Renewable gasoline from aqueous phase hydrodeoxygenation of aqueous sugar solutions prepared by hydrolysis of maple wood†
Ning
Li
a,
Geoffrey A.
Tompsett
a,
Taiying
Zhang
b,
Jian
Shi
b,
Charles E.
Wyman
b and
George W.
Huber
*a
aDepartment of Chemical Engineering, University of Massachusetts-Amherst, 686 North Pleasant Street, Amherst, MA 01003, USA. E-mail: huber@ecs.umass.edu; Fax: +1 413-545-1647; Tel: +1 413-545-0276
bCenter for Environmental Research and Technology (CE-CERT), Chemical and Environmental Engineering Department, Bourns College of Engineering, University of California, Riverside, CA 92507, USA. E-mail: cewyman@engr.ucr.edu; Fax: +1 951-827-5696; Tel: +1 951-827-5703
First published on 19th November 2010
Abstract
In this paper we demonstrate an integrated process for the production of high octane gasoline from maple wood by hydrolysis of maple wood into aqueous carbohydrate solutions followed by aqueous phase hydrodeoxygenation of the sugar solutions. The aqueous carbohydrate solutions were prepared by both hydrolysis in hot water and hydrolysis with dilute acids (H2SO4, oxalic acid). The aqueous carbohydrate solutions were a mixture of xylose, water soluble hemicellulose oligomers, acetic acid, glucose, glucose oligomers, and probably some lignin polymers. Hydrolysis with hot water produced primarily hemicellulose oligomers whereas hydrolysis with acids produced mainly xylose and acetic acid. The hydrolysis co-product was a solid enriched in cellulose and lignin. The aqueous streams were hydrodeoxygenated by a two step catalytic process in which the first catalyst bed contained a Ru/C catalyst at 393 K and the second catalyst bed contained a Pt/zirconium phosphate (Pt/ZrP) catalyst at 518 K. The Ru/C catalyst was able to selectively hydrogenate xylose into xylitol but could not selectively hydrogenate the xylose oligomers. The two stage process was able to convert the aqueous carbohydrate streams derived from maple wood into gasoline range products with carbon yields of up to 57% and an estimated octane number of 96.5. No significant catalyst deactivation was observed indicating that the catalysts are very stable. The highest gasoline yield from this two stage process was obtained from the stream produced by acid hydrolysis of maple wood with 0.5 wt% oxalic acid at 433 K for 0.5 h. These results suggest that aqueous phase processing of sugars obtained by hydrolysis is a promising option for the production of fuels and chemicals from lignocellulosic biomass.
1. Introduction
Today, as petroleum is depleted and domestic oil supplies drop, it is imperative to develop economic and highly effective processes for conversion of renewable biomass to fuels and chemicals.1 Lignocellulosic biomass is the most abundant renewable source of organic carbon on Earth and the only one of low enough cost and adequate availability for large scale sustainable production of liquid fuels. It is primarily composed of three polymeric components: cellulose, hemicellulose, and lignin. However, these polymeric components must be deconstructed into reactive intermediates which can then be used as building blocks for fuels and chemicals.2–4Hydrolysis of the hemicellulose and cellulose in biomass is a promising option for production of aqueous carbohydrate solutions that can be used as fuel precursors.3,5–9 Biomass hydrolysis is often performed in two steps. The first “pretreatment” primarily depolymerizes the hemicellulose fraction to sugars (mostly xylose and other 5-carbon sugars for many types of biomass) in an aqueous solution that will likely also include oligomers, acetic acid, and some 6-carbon sugars. Cellulose is often hydrolyzed in a second step to produce aqueous solutions of glucose as the primary target. Biomass hydrolysis options for the first step include hydrolysis with just hot water,7,10–11hydrolysis in dilute acid,12–15 autocatalytic steam explosion,12 treatment with ammonia,16–17 and treatment with ionic liquids.18–21 Three major options are typically considered for cellulose hydrolysis in the second step: dilute acid, concentrated acid, and enzymatic hydrolysis. Pretreatment and hydrolysis of lignocellulosic biomass have primarily been developed for fermentation of aqueous carbohydrate solutions to ethanol.6,9Aqueous phase processing (APP) is an exciting new technology for coupling solubilization of biomass with catalytic conversion of the resulting dissolved carbohydrate compounds into hydrocarbons and a variety of fuels and chemicals. Dumesic and co-workers have shown that APP can convert dissolved biomass-derived compounds (including sugars, sugar alcohols, bio-oils, cellulose or even lignin) into hydrogen,22–31 light alkanes,28,32 liquid alkanes,33–40 and oxygenates.41–47 We have recently identified the reaction chemistry for aqueous phase hydrodeoxygenation (APHDO) of sorbitol into C1 to C6 alkanes with bifunctional acid base catalysts.48–49 We have also shown how through modifying the reaction chemistry and the acid concentration of sites on the catalyst we can selectively produce a high octane gasoline blend from aqueous sorbitol solutions with a specially designed Pt/Zirconium phosphate (Pt/ZrP) catalyst.49
While aqueous phase processing has been shown to be promising with pure carbohydrate solutions, it is vital to use less expensive aqueous solutions derived from lignocellulosic biomass, but we know of no study that has applied aqueous phase processing to streams resulting from biomass hydrolysis. As mentioned above, a variety of process options are available for production of aqueous carbohydrate solutions from lignocellulosic biomass. However, these options produce carbohydrates with different characteristics, for examples carbohydrate monomers and carbohydrate oligomers. The resulting aqueous solutions may also contain impurities that could poison heterogeneous catalysts. Alternatively, heterogeneous catalysts may be able to easily convert molecules like furfural and HMF that would inhibit biological catalysts into fuels and chemicals, thereby deriving value from both sugars and their degradation products. Therefore, it would be highly desirable to couple hydrolysis processes with aqueous phase hydrodeoxygenation to be able to use low cost lignocellulosic feedstocks for making hydrocarbon fuels.
The objective of this paper is to show how hydrolysis can be combined with APHDO to produce fuels from lignocellulosic biomass using maple wood as a representative feedstock. In this study, we seek to understand how to optimize the hydrolysis step to produce solutions that can be effectively upgraded to biofuels. Importantly, we will identify the key intermediates in the hydrolysis residues that are desirable and undesirable for APHDO and also determine whether hydrolysis residues can be processed using APHDO without significant catalyst deactivation.
Fig. 1 shows a block flow diagram for the production of gasoline from maple wood based on hydrolysis coupled with aqueous phase hydrodeoxygenation of the hydrolysis solution. In the first step, hemicellulose in maple wood is hydrolyzed to water soluble C5 and C6 carbohydrate monomers and oligomers. This process also produces a solid containing most of the cellulose and lignin but little hemicellulose. The cellulose can be hydrolyzed to glucose50,51 or used as a feedstock to make levulinic acid which is a promising building block for diesel or jet fuels.38,53,53 The lignin can be burned or gasified to generate heat and electricity for the process or used to produce hydrogen (reforming) for use in subsequent steps. The sugars in the aqueous hemicellulose solutions can be hydrogenated at low temperature to sugar alcohols that can then be fed to a second reactor with hydrogen for hydrodeoxygenation to three major products: gasoline range molecules, light gases, and water soluble products.
