injected (n = 3) under the different chromatographic conditions. The results are presented in Table 5. The recoveries obtained (90–113%) are very satisfactory for the experimental conditions including CDs as MPA.
| Compound | Intra-day precisiona (%RSD) | Inter-day precisiona (%RSD) | ||
|---|---|---|---|---|
| MeOH–buffer 50:50 | EtOH–buffer 30:70 | MeOH–buffer 50:50 | EtOH–buffer 30:70 | |
| a n = 6. b 0.1 μM. c 1.0 μM | ||||
| Without CDs in the mobile phase | ||||
| Norharmaneb | 3.31 | 3.09 | 1.55 | 9.20 |
| Harmaneb | 2.87 | 11.53 | 1.91 | 4.29 |
| Harmine b | 4.97 | 11.42 | 1.50 | 3.60 |
| Norharmanec | 3.55 | 4.31 | 1.85 | 6.27 |
| Harmanec | 2.30 | 1.19 | 1.94 | 6.06 |
| Harmine c | 4.32 | 7.74 | 1.35 | 5.53 |
| 3 mM β-CD in the mobile phase | ||||
| Norharmaneb | 3.10 | 4.14 | 1.82 | 7.39 |
| Harmaneb | 1.79 | 2.75 | 2.95 | 8.15 |
| Harmine b | 6.09 | 9.73 | 4.76 | 5.97 |
| Norharmanec | 1.85 | 2.40 | 3.85 | 3.79 |
| Harmanec | 1.62 | 2.20 | 4.53 | 3.91 |
| Harmine c | 2.61 | 4.02 | 5.16 | 4.04 |
| 15 mM HPβ-CD in the mobile phase | ||||
| Norharmaneb | 5.25 | 7.64 | 5.42 | 8.27 |
| Harmaneb | 7.23 | 5.86 | 6.33 | 8.51 |
| Harmine b | 4.52 | 6.40 | 8.53 | 6.73 |
| Norharmanec | 4.72 | 5.49 | 3.83 | 3.10 |
| Harmanec | 4.02 | 5.34 | 2.69 | 2.88 |
| Harmine c | 4.08 | 6.37 | 3.77 | 6.19 |
| Compound | Recoveries (%)a | |
|---|---|---|
| MeOH–buffer 50:50 | EtOH–buffer 30:70 | |
| a β-Carboline alkaloids were added (c = 0.250 μM) to the previously extracted spiked serum samples containing the alkaloids in concentrations 0.500 μM. | ||
| Without CDs in the mobile phase | ||
| Norharmane | 101.3 | 113.0 |
| Harmane | 108.8 | 107.9 |
| Harmine | 106.2 | 103.6 |
| 3 mM β-CD in the mobile phase | ||
| Norharmane | 98.5 | 100.0 |
| Harmane | 99.1 | 102.1 |
| Harmine | 89.8 | 92.7 |
| 15 mM HPβ-CD in the mobile phase | ||
| Norharmane | 96.4 | 106.6 |
| Harmane | 101.6 | 107.7 |
| Harmine | 91.7 | 92.6 |
In order to verify the accuracy of the methodology (extraction and chromatographic procedures) for the determination of β-carboline derivatives in biological samples, absolute recoveries were calculated. Thus, a known amount of each β-carboline studied was added to the spiked serum samples (0.500 μM concentration) and then the complete analytical protocol was applied. The recoveries obtained are presented in Table 6. The chromatographic recoveries were close to 100%, while for the overall method the recoveries present a greater dispersion. The values obtained for the absolute recoveries are in a range similar to or better than those described in the literature for different analytes in real samples, i.e. β-carbolines in serum samples (70–130%),20aphenotiazine derivatives in human urine (67–105%)43 and anti-depressant drugs in plasma (52–110%).44
| Compound | Liquid–liquid extraction | Solid-phase extraction | ||
|---|---|---|---|---|
| MeOH–buffer 50:50 | EtOH–buffer 30:70 | MeOH–buffer 50:50 | EtOH–buffer 30:70 | |
| a The alkaloid was added up to a final 0.125 μM concentration. b The alkaloid was added up to a final 0.250 μM concentration. | ||||
| Without CDs in the mobile phase | ||||
| Norharmanea | 133.0 | 109.2 | 94.8 | 91.7 |
| Harmanea | 114.2 | 109.1 | 107.5 | 90.1 |
| Harmine a | 112.1 | 111.9 | 102.6 | 115.5 |
| Norharmaneb | 89.0 | 107.8 | 82.1 | 101.4 |
| Harmaneb | 110.4 | 112.9 | 95.0 | 95.6 |
| Harmine b | 100.7 | 134.7 | 87.2 | 111.8 |
| 3 mM β-CD in the mobile phase | ||||
| Norharmanea | 115.8 | 146.8 | 102.7 | 70.7 |
| Harmanea | 92.4 | 122.7 | 109.2 | 87.7 |
| Harmine a | 84.3 | 119.2 | 84.8 | 92.3 |
| Norharmaneb | 74.5 | 144.4 | 107.3 | 91.8 |
| Harmaneb | 99.8 | 149.5 | 105.2 | 83.0 |
| Harmine b | 87.9 | 134.7 | 97.4 | 95.0 |
| 15 mM HPβ-CD in the mobile phase | ||||
| Norharmanea | 123.5 | 146.9 | 77.3 | 85.3 |