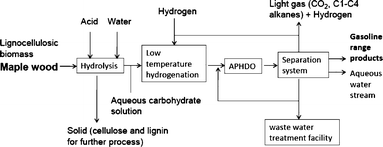 | ||
| Fig. 1 Block flow diagram for the production of gasoline from lignocellulosic biomass by aqueous phase hydrodeoxygenation of an aqueous hydrolysis solution. | ||
2. Experimental
2.1. Hydrolysis of maple wood hemicellulose
Maple wood was provided by Mascoma Corporation, NH. and air dried to a moisture content of less than 7% before being ground using a laboratory mill with an internal sieve (model 4, Arthur H. Thomas Company, Philadelphia, PA) to a particle size of less than 0.5 mm. The ground maple wood was sealed in heavy duty zipped freezer bags and stored at 255 K in a laboratory freezer. The composition of dry maple wood was analyzed following NREL standard procedures54,55 and is shown in Table 1.Fresh maple wood with 34% moisture content was directly used for hydrothermal steam treatment with no added acid. An amount of acid solution was added to the fresh maple fiber to achieve the target final acid and solid concentrations and allowed to sit overnight before steam hydrolysis. Subsequently, approximately 400 g (dry weight) of fresh or pre-impregnated maple wood was loaded into a 4 L Hastelloy C steam reactor with steam provided by a Fulton FB-075-L electric steam boiler (Fulton Boiler Works, Inc. Pulaski, NY). The hydrolysis temperature was controlled by setting the boiler pressure at the corresponding saturated steam pressure for the target temperature. At the end of the reaction time, the reaction pressure was suddenly dropped by opening a valve in the bottom of the vessel to allow discharge to a receiver and rapid cooling as the water flashed off.56
The liquid hydrolyzate fraction was separated from the cellulose and lignin enriched solid by a small Hydraulic Shop Press (12 ton, model 14590, Northern™ Industrial Tools, Burnsville, Minnesota) and collected for hydrogenation. Detailed hydrolysis conditions, including reaction time and temperature are listed in Table 3.
2.2. Preparation and characterization of catalysts
The Ru/C (Ru content 5 wt%) catalyst for this work was supplied by Strem Chemicals. Zirconium phosphate (ZrP) was prepared according to Okuhara et al.57 by precipitation of 1.0 mol L−1 ZrCl2O·8H2O (Aldrich) and 1.0 mol L−1NH4H2PO4 (Aldrich) aqueous solution at a molar ratio of P/Zr = 2.0. The precipitate was filtered, washed with water, dried at 373 K overnight, and calcined at 673 K for 4 h. Pt was loaded on the ZrP support by incipient wetness impregnation with tetra-amine platinum nitrate (Strem Chemicals) aqueous solution. The Pt content in the catalysts was kept at 4 wt%. The mixture was then dried in an oven overnight at 373 K and calcined at 533 K for 3 h in air.XRD patterns of the catalysts were obtained with a Philips X'PERT powder diffractometer equipped with an on-line computer. Ni-filtered Cu-Kα radiation was used and detected using an X'cellerator detector. An accelerating voltage of 45 keV was used at 40 mA, and X-ray patterns were obtained at a scan speed of 0.1°(2θ) s−1 averaged for 10 s per step over a range of 5 to 80° 2θ.
Specific BET surface areas of the Ru/C catalyst and Pt/ZrP catalysts were determined by nitrogen adsorption at 77 K with a Quantachrome Autosorb® MC-1 automated gas sorption system. Before each experiment, the samples were evacuated at 573 K for 24 h.
H2 uptake of the catalysts was measured on the Quantachrome Autosorb instrument by irreversible H2 adsorption at 303 K. Metal dispersion was calculated by the molar ratio of surface metal atoms as determined by hydrogen uptake divided by the total metal atoms.
Ammonia-temperature programmed desorption (NH3-TPD) of the ZrP catalyst as prepared was conducted using a TA Instruments SDT Q600 system coupled with a quadrupole mass spectrometer (ExTorr XT 300) to analyze the ammonia desorbed from the sample. A 20 mg sample was placed in an alumina crucible and heated at 873 K for 0.5 h with flowing helium to remove physically absorbed water. Ammonia adsorption was performed at 373 K, and after saturation, heating from 373 K to 923 K was done at a linear rate of 10 K min−1 with a constant helium flow rate of 100 mL min−1. Desorbed ammonia was detected with the mass spectrometer with mass signals recorded by a computer (ExTorr software).
The FT-IR spectra with ammonia as a probe molecule were recorded on a Bruker Equinox 55 spectrometer at a resolution of 4 cm−1 (averaging 50 scans). A Harrick Scientific “Praying Mantis” Diffuse Reflectance Infrared cell (DRIFTS) allowed in situ recording of the spectra at ambient temperature and catalyst activation at higher temperatures. The cell was equipped with a heater and connected to a gas flow system. The temperature was monitored with a thermocouple placed in direct contact with the sample. Powder samples (∼20 mg) were loaded into the DRIFTS cell for infrared studies with a KBr spectrum (taken at ambient temperature beforehand) used as a background reference. Before the surface characterization was performed, the samples were activated by heating at 773 K for 2 h with a helium (Airgas, UHP) flow of 10 mL min−1, cooled down to 373 K and saturated with ammonia (Airgas, anhydrous 99.99%) for 15 min. The gas flow was then switched back to helium (10 mL min−1) to remove physically adsorbed ammonia, and the spectrum monitored until no change was observed (∼20 min). Then, the samples were heated with flowing helium (10 mL min−1) to various temperatures, and the spectra were recorded at temperatures up to 868 K. All of the spectra presented were obtained by subtraction of the corresponding background reference, with data analysis and peak fitting by GRAMS/AI® software (ThermoScientific).
2.3. Low-temperature hydrogenation and aqueous phase hydrodeoxygenation
Both low temperature hydrogenation and APHDO of the aqueous hydrolysis samples were carried out in a stainless steel tubular flow reactor described in our previous work.48 For low temperature hydrogenation, 2.0 g of Ru/C catalyst was used, while for APHDO, 3.3 g of Pt/ZrP catalyst was employed. Before the reaction, the Ru/C and Pt/ZrP catalysts were reduced in the reactor by flowing hydrogen from the bottom at about 160 mL min−1 at 433 K and 723 K, respectively. The temperature program applied for reduction was to ramp up from room temperature to the set temperatures at a heating rate of 1 K min−1, soak the catalyst at the set temperatures for 2 h, and then cool down to reaction temperature along with slowly increasing the pressure to 6.21 MPa. A JASCO PU980 HPLC pump fed liquid to the reactor bottom along with hydrogen at a flow rate of 40 mL min−1. The product from the reactor tube passed through a gas-liquid separator, and the gaseous products flowed through a back pressure regulator to maintain the pressure in the reaction system. The gaseous products were analyzed by two online gas chromatographs (HP 5890 series II). In addition, CO2 in the gaseous product was measured with a Thermal Conductivity Detector (TCD) employing an Alltech HAYESEP DB 100/120 packed column (part no. 2836PC) with the oven temperature held constant at 348 K, the TCD and injection port held at 433 K and 393 K, respectively, and 41 mL min−1 of helium carrier gas controlling the column flow rate. Alkanes in the gaseous product were analyzed using a GC with a flame ionization detector (FID) and an Alltech AT-Q capillary column (part no. 13950) with helium carrier gas at a column flow rate of 1 mL min−1. Both the injection port and the detector were held at 473 K. The following GC oven temperature history was used: hold at 313 K for 6 min, ramp to 453 K at 5 K min−1, and hold at 453 K for 25 min.Liquid products were drained periodically from the gas-liquid separator and analyzed by GC-MS, GC with FID detector, HPLC, and the TOC analyzer. The GC-MS was a Shimadzu GC-2010 with an Rtx-VMS capillary column, helium carrier gas at a column flow rate of 1.57 mL min−1, and injection port and detector were held at 513 K. Following injection of 1 μL of liquid sample, the column temperature was held at 308 K for 5 min, ramped up linearly to 323 K at 5 K min−1, then ramped up to 513 K at 20 K min−1, and then kept at 513 K for 7.5 min. Organic species in the liquid were analyzed with another GC (Agilent 7890A) with an FID detector and auto sampler that separated organic compounds using a Rtx-VMS capillary column through which passed helium at a column flow rate of 3 mL min−1 as the carrier gas. Both the injection port and the detector were held at 513 K. For each analysis, 1 μL of liquid sample was injected, and the column temperature was held at 313 K for 5 min, ramped up to 513 K at 7.5 K min−1 and kept at 513 K for 15 min. A Shimadzu HPLC with UV-Vis (SPD-20AV) and RID (RID-10A) detectors, was also used to analyze liquid products from injection of 1 μL of liquid sample through a BIO-RAD Aminex HPX-87H column (catalog no. 125-0140) maintained at 303 K with 0.005 M H2SO4 as the mobile phase flowing at a rate of 0.6 mL min−1. The carbohydrate oligomers in the liquid sample were analyzed following NREL standard procedures.54,55
3. Results and discussion
3.1. Characterization of catalysts
The XRD patterns of the Ru/C and Pt/ZrP catalysts used in this research are shown in Fig. 2. We can see that the Pt/ZrP is amorphous indicating that the Pt is well dispersed on the support (particle size less than 5 nm). Ru peaks can also be observed for the Ru/C catalyst, with the particle size of Ru particles estimated as 21.5 nm by Scherrer's equation.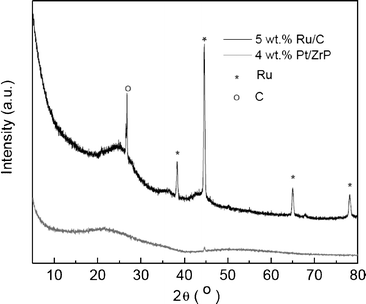 | ||
| Fig. 2 XRD patterns of 5 wt% Ru/C and 4 wt% Pt/ZrP catalysts. | ||
The specific BET surface areas and metal dispersions of the Ru/C and Pt/ZrP catalysts are shown in Table 2. The metal dispersion of the Ru/C catalyst (6.7%) is obviously lower than that of the Pt/ZrP catalyst (72%), consistent with the XRD results and consistent with the bigger particle size of Ru.
| Catalyst | BET surface area/m2 g−1 | Metal dispersion (%) |
|---|---|---|
| 5 wt% Ru/C | 635 | 6.7 |
| 4 wt% Pt/ZrP | 102 | 72.0 |
The NH3-TPD results for the ZrP support are shown in Fig. 3. The wide peak from 400 to 900 K indicates weak, moderate, and strong acid sites on the ZrP surface. From the area of the ammonia peak, the density of acid sites on ZrP was estimated as 0.84 mmol g−1.
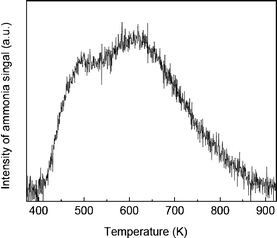 | ||
| Fig. 3 NH3-TPD results for ZrP. | ||
As we reported in our previous work,49 bands at 1430 cm−1 and 1674 cm−1 were observed, even at high temperature in the NH3-DRIFTS spectra for ammonia adsorption over ZrP (cf. Fig. S1 in supplementary information†), indicating the presence of both strong Brønsted acid and Lewis acid sites on the ZrP surface, respectively.58 The Brønsted acid site band is 27 times that of the Lewis site (B/L = 27), and the band due to Brønsted acid sites is stable at temperatures up to 773 K. These results indicate that this catalyst has both Brønsted and Lewis sites with the stronger Brønsted sites dominating.
3.2. Steam hydrolysis of maple wood
The liquid samples from maple wood hydrolysis at different treatment conditions were analyzed following the NREL standard procedures,54,55 with the results shown in Table 3. For hydrothermal treatment at 473 K (Sample 1), the pH of the liquid samples were lower than 7, indicating that the liquid samples were acidic due to the generation of organic acids including acetic, formic, and levulinic acids. Acetic acid is known to be released by partial deacetylation of hemicellulose during hydrolysis,1,53 while formic and levulinic acids are formed by the dehydration of C6 sugars.59 These acids catalyze hydrolysis. Small amounts of furfural and hydroxymethylfurfural (HMF) were also identified.41,60,61| Sample | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| a Carbon extracted (%) = Carbon in the liquid sample × 100%/Carbon in the maple wood. b Selectivity (%) = Carbon in the specific compound × 100%/Carbon in the solution measured by TOC analyzer. | |||||||
| Catalyst | Hydrothermal | H2SO4 | Oxalic acid | ||||
| T/K | 473 | 473 | 473 | 433 | 433 | 453 | 433 |
| Solid loading (%) | 66 | 66 | 66 | 60 | 60 | 50 | 50 |
| Acid loading (%) | 0 | 0 | 0 | 0.5 | 0.5 | 0.5 | 0.5 |
| Reaction time/min | 5 | 15 | 30 | 30 | 30 | 10 | 30 |
| pH of solution | 3.3 | 3.1 | 3.0 | 1.8 | 2.0 | 2.4 | 2.5 |
| Carbon extracted (%)a | 15.0 | 30.2 | 27.2 | 31.6 | 38.1 | 35.2 | 38.0 |
| Carbon concentration/mol L−1 | 4.3 | 3.1 | 2.0 | 2.4 | 4.2 | 6.2 | 3.2 |
| Carbon Selectivityb | |||||||
| C5 monomers | 5.0 | 17.7 | 15.8 | 34 | 36.1 | 41.4 | 37.2 |
| C6 monomers | 1.8 | 2.1 | 3.8 | 4.8 | 4.9 | 5.4 | 4.4 |
| C5 oligomers | 51.6 | 22.1 | 4.8 | 16.5 | 12.9 | 14.1 | 20.5 |
| C6 oligomers | 4.8 | 4.1 | 2.8 | 1.4 | 1.3 | 1.5 | 2.0 |
| Furfural | 0.5 | 3.5 | 6.4 | 0.8 | 1.0 | 0.9 | 0.8 |
| HMF | 0.8 | 2.1 | 5.2 | 1.3 | 1.2 | 0.6 | 0.9 |
| Formic acid | 1.7 | 3.2 | 6.3 | 1.4 | 1.3 | 1.9 | 1.0 |
| Acetic acid | 3.6 | 11.8 | 21.3 | 8.8 | 9.3 | 7.8 | 9.2 |
| Levulinic acid | 0.6 | 1.1 | 1.6 | 0.3 | 0.2 | 0.4 | 0.9 |
| Carbon identified (%) | 70.4 | 67.6 | 67.9 | 69.2 | 68.1 | 74.0 | 77.0 |
Increasing the reaction time of the pretreatment from 5 min to 15 min decreased the solution pH, increased the amount of carbon extracted from the maple wood, and increased the selectivity towards sugar monomers. Some carbonaceous species (∼30 wt%) could not be identified in the liquid samples and may be soluble lignin polymers or humins that are difficult to characterize. Increasing the reaction time from 15 to 30 min (samples 2–3) for the hydrothermal hydrolysis samples led to a decrease in both carbon extraction yield and carbohydrate selectivity, a result that can be explained by an increase in the formation of organic acid, furfural, HMF, and water soluble humins, with the latter difficult to analyze.41,61
During steam hydrolysis, condensation diluted the hydrolyzate and made it more difficult to compare the total carbon concentrations. However, the acidic steam treatments extracted more carbon with higher selectivity to monomeric sugars. Due to potential catalyst deactivation by sulfuric acid, oxalic acid steam treatment was chosen to provide hydrolyzates with different product distributions. Samples 4 and 5 revealed the different product distributions resulting from use of sulfuric acid and oxalic acid at the same temperature and time. Although sulfuric acid was more acidic and resulted in a lower pH, oxalic acid realized a higher selectivity of xylose monomer. Higher temperature and shorter reaction time was applied to prepare sample 6 to reduce steam condensation and resulted in the highest carbon concentration with the highest xylose monomer selectivity among the 7 samples. Sample 7 was prepared at the same reaction conditions as sample 5 except with a lower solids loading of 50% to investigate the effect of solid loading on the hydrolyzate. As shown in Table 3, sample 7 had slightly more xylose monomers and significantly more xylooligomers with more carbon identified.
Dilute sulfuric or oxalic acid increased the selectivity to monomers and also the amount of carbon extracted from maple wood at lower temperatures than for hydrothermal pretreatment. Thus, although dilute sulfuric acid is often used to catalyze the hydrolysis reaction,62,63oxalic acid produced a higher extracted carbon yield than sulfuric acid.
3.3. Low temperature hydrogenation
A low temperature hydrogenation is the first step in the conversion of aqueous carbohydrates solutions into gasoline.29 Converting carbohydrates into sugar alcohols in this step avoids undesired decomposition reactions that would occur with glucose at higher temperatures.Prior to investigating the hydrogenation of sugar solutions from maple wood, we studied the hydrogenation of the pure carbohydrate compounds of glucose, xylose, sucrose, and cellobiose as examples of C6 carbohydrates, C5 carbohydrates, and carbohydrate oligomers, respectively.