| Harmanea | 115.5 | 153.1 | 83.0 | 96.6 |
| Harmine a | 112.0 | 159.5 | 92.1 | 97.6 |
| Norharmaneb | 76.9 | 136.8 | 78.7 | 81.8 |
| Harmaneb | 100.1 | 151.4 | 81.0 | 78.6 |
| Harmine b | 92.3 | 147.4 | 82.3 | 93.0 |
Regarding the extraction procedures employed, it can be appreciated that the recoveries obtained for the aliquots extracted by SPE are better than those obtained from the LLE using the method described by Zheng.20a The absolute recoveries obtained for the SPE method were in the range 81–115% in 33 out of 36 cases, but the recoveries for LLE-extracted samples were less adequate. These results can be explained considering that the extracts obtained by both extraction procedures are in different solvents, since the eluates obtained from SPE are in a methanol–acidic aqueous solution and the acid pH assures the protonation of the alkaloids, thus increasing their solubilities and allowing the use of a reduced volume of organic solvent. On the other hand, liquid–liquid extraction according to the Zheng protocol20a requires tedious evaporation of each individual fraction and subsequent re-dissolution of the dry residues in a small volume of methanol to avoid dilution of the analytes, and in these conditions the residue is not completely dissolved. Since these methanol solutions are then directly injected into the chromatograph, these problems lead to an increase in the width of the chromatographic peaks for the case of the LLE-extracted samples (Fig. 7), for both methanolic and ethanolic mobile phases. This behaviour was not observed for the SPE extracts, explaining why accuracy values obtained in the validation for SPE are better than for LLE. As an additional advantage, SPE results in a lower volume of extractive mixtures, which have a higher proportion of water; this is possible due to the ionic exchange retention mechanism. Besides, SPE requires less operator time and simpler experimental conditions.
Application of the developed methodology to the analysis of β-carbolines in human serum samples
The pooled serum samples spiked with β-carbolines at 0.250 μM and 0.500 μM concentrations were analyzed by the proposed RP-HPLC procedures after sample clean-up (LLE or SPE). The experimental concentrations of β-carbolines were calculated by considering the areas under the peaks and comparing them to those obtained from the standard calibration curves. The values of alkaloid concentrations found in the samples are shown in Table 7. According to the figures of merit obtained, the differences among the theoretical values of β-carbolines and the experimental values obtained are related to the extraction methodology employed. In conclusion, these chromatographic procedures can be successfully applied to the quantitation of β-carbolines in biological samples.| Compound | Liquid–liquid extraction | Solid-phase extraction | ||
|---|---|---|---|---|
| MeOH–buffer 50:50 | EtOH–buffer 30:70 | MeOH–buffer 50:50 | EtOH–buffer 30:70 | |
| a The theoretical value was 0.250 μM. b The theoretical value was 0.500 μM. | ||||
| Without CDs in the mobile phase | ||||
| Norharmanea | 0.248 | 0.285 | 0.205 | 0.243 |
| Harmanea | 0.285 | 0.261 | 0.231 | 0.217 |
| Harmine a | 0.290 | 0.294 | 0.230 | 0.258 |
| Norharmaneb | 0.435 | 0.457 | 0.379 | 0.458 |
| Harmaneb | 0.494 | 0.448 | 0.419 | 0.436 |
| Harmine b | 0.482 | 0.469 | 0.405 | 0.460 |
| 3 mM β-CD in the mobile phase | ||||
| Norharmanea | 0.240 | 0.255 | 0.261 | 0.214 |
| Harmanea | 0.272 | 0.271 | 0.278 | 0.234 |
| Harmine a | 0.237 | 0.272 | 0.253 | 0.210 |
| Norharmaneb | 0.408 | 0.422 | 0.523 | 0.418 |
| Harmaneb | 0.460 | 0.473 | 0.562 | 0.449 |
| Harmine b | 0.418 | 0.488 | 0.504 | 0.413 |
| 15 mM HPβ-CD in the mobile phase | ||||
| Norharmanea | 0.231 | 0.233 | 0.202 | 0.198 |
| Harmanea | 0.255 | 0.256 | 0.229 | 0.213 |