The reaction conditions applied in this study effectively hydrogenated both glucose and xylose to sorbitol and xylitol, respectively, in high yields, as shown in Table 4. High yields of sorbitol and mannitol were also obtained for sucrose hydrogenation. However, these reaction conditions were not able to convert cellobiose to sorbitol with high yields but instead primarily made 3-β-D-glucopyra-nosyl-D-glucitol. This indicates that the β-1,4 linkage of two D-glucopyranose monomers63 is difficult to hydrolyze under our reaction conditions, consistent with results observed by Yan et al.64 Thus, the C–O–C bond (1,4-β-glucoside bond) connecting two adjacent glucose monomers in cellobiose is more difficult to cleave than the one in the glucose ring. By comparison, hydrolysis of β-D-fructofuranosyl-(2→1)-α-D-glucopyranoside, which is the C–O–C linkage between carbohydrate molecules in sucrose, occurs rapidly at our reaction conditions.74 In addition, a small amount of carbon was converted into gas phase alkanes, mainly methane (carbon selectivity ∼95%). These results indicate that our current reaction conditions are sufficient to hydrogenate sugars into sugar alcohols and also to hydrolyze some types of C–O–C bonds. However, other C–O–C bonds are not hydrogenated rapidly at the same reaction conditions.
| Feedstock | 15 wt% Glucose | 15 wt% Xylose | 15 wt% Sucrose | 10 wt% Cellobiose | ||
|---|---|---|---|---|---|---|
| a WHSV = weight hourly space velocity b Gas phase carbon selectivity (%) = Carbon in specific compounds × 100%/Sum of carbon identified by GC. c Liquid phase carbon selectivity (%) = Carbon in the specific compounds × 100%/Carbon in the liquid phase product measured by TOC. | ||||||
| WHSV/h−1 a | 1.2 | 2.4 | 1.2 | 2.4 | 1.2 | 1.2 |
| Carbon to gas phase (%) | 2.6 | 2.0 | 3.2 | 2.1 | 3.0 | 1.7 |
| Gas phase carbon selectivity (%)b | ||||||
| Methane | 95.5 | 95.4 | 95.7 | 94.5 | 95.5 | 96.2 |
| Ethane | 1.1 | 1.3 | 1.1 | 1.5 | 0.8 | 0.6 |
| Propane | 1 | 1.3 | 1.2 | 1.7 | 1.1 | 0.6 |
| Butane | 0.9 | 1.2 | 1.1 | 1.3 | 1.3 | 1.1 |
| Pentane | 0.4 | 0.9 | 0.9 | 1 | 0.9 | 0.9 |
| Hexane | 1.1 | 0.1 | 0 | 0 | 0.3 | 0.3 |
| Carbon in liquid by TOC (%) | 99.4 | 99.0 | 99.8 | 99.5 | 92.8 | 94.5 |
| Liquid phase carbon selectivity (%)c | ||||||
| Sorbitol and mannitol | 94.1 | 96.8 | 0 | 0 | 95.4 | 8.2 |
| Xylitol | 0 | 0 | 85.4 | 91.9 | 0 | 0 |
| Sucrose | 0 | 0 | 0 | 0 | 4.6 | 0 |
| 3-β-D-Glucopyra-nosyl-D-glucitol | 0 | 0 | 0 | 0 | 0 | 90.7 |
| Carbon identified (%) | 96.1 | 97.8 | 88.4 | 93.5 | 94.8 | 95.2 |
Table 5 shows the results from the low temperature hydrogenation of sample 7 at different reaction temperatures. Sample 7 was the hydrolysis product prepared from maple wood using 0.5 wt% oxalic acid at 433 K for 30 min, as shown in Table 3. No evident change in activity of the Ru/C catalyst was observed for the results of the experiments from 393–413 K. The gas phase carbon yield increased with increasing temperature with methane being the primary product in the gas phase. The yield of liquid phase products decreased as the temperature was increased.
| Reaction T/K | 393 | 403 | 413 | 433 | 453 | 473 |
|---|---|---|---|---|---|---|
| a Gas phase carbon selectivity (%) = Carbon in specific compounds × 100%/Sum of carbon identified by GC. b Liquid phase carbon selectivity (%) = Carbon in the specific compounds × 100%/Carbon in the liquid phase product measured by TOC. | ||||||
| Carbon in gas phase by GC (%) | 2.1 | 2.9 | 4.8 | 15.7 | 35.1 | 59.2 |
| Gas phase carbon selectivity (%)a | ||||||
| Methane | 87.0 | 87.6 | 88.7 | 87.9 | 86.4 | 79.7 |
| Ethane | 6.8 | 6.5 | 5.7 | 5.1 | 5.7 | 7.1 |
| Propane | 1.5 | 1.4 | 1.6 | 3.2 | 4.4 | 7.9 |
| Butane | 2.2 | 2.2 | 1.8 | 2.2 | 2.2 | 3.3 |
| Pentane | 1.2 | 1 | 1.2 | 1.3 | 1 | 1.4 |
| Hexane | 1.3 | 1.3 | 1.0 | 0.3 | 0.3 | 0.6 |
| Carbon in liquid by TOC (%) | 96.2 | 92.3 | 88.9 | 76.0 | 49.5 | 41.2 |
| Liquid phase carbon selectivity (%)b | ||||||
| Methanol | 0.7 | 0.8 | 0.8 | 0.6 | 1.0 | 0.3 |
| Ethanol | 3.5 | 4.3 | 4.6 | 3.5 | 3.1 | 0.9 |
| Acetic acid | 10.3 | 9.0 | 12.1 | 12.9 | 10.8 | 3.5 |
| Propanol | 0.1 | 0.1 | 0.1 | 0.6 | 2.5 | 2.6 |
| Butanol | 0 | 0 | 0 | 0 | 0 | 0.7 |
| Tetra-hydrofuran | 0.1 | 0.2 | 0.3 | 1.0 | 4.0 | 13.3 |
| Butanediol | 0.1 | 0.3 | 0.4 | 2.9 | 5.1 | 3.5 |
| Pentanol | 0.3 | 0.5 | 0.6 | 1.6 | 4.8 | 9.6 |
| Methyl-tetrahydro-furan | 4.6 | 6.1 | 6.2 | 5.5 | 6.6 | 6.1 |
| Tetra-hydrofurfural alcohol | 0.4 | 0.4 | 0.8 | 2.2 | 5.0 | 5.6 |
| Pentanediol | 0.2 | 0.3 | 0.3 | 0.5 | 0.5 | 0.5 |
| Levulinic acid | 0.5 | 0.6 | 1.9 | 0.9 | 2.8 | 2.8 |
| Xylitol | 43.9 | 48.2 | 50.6 | 38.5 | 25.1 | 0.6 |
| Sorbitol | 4.3 | 5.2 | 6.3 | 6.2 | 3.3 | 0.6 |
| C5 oligomer | 13.7 | 13.7 | 11.7 | 5.0 | 0.9 | 0 |
| C6 oligomer | 2.9 | 3.0 | 2.7 | 1.2 | 0.3 | 0 |
| Carbon identified (%) | 84.3 | 88.3 | 93.1 | 78.9 | 72.5 | 79.6 |
A range of reactions occur in the low temperature hydrogenation step, as shown in Fig. 4. Sugar oligomers were converted into sugar monomers and acetic acid, the latter being released in hemicellulose hydrolysis.1,53 The sugar monomers xylose and glucose were then hydrogenated into the sugar alcohols xylitol and sorbitol respectively. The sugars may also undergo dehydration reactions to produce furfural and HMF which can undergo hydrogenation reactions to produce tetrahydrofurfuryl alcohols, methyl-tetrahydrofuran, and potentially pentanol as well. The lower molecular weight organic acids, acetic and formic, are partially hydrogenated to the corresponding alcohols ethanol and methanol. This is consistent with our recent work on the hydrogenation of acetic acid over the same catalyst.65 However, organic acids were still present in the products after low temperature hydrogenation, indicating that hydrogenation of these organic acids is slow under the reaction conditions used in this study. Levulinic acid is also probably partially hydrogenated to pentanediol. One possible route for this reaction is that levulinic acid can be hydrogenated to γ-valerolactone66–68 which may be further hydrogenated to pentanediol by a ring opening reaction. Then, the pentanediol can be hydrogenated to pentanol or methyl tetrahydrofuran. Pentanol can also be generated by ring opening of methyltetrahydrofuran. The desired reactions that occur in this low temperature hydrogenation reaction include: hydrolysis of sugar oligomers, hydrogenation of sugars into sugar alcohols, hydrogenation of organic acids into their corresponding alcohols, and hydrogenation of furans into their saturated form. Undesired reactions include dehydration reactions which produce furfural and levulinic acid and C–C bond cleavage to produce undesired gas range alkanes. Higher temperatures increased the amount of these undesired products. Therefore, it seems desirable to operate the hydrogenation at a temperature as low as possible while still realizing high rates of the desired reactions. Therefore, for the remaining work in this paper, we focused on hydrogenation reactions at a lower temperature of 393 K.
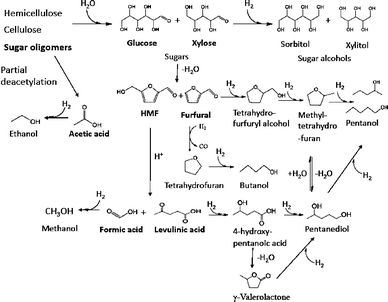 | ||
| Fig. 4 Proposed reactions network in the low-temperature hydrogenation of aqueous maple wood hydrolysis products. | ||
Table 6 shows the results from the low temperature aqueous phase hydrogenation of maple wood hydrolysis samples 1–6 at 393 K. As shown in Table 6 less than 2.5% of the carbon was converted into gas phase products. However, the liquid phase selectivity changed significantly for the different samples. Xylitol and sorbitol selectivity were much higher for samples 4–6 which contained more carbohydrate monomers. Samples 1–3 were prepared by hydrothermal hydrolysis which primarily produced sugar oligomers. In contrast, samples 4–6 produced by hydrolysis with oxalic and sulfuric acids contained primarily sugar monomers. These results indicate that our hydrogenation reaction conditions did not breakdown hemicellulose oligomers. Acid catalysts are required to decompose these oligomers so that they can be hydrogenated. Future work in catalyst design for this low temperature hydrogenation step should focus on the development of solid catalysts that can perform both oligomer hydrolysis and the hydrogenation of the resulting sugars.
| Samplea | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| a See Table 3 for details of the feedstock samples. b Gas phase carbon selectivity (%) = Carbon in specific compounds × 100%/Sum of carbon identified in gas phase by GC. c Liquid phase carbon selectivity (%) = Carbon in the specific compounds × 100%/Carbon in the liquid phase product measured by TOC. | ||||||
| Catalyst | Hydrothermal | H2SO4 | Oxalic acid | |||
| T/K | 473 | 473 | 473 | 433 | 433 | 453 |
| Reaction time/min | 5 | 15 | 30 | 30 | 30 | 10 |
| Carbon concentration/mol L−1 | 4.3 | 3.1 | 2.0 | 2.4 | 4.2 | 6.2 |
| Carbon in gas phase by GC (%) | 0.5 | 1.1 | 2.4 | 0.3 | 0.6 | 0.4 |
| Gas phase carbon selectivityb (%) | ||||||
| Methane | 93.9 | 89.1 | 86.2 | 86.6 | 88.8 | 89 |
| Ethane | 1.5 | 2.9 | 4.1 | 4.8 | 2.5 | 1.5 |
| Propane | 0 | 1.8 | 2 | 0 | 1.2 | 1.6 |
| Butane | 2.3 | 1.6 | 2 | 0 | 1.7 | 1.5 |
| Pentane | 0 | 0.9 | 1.5 | 0 | 1.3 | 1.7 |
| Hexane | 2.3 | 3.6 | 4.1 | 8.5 | 4.5 | 4.8 |
| Carbon in liquid by TOC (%) | 98.3 | 89.2 | 75.6 | 96.9 | 93.1 | 93.7 |
| Liquid phase carbon selectivityc (%) | ||||||
| Methanol | 0.4 | 1.0 | 2.4 | 1.3 | 1.2 | 0.9 |
| Ethanol | 1.8 | 1.2 | 0.5 | 0.4 | 0.4 | 0.1 |
| Acetic acid | 4.0 | 12.2 | 21.0 | 11.9 | 10.3 | 9.2 |
| Propanol | 0.1 | 0 | 0 | 0 | 0 | 0 |
| Butanediol | 0.4 | 0 | 0 | 0.3 | 0 | 0 |
| Pentanol | 0.4 | 0.4 | 0.2 | 0 | 0 | 0 |
| Methyl-tetrafuran | 0.2 | 0.3 | 0.2 | 0.1 | 0.1 | 0.1 |
| Tetra-hydrofurfural alcohol | 1.6 | 2.0 | 1.3 | 0.6 | 0.6 | 0.5 |
| Pentanediol | 0.5 | 0.3 | 0.2 | 0 | 0 | 0 |
| Xylitol | 8.5 | 20.2 | 15.7 | 41.0 | 41.7 | 42.4 |
| Sorbitol | 3.8 | 9.4 | 13.7 | 16.7 | 15.5 | 14.9 |
| C5 oligomers | 48.9 | 18.9 | 6.6 | 1.7 | 2.2 | 2.3 |
| C6 oligomers | 9.2 | 5.5 | 3.5 | 0.6 | 1.0 | 1.2 |
| Carbon identified (%) | 78.8 | 64.8 | 51.8 | 68.3 | 67.9 | 72.8 |
Several recent papers have shown that it is indeed possible to produce sugar alcohols from sugar polymers with heterogeneous catalysts [Table S2, supporting material†]. For example, Luo et al. found that cellulose can be hydrogenated to sorbitol at 518 K.69 In addition, Yan et al. found that cellobiose can be hydrogenated over nano particles of Ru to sorbitol at 393 K and pH = 2.64 Robinson et al. also reported the hydrogenolysis of cellulose and hemicellulose over Ru/C in the presence of mineral acid.70,71
3.4. Aqueous phase hydrodeoxygenation in a two bed reactor system
As the final aspect of this research, we applied a two-bed reactor system to produce gasoline compounds by APHDO of aqueous sugar solutions and aqueous solutions prepared by maple wood hydrolysis, as shown in Table 7. The first bed contained the Ru/C catalyst at 393 K, followed by a second bed filled with the Pt/ZrP catalyst at 518 K. No catalyst deactivation was observed for these reactions in the reaction times of this study. Analogous to our observations for APHDO of sorbitol and xylitol,48 three categories of products were generated: 1) light gases such as CO2, methane, ethane, propane, and butane; 2) gasoline range products including pentane, hexane, and C2–C6 monofunctional compounds such as alcohols, ketones, cyclic ethers, and small amount of carboxylic acids; and 3) aqueous phase products including methanol, 1,4-sorbitan, isosorbide, propanediol, butanediol, pentanediol, and hexanediol.| Feedstock | 15 wt% Glucose | 15 wt% Xylose | Maple wood samplea | ||
|---|---|---|---|---|---|
| 4 | 5 | 6 | |||
| a See Table 3 for details of the feedstock samples. b C1–C4 light gas carbon selectivity (%) = Carbon in specific compounds × 100%/Sum of carbon identified in light gas cut. c Gasoline products carbon selectivity (%) = Carbon in the specific compounds × 100%/Sum of carbon identified in gasoline range products. d Research octane number (RON) of the gasoline range products were estimated according to the method introduced in the literature.72 See supplementary material for details.† e Aqueous phase cut carbon selectivity (%) = Carbon in the specific compounds × 100%/Sum of carbon identified in aqueous cut. | |||||
| Catalyst | — | — | H2SO4 | Oxalic acid | |
| T/K | — | — | 433 | 433 | 453 |