| Harmine a | 0.243 | 0.246 | 0.206 | 0.179 |
| Norharmaneb | 0.387 | 0.424 | 0.386 | 0.423 |
| Harmaneb | 0.438 | 0.461 | 0.439 | 0.438 |
| Harmine b | 0.401 | 0.416 | 0.385 | 0.381 |
Comparison to other chromatographic separations (RP-HPLC) of β-carbolines
The proposed method was compared to some of the most representative HPLC quantitation methods for β-carboline alkaloids14,18b,20a,26a in terms of solvent consumption. The methodology described here avoided the use of acetonitrile and also allowed a considerable reduction of the proportion of methanol in the mobile phase (from 70% to 50%). It also allowed the use of ethanol as mobile phase, which was unprecedented. Furthermore, the presence of CDs as MPA increased the sensitivity for detection of the analytes and did not reduce the accuracy or precision of the methodology.Conclusions
The inclusion complexes from norharmane, harmane and harmine with β-CD and HPβ-CD have been characterized by 1D- and 2D-NMR experiments. For the cases of harmane and harmine, and for inclusion complexes with both 1:1 and 1:2 alkaloid–CD stoichiometries, these studies show that inclusion takes place through the benzene ring of the host molecules, and the pyridine moiety remains partially outside the CD cavities. In the case of norharmane, either the benzene or the pyridine rings at the ends of the molecule can enter the CD cavities. Interestingly, these structural studies are in good agreement with the association constant values obtained by RP-HPLC, where the highest Kass values were those obtained for the case of norharmane because the more effective interactions with the CDs reduce the interaction with the stationary phases and hence the retention times. This correlation between structural data obtained by NMR and chromatographic behaviour constitutes another innovative aspect of our work. The addition of CDs allows a reduction of the organic solvent content in the HPLC mobile phases. Furthermore, it also allows the use of renewable solvents (methanol and ethanol) as the organic component, avoiding the use of the more toxic acetonitrile, the most frequent organic component in the analysis of β-carbolines by RP-HPLC. It has been demonstrated that the presence of β-CD and HPβ-CD as MPA does not exert a negative influence on the analytical performance of the chromatographic procedure. On the contrary, the sensitivity is increased in the presence of CDs and the accuracy and precision of the method are very satisfactory for the analysis of these interesting compounds in complex biological matrices. The use of SPE pre-treatment was compared with LLE and had the advantage of a lower use of organic solvents, while it led to better analytical results and a good compatibility of the extraction solvents with the mobile phase composition. Therefore the presence of CDs in the mobile phases potentially allows the traditional HPLC technique to become an attractive eco-separation technique using conventional and inexpensive columns and stationary phases, under simple and user-friendly experimental conditions.Acknowledgements
Financial support from Ministerio de Ciencia e Innovación (Spain) through grants CTQ 2009-11312-BQU and CTQ 2009-12320-BQU as well as from Grupos de Investigación UCM (920234) is gratefully acknowledged. The authors are also grateful to MEC for the award of a FPU research fellowship to V. González-Ruiz.References
- J. Sezjtli, Chem. Rev., 1998, 98, 1743–1753 CrossRef CAS.
- (a) H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Sustainability, 2009, 1, 924–945 Search PubMed; (b) B. Chankvetadze, in: Cyclodextrins and their complexes, ed. H. Dodziuk, Wiley-VCH, Weinheim, 2006, pp. 119–146 Search PubMed; (c) S. Tilloy, H. Bricout and E. Monflier, Green Chem., 2002, 4, 188–193 RSC.