| Reaction time/min | — | — | 30 | 30 | 10 |
| Carbon concentration/mol L−1 | 5.0 | 5.0 | 2.4 | 4.2 | 6.2 |
| Light gas yield (%) | 10.3 | 9.8 | 3.1 | 9.1 | 4.3 |
| C1–C4 light gas carbon selectivity (%)b | |||||
| CO2 | 33.3 | 37.8 | 73.8 | 64.5 | 77.1 |
| Methane | 43.0 | 45.9 | 15.9 | 20.4 | 16.4 |
| Ethane | 5.8 | 2.9 | 2.1 | 5.5 | 1.4 |
| Propane | 9.1 | 4.2 | 2.7 | 4.5 | 1.8 |
| Butane | 8.8 | 9.2 | 5.5 | 5.2 | 3.3 |
| Gasoline range products yield (%) | 36.1 | 35.7 | 42.9 | 57.0 | 25.6 |
| Gasoline products carbon selectivity (%)c | |||||
| Pentane | 4.3 | 6.5 | 1.1 | 3.3 | 2.0 |
| Hexane | 11.2 | 0 | 2.5 | 2.4 | 1.9 |
| Ethanol | 3.1 | 3.3 | 0.6 | 12.0 | 4.7 |
| Propanol | 14.2 | 10.9 | 4.0 | 10.7 | 2.0 |
| Acetone | 0.9 | 0.6 | 0.6 | 0.6 | 0.9 |
| Butanol | 3.5 | 6.6 | 2.2 | 5.3 | 4.0 |
| Butanone | 1.2 | 1.5 | 0.7 | 1.2 | 0.2 |
| Tetrahydrofuran | 0.7 | 2.5 | 1.3 | 2.2 | 1.7 |
| Butanoic acid | 0.5 | 1.8 | 0 | 0 | 0 |
| Pentanol | 7.7 | 14.5 | 5.6 | 8.7 | 8.2 |
| Pentanone | 5.5 | 8.0 | 2.5 | 5.2 | 2.3 |
| Tetrahydropyran | 0.7 | 0.9 | 0.4 | 0.9 | 0.2 |
| Methyl-tetrahydrofuran | 2.3 | 5.4 | 6.5 | 4.3 | 5.3 |
| Tetrahydro-furfuryl alcohol | 14.2 | 29.8 | 47.3 | 23.1 | 28.5 |
| Pentanoic acid | 0 | 7.8 | 0 | 0 | 0 |
| Hexanol | 4.9 | 0 | 5.7 | 4.0 | 1.0 |
| Hexanone | 9.8 | 0 | 17.8 | 6.4 | 10.9 |
| 2,5-Dimethyl-tetrahydrofuran | 1.7 | 0 | 0.6 | 4.4 | 18.7 |
| Tetrahydropyran alcohol | 4.0 | 0 | 0.6 | 5.2 | 7.7 |
| Hexanoic acid | 9.6 | 0 | 0 | 0 | 0 |
| Estimated research octane number (RON)d | 81.8 | 96.3 | 89.0 | 96.5 | 91.1 |
| Aqueous phase cut yield (%) | 38.8 | 35.4 | 56.4 | 15.0 | 41.5 |
| Aqueous cut carbon selectivity (%)e | |||||
| Methanol | 1.4 | 1.7 | 0.3 | 1.9 | 1.4 |
| Propanediol | 0 | 11.1 | 0 | 0 | 0 |
| Butanediol | 3.9 | 25.1 | 2.0 | 1.8 | 3.0 |
| Pentanediol | 0 | 9.8 | 4.7 | 0 | 1.1 |
| Hexanediol | 5.3 | 0 | 8.9 | 3.9 | 3.2 |
| Xylitol | 0 | 52.3 | 34.2 | 3.8 | 25.3 |
| Sorbitan and isosorbide | 83.6 | 0 | 8.8 | 2.4 | 4.2 |
| Sorbitol | 5.9 | 0 | 2.3 | 1.2 | 3.3 |
| Carbon identified (%) | 85.2 | 80.9 | 102.4 | 81.1 | 71.4 |
As shown in Table 7, most of the carbon was still in the liquid phase after the two-stage hydrodeoxygenation process, with a light gas carbon yield of less than 10%. The estimated research octane number (RON) of the gasoline range products from the aqueous phase hydrodeoxygenation of glucose, xylose and maple wood hydrolysis is 82 to 96, which is similar to the RON of gasoline in the US market today. Thus, the majority of the monofunctional compounds could be used as high octane gasoline additives or as precursors to produce longer chain diesel and jet fuel molecules. For glucose and xylose, the carbon yields of both the gasoline and aqueous products were about 36%, and similar (or higher) to gasoline and aqueous yields were observed when maple wood hydrolysis samples were used as feedstock. Future work is needed to increase gasoline yields through better catalyst design.
The light gases were mainly composed of CO2 or methane (>80%). These light alkanes and CO2 could be recycled back into the reactor with unreacted hydrogen or reformed into hydrogen.73,74 The cyclic ether produced could be used as a solvent, and products left in the aqueous phase could be recycled back to the reactor with any unreacted sorbitol or xylitol.
A higher gasoline yield of 57% was obtained with sample 5. This is higher than the 48% yield of monofunctional (or gasoline) range compounds obtained by Dumesic et al. over Pt–Re/C catalyst with sorbitol or glucose solutions.43,75 Dumesic et al.'s catalyst had higher loadings of precious metals (5.1 wt% Pt and 4.9 wt% Re). We have also been able to obtain higher yields (70 carbon%) of gasoline range products from APHDO of 20 wt% sorbitol and xylitol over Pt/ZrP catalyst.49 This indicates that this process could be further optimized to achieve higher yields. The results from this paper suggest that aqueous phase catalytic processing can be used to efficiently produce gasoline from low cost carbohydrate solutions obtained from hydrolysis of cellulosic biomass that have an equivalent unit energy cost to petroleum at only $20/barrel.76,77 However, for this technology to become commercially viable processing costs must be further reduced.
Fig. 5 shows the 3 key classes of reactions that occur during the APHDO of aqueous carbohydrate containing solutions:48 1) C–C bond cleavage on metal sites; 2) C–O cleavage reaction on acid sites; and 3) hydrogenation on metal sites. Carbon–carbon bond cleavage reactions happen by two different pathways: 1) dehydrogenation followed by decarbonylation and 2) dehydrogenation followed by retro-adol condensation. In the APHDO process, sorbitol and xylitol undergo a series of dehydration/hydrogenation reactions to form hexanetriol and tetrahydrofurfuryl alcohol, respectively, and these products undergo further dehydration/hydrogenation to form hexanol and pentanol followed by hexane and pentane. The oxygenated intermediates can undergo C–C bond cleavage to form smaller oxygenates, CO2, and smaller alkanes. Fig. 5 shows the rich reaction chemistry involved in APHDO of biomass derived oxygenates that can be further tuned by adjusting the relative reaction pathways through further catalyst design and optimization of the reactor conditions.
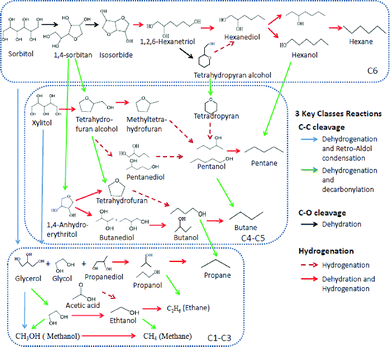 | ||
| Fig. 5 Reaction pathways for the hydrodeoxygenation of hydrogenated aqueous maple wood hydrolysis products. | ||
3.5. Overall carbon yield and mass yield of gasoline range products for the complete process
In Table 8 we show the overall mass and carbon yields for producing gasoline from maple wood by hydrodeoxygenation of the hemicellulose sugars from 3 different maple wood samples. The hydrolysis reaction also produces a solid residue that contains most of the cellulose and lignin that was originally in the maple wood. This hydrolysis residue could be further processed to convert the cellulose and lignin into fuels, chemicals, or pulp. Between 31 to 38% of the carbon in the maple wood is extracted into aqueous solutions which are then fed into the APHDO reactor. The APHDO then converts between 26 to 57% of the carbon in these aqueous solutions into gasoline range products. The remainder of the carbon is converted into light gases or carbon that stays in the aqueous phase. It should be noted that we have not tried to optimize the amount of carbon that can be converted into gasoline range products in this paper and future development in catalysis and reactor design could help us improve the yield further. The overall carbon yield of gasoline from maple wood ranges from 9 to 22%. This process thus, currently produces between 6 to 15 wt% yield of gasoline from wood with solid cellulose and lignin as a by-product.| Feedstock | Maple wood samplea | ||
|---|---|---|---|
| 4 | 5 | 6 | |
| a See Table 3 for details of the feedstock samples. b Overall carbon yield of gasoline range products from maple wood (%) = Carbon extracted by hydrolysis × Gasoline range products yield from APHDO of hydrolysis sample. c Overall mass yield of gasoline products from 100 kg maple wood (kg) = 45.5 × Overall carbon yield of gasoline range products from maple wood/Carbon percentage in the gasoline products deduced from carbon selectivity. (Carbon percentage in the gasoline range products (%) = 100 × 12.01/(Sum of specific selectivity × molecular weight/carbon number).) | |||
| Catalyst | H2SO4 | Oxalic acid | |
| Hydrolysis T/K | 433 | 433 | 453 |
| Hydrolysis reaction time/min | 30 | 30 | 10 |
| Carbon extracted by hydrolysis (%) | 31.6 | 38.1 | 35.2 |
| Gasoline range products carbon yield from APHDO of hydrolysis sample (carbon%) | 42.9 | 57.0 | 25.6 |
| Overall carbon yield of gasoline range products from maple wood (carbon%)b | 13.6 | 21.7 | 9.0 |
| Overall mass yield of gasoline products (wt%)c | 9.6 | 15.3 | 6.3 |
4. Conclusions
In this work, we demonstrated that gasoline range compounds can be produced by aqueous phase hydrodeoxygenation of aqueous carbohydrate solutions obtained from maple wood hydrolyzates that were a mixture of xylose, water soluble hemicellulose oligomers, acetic acid, glucose, glucose oligomers, and some lignin or humin polymers. Hydrothermal processing with hot water produced a large fraction of xylo-oligomers, whereas hydrolysis with dilute sulfuric or oxalic acid produced much higher yields of xylose monomers. In low temperature hydrogenation with Ru/C as the catalyst for conversion of the aqueous carbohydrates into gasoline, the Ru/C catalyst selectively hydrogenated xylose into xylitol and glucose into sorbitol but could not selectively hydrogenate xylose and glucose oligomers to xylitol and sorbitol. This result demonstrates that hydrolysis did not occur during low temperature hydrogenation. For hydrodeoxygenation in a two-stage reactor in which the first catalyst bed contained a Ru/C catalyst at 393 K and the second catalyst bed contained a Pt/zirconium phosphate (Pt/ZrP) catalyst at 518 K, up to 57% carbon yields of gasoline range products were obtained. The research octane number of these products was estimated to be 82–96 which is similar to octane number in the US gasoline markets. The Ru/C and Pt/ZrP catalysts were very stable at the conditions investigated in that no significant catalyst deactivation was observed even after 200 h of on-stream testing. Thus, aqueous phase hydrodeoxygenation of biomass derived hydrolysis residues is a promising option for the production of fuels and chemicals from lignocellulosic biomass that merits further research.Acknowledgements
This work was supported by a NSF-MRI grant (CBET-0722802) and by the Defense Advanced Research Project Agency and Army Research Lab through the Defense Science Office Cooperative Agreement W911NF-09-2-0010 (Surf-Cat: Catalysts for production of JP-8 range molecules from lignocellulosic Biomass). The views, opinions, and/or findings contained in this article are those of the authors and should not be interpreted as representing the official views or policies, either expressed or implied, of the Defense Advanced Research Projects Agency or the Department of Defense. We also appreciate the help of Ieva Narkeviciute for infrared experiments. The UCR team is also grateful to the Center for Environmental Research and Technology of the Bourns College of Engineering (CE-CERT) at for providing key equipment and facilities and the Ford Motor Company for funding the Chair in Environmental Engineering at CE-CERT that augments support for projects such as this.Notes and references
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS.
- M. Stocker, Angew. Chem., Int. Ed., 2008, 47, 9200–9211 CrossRef CAS.
- J. Jae, G. A. Tompsett, Y. C. Lin, T. R. Carlson, J. C. Shen, T. Y. Zhang, B. Yang, C. E. Wyman, W. C. Conner and G. W. Huber, Energy Environ. Sci., 2010, 3, 358–365 RSC.
- Y.-C. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 RSC.
- C. E. Wyman, B. E. Dale, R. T. Elander, M. Holtzapple, M. R. Ladisch, Y. Y. Lee, C. Mitchinson and J. N. Saddler, Biotechnol. Prog., 2009, 25, 333–339 CrossRef CAS.
- S. Brethauer and C. E. Wyman, Bioresour. Technol., 2010, 101, 4862–4874 CrossRef CAS.
- M. Laser, D. Schulman, S. G. Allen, J. Lichwa, M. J. Antal and L. R. Lynd, Bioresour. Technol., 2002, 81, 33–44 CrossRef CAS.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673–686 CrossRef CAS.
- F. M. Girio, C. Fonseca, F. Carvalheiro, L. C. Duarte, S. Marques and R. Bogel-Lukasik, Bioresour. Technol., 2010, 101, 4775–4800 CrossRef CAS.
- B. Yang and C. E. Wyman, Bioresour. Technol., 2008, 99, 5756–5762 CrossRef CAS.
- S. G. Allen, D. Schulman, J. Lichwa, M. J. Antal, M. Laser and L. R. Lynd, Ind. Eng. Chem. Res., 2001, 40, 2934–2941 CrossRef CAS.
- T. A. Lloyd and C. E. Wyman, Bioresour. Technol., 2005, 96, 1967–1977 CrossRef CAS.
- G. P. van Walsum and H. Shi, Bioresour. Technol., 2004, 93, 217–226 CrossRef CAS.
- Q. Jin, H. M. Zhang, L. S. Yan and H. Huang, Progress in Chemistry, 2010, 22, 654–662 Search PubMed.
- S. Monavari, M. Galbe and G. Zacchi, Biotechnol. Biofuels, 2009, 2, 6, DOI:10.1186/1754-6834-2-6.
- T. H. Kim and Y. Y. Lee, Bioresour. Technol., 2005, 96, 2007–2013 CrossRef CAS.
- T. H. Kim and Y. Y. Lee, Bioresour. Technol., 2006, 97, 224–232 CrossRef CAS.
- T. C. R. Brennan, S. Datta, H. W. Blanch, B. A. Simmons and B. M. Holmes, BioEnergy Res., 2010, 3, 123–133 CrossRef.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672–679 RSC.
- S. D. Zhu, Y. X. Wu, Q. M. Chen, Z. N. Yu, C. W. Wang, S. W. Jin, Y. G. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- C. Sievers, M. B. Valenzuela-Olarte, T. Marzialetti, D. Musin, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2009, 48, 1277–1286 CrossRef CAS.
- G. W. Huber, and J. A. Dumesic, An overview of aqueous-phase catalytic processes for production of hydrogen and alkanes in a biorefinery, Hornbaek, Denmark, 2005 Search PubMed.
- G. W. Huber, J. W. Shabaker and J. A. Dumesic, Science, 2003, 300, 2075–2077 CrossRef CAS.
- R. D. Cortright, R. R. Davda and J. A. Dumesic, Nature, 2002, 418, 964–967 CrossRef CAS.
- J. W. Shabaker, D. A. Simonetti, R. D. Cortright and J. A. Dumesic, J. Catal., 2005, 231, 67–76 CrossRef CAS.
- J. W. Shabaker, G. W. Huber and J. A. Dumesic, J. Catal., 2004, 222, 180–191 CrossRef CAS.
- J. W. Shabaker, R. R. Davda, G. W. Huber, R. D. Cortright and J. A. Dumesic, J. Catal., 2003, 215, 344–352 CrossRef CAS.
- T. P. Vispute and G. W. Huber, Green Chem., 2009, 11, 1433–1445 RSC.
- R. R. Davda and J. A. Dumesic, Chem. Commun., 2004, 36–37 RSC.
- D. L. King, L. Zhang, G. Xia, A. M. Karim, D. J. Heldebrant, X. Wang, T. Peterson and Y. Wang, Appl. Catal. B, 2010, 99(1–2), 206–213 CrossRef CAS.