- T. Cserháti and E. Forgacs, Cyclodextrins in Chromatography, Royal Society of Chemistry, Cambridge, 2003 Search PubMed.
- C. Gazpio, M. Sánchez, I. X. García-Zubiri, I. Velaz, C. Martínez-Ohárriz, C. Martín and A. Zornoza, J. Pharm. Biomed. Anal., 2005, 37, 487–492 CrossRef CAS.
- P. K. Zarzycki, H. Ohta, Y. Saito and K. Jinno, Anal. Bioanal. Chem., 2008, 391, 2793–2801 CrossRef CAS.
- J. Ye, G. Chen and S. Zeng, J. Chromatogr., B, 2006, 843, 289–294 CrossRef CAS.
- D. Chen, S. Jiang, Y. Chen and Y. Hu, J. Pharm. Biomed. Anal., 2004, 34, 239–245 CrossRef.
- A. Kwaterczak, K. Duszczyk and A. Bielejewska, Anal. Chim. Acta, 2009, 645, 98–104 CrossRef CAS.
- (a) A. El-Gindy, S. Emara, M. K. Mesbah and G. M. Hadad, J. Assoc. Off. Anal. Chem., 2006, 89, 65–70 CAS; (b) N. F. S. de Melo, R. Grillo, A. H. Rosa and L. F. Fraceto, J. Pharm. Biomed. Anal., 2008, 47, 865–869 CrossRef CAS; (c) R. Grillo, N. F. S. de Melo, C. M. Moraes, R. Lima, C. M. S. Menezes, E. I. Ferreira, A. H. Rosa and L. F. Fraceto, J. Pharm. Biomed. Anal., 2008, 47, 295–302 CrossRef CAS; (d) J. M. López-Nicolás and F. García-Carmona, Food Chem., 2008, 109, 868–875 CrossRef CAS; (e) P. Rodríguez-Bonilla, J. M. López-Nicolás and F. García-Carmona, J. Chromatogr., B, 2010, 878, 1569–1575 CrossRef CAS; (f) J. Guan, J. Yang, Y. Bi, S. Shi, F. Yan and F. Li, J. Sep. Sci., 2008, 31, 288–293 CrossRef CAS.
- (a) J. M. López-Nicolás, E. Núñez-Delicado, A. J. Pérez-López, A. Carbonell-Barrachina and P. Cuadra-Crespo, J. Chromatogr., A, 2006, 1135, 158–165 CrossRef CAS; J. Pan, G. Kai, C. Yuan and R. Jin, Chromatographia, 2007, 66, 121–123 CrossRef CAS; J. Pan, G. Kai, C. Yuan, B. Zhou, R. Jin and Y. Yuan, Chin. J. Chromatogr., 2007, 25, 316–318 Search PubMed.
- (a) A. M. Soban, S. A. Ebrahimi and M. Mahmoudian, J. Pharm. Sci., 2002, 5, 19–23; (b) Y. Totsuka, H. Ushiyama, J. Ishihara, R. Sinha, S. Goto, T. Sugimura and K. Wakabayashi, Cancer Lett., 1999, 143, 139–143 CrossRef CAS.
- R. A. Glennon, B. Grella, R. J. Tyacke, A. Lau, J. Westaway and A. L. Hudson, Bioorg. Med. Chem. Lett., 2004, 14, 999–1002 CrossRef CAS.
- (a) D. Fekkes, A. Tuite, I. Bom and L. Pepplinkhuizen, Life Sci., 2001, 69, 2113–2121 CrossRef CAS; (b) A. Greube and H. Rommelspacher, J. Chromatogr., B, 2003, 784, 155–168 CrossRef CAS.
- E. D. Louis, W. Jiang, K. M. Pellegrino, E. Ríos, P. Factor-Litvak, C. Henchcliffe and W. Zheng, NeuroToxicology, 2008, 29, 294–300 CrossRef CAS.
- N. Wodarz, G. A. Wiesbeck, H. Rommelspacher, P. Riederer and J. Böning, Alcohol.: Clin. Exp. Res., 1996, 20, 706–710 CrossRef CAS.
- R. Stohler, H. Rommelspacher and D. Ladewig, Eur. Psychiat., 1995, 10, 56–58 Search PubMed.
- U. Breyer-Pfaff, G. Wiatr, I. Stevens, H. J. Gaertner, G. Mundle and K. Mann, Life Sci., 1996, 58, 1425–1432 CrossRef CAS.
- (a) T. Herraiz, J. Chromatogr., A, 2000, 881, 483–499 CrossRef; (b) M. Kartal, M. Altun and S. Kurucu, J. Pharm. Biomed. Anal., 2003, 31, 263–269 CrossRef CAS.
- (a) J. Adachi, Y. Mizoi, T Naito, K. Yamamoto, S. Fujiwara and I. Ninomiya, J. Chromatogr., A, 1991, 538, 331–339 CrossRef CAS; (b) T. Herraiz and C. Chaparro, J. Chromatogr., A, 2006, 1120, 237–243 CrossRef CAS.
- (a) W. Zheng, S. Wang, L. F. Barnes, Y. Guan and E. D. Louis, Anal. Biochem., 2000, 279, 125–129 CrossRef CAS; (b) E. D. Louis, W. Zheng, E. C. Jurewicz, D. Watner, J. Chen, P. Factor-litvak and M. Parides, Neurology, 2002, 59, 1940–1944 CAS; (c) Y. Guan, E. D. Louis and W. Zheng, J. Toxicol. Environ. Health, Part A, 2001, 64, 645–660 Search PubMed; (d) T. R. Bosin and K. F. Faull, J. Chromatogr., B, 1988, 428, 229–236 CrossRef CAS.
- H. Tsuchiya, J. Chromatogr. A, 2004, 1031, 325–330 CrossRef CAS.
- L. Agüí, C. Peña-Farfal, P. Yánez-Sedeño and J. M. Pingarrón, Anal. Chim. Acta, 2007, 585, 323–330 CrossRef.
- (a) C. J. Smith, S. Qian, Q. Zha and S. C. Moldoveanu, J. Chromatogr. A, 2004, 1046, 211–216 CrossRef CAS; (b) S. Casal, E. Mendes, J. O. Fernandes, M. B. P. P. Oliveira and M. A. Ferreira, J. Chromatogr., A, 2004, 1040, 105–114 CrossRef CAS; (c) T. Herráiz and C. Chaparro, Biochem. Biophys. Res. Commun., 2005, 326, 378–386 CrossRef CAS.
- J. Stöckigt, Y. Sheludko, M. Unger, I. Gerasimenko, H. Warzecha and D. Stöckigt, J. Chromatogr., A, 2002, 967, 85–113 CrossRef.
- (a) T. Herraiz, Food Chem., 1999, 66, 313–321 CrossRef CAS; (b) T. Herraiz, Food Addit. Contam., Part A, 2002, 19, 748–754 Search PubMed; (c) N. Rosales-Conrado, M. E. León-Gonzáles, L. V. Pérez-Arribas and L. M. Polo-Díez, Anal. Bioanal. Chem., 2008, 391, 1433–1442 CrossRef CAS; (d) T. Herraiz, H. Guillén and V. J. Arán, Chem. Res. Toxicol., 2008, 21, 2172–2180 CrossRef CAS; (e) Y. M. Back, J. H. Lee, H. S. Shin and K. G. Lee, Food Addit. Contam., Part A, 2009, 26, 298–305 Search PubMed; (f) T. Herraiz, D. González, C. Ancín-Azpilicueta, V. J. Arán and H. Guillén, Food Chem. Toxicol., 2010, 48, 839–845 CrossRef CAS.
- (a) M. Yritia, J. Riba, J. Ortuño, A. Ramírez, A. Castillo, Y. Alfaro, R. de la Torre and M. J. Barbanoj, J. Chromatogr., B, 2002, 779, 271–281 CrossRef CAS; (b) Moncrieff, J. Chromatogr., B, 1989, 496, 269–278 CrossRef.
- F. M. Kerton, Alternative Solvents for Green Chemistry, Royal Society of Chemistry, Cambridge, 2009, pp. 97–117 Search PubMed.
- (a) C. Capelo, U. Fischer and K. Hungerbühler, Green Chem., 2007, 9, 927–934 RSC; (b) C. J. Welch, T. Brkovic, W. Shafer and X. Gong, Green Chem., 2009, 11, 1232–1238 RSC.
- J.-F. Jen, S.-L. Hsiao and K. H. Liu, Talanta, 2002, 58, 711–717 CrossRef CAS.