- M. B. Valenzuela, C. W. Jones and P. K. Agrawal, Energy Fuels, 2006, 20, 1744–1752 CrossRef CAS.
- G. W. Huber, R. D. Cortright and J. A. Dumesic, Angew. Chem., Int. Ed., 2004, 43, 1549–1551 CrossRef CAS.
- G. W. Huber, J. N. Chheda, C. J. Barrett and J. A. Dumesic, Science, 2005, 308, 1446–1450 CrossRef CAS.
- S. Crossley, J. Faria, M. Shen and D. E. Resasco, Science, 2010, 327, 68–72 CrossRef CAS.
- R. M. West, Z. Y. Liu, M. Peter and J. A. Dumesic, ChemSusChem, 2008, 1, 417–424 CrossRef CAS.
- N. Lohitharn and B. H. Shanks, Catal. Commun., 2009, 11, 96–99 CrossRef CAS.
- C. Zhao, Y. Kou, A. A. Lemonidou, X. B. Li and J. A. Lercher, Chem. Commun., 2010, 46, 412–414 RSC.
- D. M. Alonso, J. Q. Bond, J. C. Serrano-Ruiz and J. A. Dumesic, Green Chem., 2010, 12, 992–999 RSC.
- D. Y. Hong, S. J. Miller, P. K. Agrawal and C. W. Jones, Chem. Commun., 2010, 46, 1038–1040 RSC.
- D. C. Elliott, T. R. Hart, G. G. Neuenschwander, L. J. Rotness and A. H. Zacher, Environ. Prog. Sustainable Energy, 2009, 28, 441–449 Search PubMed.
- Y. Roman-Leshkov, C. J. Barrett, Z. Y. Liu and J. A. Dumesic, Nature, 2007, 447, 982–985 CrossRef CAS.
- J. N. Chheda, Y. Roman-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342–350 RSC.
- R. M. West, E. L. Kunkes, D. A. Simonetti and J. A. Dumesic, Catal. Today, 2009, 147, 115–125 CrossRef CAS.
- N. Ji, T. Zhang, M. Y. Zheng, A. Q. Wang, H. Wang, X. D. Wang and J. G. G. Chen, Angew. Chem., Int. Ed., 2008, 47, 8510–8513 CrossRef CAS.
- E. P. Maris, W. C. Ketchie, M. Murayama and R. J. Davis, J. Catal., 2007, 251, 281–294 CrossRef CAS.
- E. P. Maris and R. J. Davis, J. Catal., 2007, 249, 328–337 CrossRef CAS.
- D. C. Elliott and T. R. Hart, Energy Fuels, 2009, 23, 631–637 CrossRef CAS.
- N. Li and G. W. Huber, J. Catal., 2010, 270, 48–59 CrossRef CAS.
- N. Li, G. A. Tompsett and G. W. Huber, ChemSusChem, 2010, 3(10), 1154–1157 CrossRef CAS.
- C. E. Wyman, S. R. Decker, M. E. Himmel, J. W. Brady, C. E. Skopec, and L. Viikari, Polysaccharides: Structural Diversity and Functional Versatility, Marcel Dekker, Inc., New York, 2004 Search PubMed.
- C. E. Wyman, ed., Handbook on Bioethanol: Production and Utilization, Taylor & Francis, Washington DC, 1996 Search PubMed.
- J. Q. Bond, D. M. Alonso, D. Wang, R. M. West and J. A. Dumesic, Science, 2010, 327, 1110–1114 CrossRef CAS.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, and D. Templeton, Laboratory Analytical Procedures, National Renewable Energy Laboratory, Golden, CO, 2005 Search PubMed.
- A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton, and D. Crocker, Laboratory Analytical Procedure, National Renewable Energy Laboratory, Golden, CO, 2006 Search PubMed.
- B. Yang, and C. E. Wyman, Methods in Molecular Biology: Biofuels, Humana Press, Totowa, NJ, 2009 Search PubMed.
- Y. Kamiya, S. Sakata, Y. Yoshinaga, R. Ohnishi and T. Okuhara, Catal. Lett., 2004, 94, 45–47 CrossRef CAS.
- M. R. Basila and T. R. Kantner, J. Phys. Chem., 1967, 71, 467 CrossRef CAS.
- J. F. Saeman, Ind. Eng. Chem., 1945, 37, 43–52 CrossRef CAS.
- H. E. Grethlein, and A. O. Converse, Food, Feed, and Fuel from Biomass, Oxford & IBH Publishing Company, New Delhi, 1991 Search PubMed.
- R. M. West, Z. Y. Liu, M. Peter, C. A. Gartner and J. A. Dumesic, J. Mol. Catal. A: Chem., 2008, 296, 18–27 CrossRef CAS.
- R. Kumar and C. E. Wyman, Carbohydr. Res., 2008, 343, 290–300 CrossRef CAS.
- S. E. Jacobsen and C. E. Wyman, Appl. Biochem. Biotechnol., 2000, 84–86, 81–96 CrossRef CAS.
- N. Yan, C. Zhao, C. Luo, P. J. Dyson, H. C. Liu and Y. Kou, J. Am. Chem. Soc., 2006, 128, 8714–8715 CrossRef CAS.
- H. Olcay, L. Xu, Y. Xu and G. Huber, ChemCatChem, 2010, 2(11), 1420–1424 Search PubMed.
- H. Heeres, R. Handana, D. Chunai, C. B. Rasrendra, B. Girisuta and H. J. Heeres, Green Chem., 2009, 11, 1247–1255 RSC.
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC.
- H. Mehdi, V. Fabos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horvath, Top. Catal., 2008, 48, 49–54 CrossRef CAS.
- C. Luo, S. Wang and H. Liu, Angew. Chem., Int. Ed., 2007, 46, 7636–7639 CrossRef CAS.
- J. M. Robinson, C. E. Burgess, M. A. Bently, C. D. Brasher, B. O. Horne, D. M. Lillard, J. M. Macias, H. D. Mandal, S. C. Mills, K. D. O'Hara, J. T. Pon, A. F. Raigoza, E. H. Sanchez and J. S. Villarreal, Biomass Bioenergy, 2004, 26, 473–483 CrossRef CAS.
- J. M. Robinson, C. E. Burgess, M. A. Bently, C. D. Brasher, B. O. Horne, D. M. Lillard, J. M. Macias, L. D. Marrufo, H. D. Mandal, S. C. Mills, K. D. O'Hara, J. T. Pon, A. F. Raigoza, E. H. Sanchez, J. S. Villarreal and Q. Xiang, ACS Symp. Ser., 2006, 921, 304–320 CAS.
- K. Owen, and T. Coley, Automotive Fuels Reference Book, Society of Automotive Engineers. Inc., Warrendale, 1995 Search PubMed.
- J. A. Satrio, B. H. Shanks and T. D. Wheelock, Ind. Eng. Chem. Res., 2005, 44, 3901–3911 CrossRef CAS.
- K. O. Albrecht, J. A. Satrio, B. H. Shanks and T. D. Wheelock, Ind. Eng. Chem. Res., 2010, 49, 4091–4098 CrossRef CAS.
- E. L. Kunkes, D. A. Simonetti, R. M. West, J. C. Serrano-Ruiz, C. A. Gartner and J. A. Dumesic, Science, 2008, 322, 417–421 CrossRef CAS.
- L. R. Lynd, C. E. Wyman and T. U. Gerngross, Biotechnol. Prog., 1999, 15, 777–793 CrossRef CAS.
- L. R. Lynd, M. S. Laser, D. Brandsby, B. E. Dale, B. Davison, R. Hamilton, M. Himmel, M. Keller, J. D. McMillan, J. Sheehan and C. E. Wyman, Nat. Biotechnol., 2008, 26, 169–172 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NH3–DRIFTS spectra of ZrPOx after adsorption of ammonia at 373 K followed by degassing under He flow at various temperatures; table of the physical properties and research octane number of different gasoline range products; table of methods of hydrogenation of mono-, di- and polysaccharides. See DOI: 10.1039/c0gc00501k |
| This journal is © The Royal Society of Chemistry 2011 |