- L. Zhu, L. Ding, Q. Zhang, L. Wang, F. Tang, Q. Liu and S. Yao, Green Chem., 2009, 11, 132–137 RSC; (a) T. Cserháti and E. Forgács, J. Chromatogr., A, 1996, 722, 33–40 CrossRef CAS; (b) O. Jiménez, M. A. García and M. L. Marina, J. Chromatogr., A, 1996, 719, 15–26 CrossRef CAS.
- A. C. Moffat, J. V. Jackson, S. M. Moss and B. Widdop, Clarke's Isolation and Identification of Drugs (2nd edn), Pharmaceutical Press, London, 1986 Search PubMed.
- A. G. León, A. I. Olives, B. del Castillo and M. A. Martín, J. Chromatogr., A, 2008, 1192, 254–258 CrossRef CAS.
- A. G. León, A. I. Olives, M. A. Martín and B. del Castillo, J. Inclusion Phenom. Macrocyclic Chem., 2007, 57, 577–583 CrossRef CAS.
- (a) L. Martín, A. León, M. A. Martín, B. del Castillo and J. C. Menéndez, J. Pharm. Biomed. Anal., 2003, 32, 991–1001 CrossRef CAS; (b) L. Liu and Q.-X. Guo, J. Inclusion Phenom. Macrocyclic Chem., 2002, 42, 1–14 CrossRef CAS.
- (a) H.-J. Schneider, F. Hacket, V. Rüdiger and H. Ikeda, Chem. Rev., 1998, 98, 1755–1785 CrossRef CAS; (b) G. Fronza, A. Mele, E. Redenti and P. Ventura, J. Org. Chem., 1996, 61, 909–914 CrossRef CAS; (c) P. K. Owens, A. F. Fell, M. W. Coleman, M. Kinns and J. C. Berridge, J. Pharm. Biomed. Anal., 1997, 15, 1603–1619 CrossRef CAS; (d) C. Danel, N. Azaroual, C. Foulon, J.-F. Goosens, G. Vermeersch, J.-P. Bonte and C. Vaccher, Tetrahedron: Asymmetry, 2006, 17, 975–983 CrossRef CAS; (e) M. C. Bergonzi, A. R. Bilia, L. Di Bari, G. Mazzi and F. F. Vincieri, Bioorg. Med. Chem. Lett., 2007, 17, 5744–5748 CrossRef CAS.
- K. G. Flood, E. R. Reynolds and N. H. Snow, J. Chromatogr., A, 2000, 903, 49–65 CrossRef CAS.
- Comprehensive Supramolecular Chemistry: Cyclodextrins, ed. J. Szejtli and T. Osa, Elsevier, Oxford, 1996, p. 228 Search PubMed.
- (a) A. Bielejewska, K. Duszczyk and D. Sybilska, J. Chromatogr., A, 2001, 931, 81–93 CrossRef CAS; (b) K. A. Connors, J. Pharm. Sci., 1995, 84, 843–848 CAS; (c) Y. Matsui and K. Mochida, Bull. Chem. Soc. Jpn., 1979, 52, 2808–2814 CAS.
- I. Clarot, D. Clédat, S. Battu and P. J. P. Cardot, J. Chromatogr., A, 2000, 903, 67–76 CrossRef CAS.
- B. Claude, Ph. Morin, M. Lafosse and P. Andre, J. Chromatogr. A, 2004, 1049, 37–42 CrossRef CAS.
- J. Zukowski, D. Sybilska and J. Jurczak, Anal. Chem., 1985, 57, 2215–2219 CrossRef CAS.
- P. Prognon, A. Kasselouri, M. C. Desroches and G. Mahuzier, Analusis, 2000, 28, 664–669 CrossRef CAS.
- M. Cruz-Vera, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr., B, 2009, 877, 37–42 CrossRef CAS.
- A. R. Chaves, S. M. Silva, R. H. C. Queiroz, F. M. Lanças and M. E. C. Queiroz, J. Chromatogr., B, 2007, 850, 295–302 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional NMR spectra and figures. See DOI: 10.1039/c0gc00456a |
| ‡ Current address: Laboratorio de Análisis de Medicamentos, Departamento de Análisis y Control, Facultad de Farmacia, Universidad de Los Andes, Mérida, Venezuela. |
| This journal is © The Royal Society of Chemistry 2011 |