Vanadate(V)-dependent bromoperoxidase immobilized on magnetic beads as reusable catalyst for oxidative bromination†
Diana
Wischang
a,
Jens
Hartung
*a,
Thomas
Hahn
b,
Roland
Ulber
b,
Tobias
Stumpf
c and
Claudia
Fecher-Trost
c
aFachbereich Chemie, Organische Chemie, Technische Universität Kaiserslautern, Erwin-Schrödinger-Straße, D-67663, Kaiserslautern, Germany. E-mail: hartung@chemie.uni-kl.de; Fax: +49 631 205 3921
bFachbereich Maschinenbau und Verfahrenstechnik, Technische Universität Kaiserslautern, Gottlieb-Daimler-Straße, D-67663, Kaiserslautern, Germany
cFachbereich Biologie, Proteinfunktion/Proteomics, Technische Universität Kaiserslautern, Paul-Ehrlich-Straße, D-67663, Kaiserslautern, Germany
First published on 2nd December 2010
Abstract
Vanadate(V)-dependent bromoperoxidase I (Ascophyllum nodosum) was immobilized on magnetic micrometre-sized particles in quantitative yields, with up to 40% retention of initial bromoperoxidase (BPO) activity. The immobilized enzyme was stable with a half-life time of about 160 days. It served as reusable catalyst for bromide oxidation with H2O2 in up to 14 consecutive experiments. Reactivity that resulted from enzymatic bromide oxidation was applicable for methyl pyrrole-2-carboxylate conversion into derivatives of naturally occurring compounds (e.g. from Agelas oroides) with product selectivity of up to 75%.
Introduction
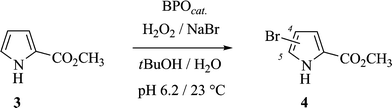
Vanadate(V)-dependent bromoperoxidase I from the brown alga Ascophyllum nodosum [VBrPO(AnI), EC 1.11.1.10]1 catalyzes the oxidation of bromide with H2O2 (Scheme 1) in weakly acidic solutions (pH 6.2–6.5).2 The turnover frequency of this process exceeds the hitherto most active non-enzymatic alternatives by 4–5 orders of magnitudes.3 The enzyme tolerates elevated temperatures and a variety of organic co-solvents without notable loss of its bromoperoxidase (BPO) activity over time spans typically needed for substrate bromination in organic synthesis.4,5 VBrPOs from other organisms than A. nodosum were discovered from screenings and purified to homogeneity.2,6,7 Some bromoperoxidases even were genetically expressed in fungi or bacteria. None of these enzymes, however, shares similar favorable characteristics for application in oxidation catalysis for the pursuit of sustainable hydrocarbon bromination than VBrPO(AnI).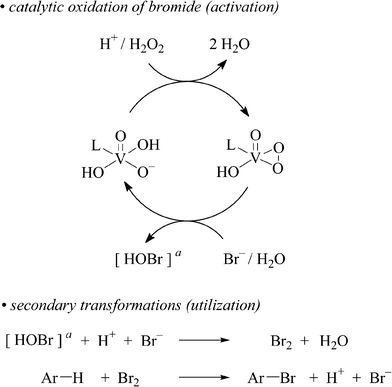 | ||
| Scheme 1 Stoichiometry for VBrPO(AnI)-catalyzed bromide oxidation with H2O2 (top) and secondary non-enzymatic reactions utilizing electrophilic bromination reagents for arene functionalization (bottom; L = apoenzyme; Ar = aryl or heteroaryl, see below). a Proposed intermediate in an early phase of the reaction.8 | ||
The debate on the nature of the electrophilic bromination reagent that is liberated from the enzymatic process has not yet been settled (cf.Scheme 1).9–11 The overall reaction, however, has attracted considerable attention. It has been associated with formation of naturally occurring organobromine compounds, although none of the proposed pathways have so far been biosynthetically proven.10,12 In terms of organic synthesis, the use of VBrPO(AnI) as catalyst for oxidative brominations in ocean water-like media probably constitutes the most sustainable version of the on-site bromination of arenes,13–16 as recently documented by bromofunctionalization of benzenoid, non-benzenoid, and heterocyclic arenes on an analytical scale.5 For further exploration of this chemistry and its development into a widely applicable method, the aspect of catalyst supply, however, requires particular attention. Since the enzyme is not yet biotechnologically available, it needs to be extracted from the genuine source. The process, although well elaborated, is time and material consuming.17BPO immobilization onto a solid support,18 in this sense would open perspectives for its repetitive use and thus a reduction of the amount needed for conducting larger scale studies. Structural complexity of surfaces,19 however, still precludes clear cut predictions on efficiency of catalyst immobilization, its durability and its activity during turnover in close proximity to the solid liquid interface.
As part of a project on the chemistry of VBrPO(AnI) in organic synthesis we chose to immobilize the BPO on magnetic beads. It was the aim to obtain a robust oxidation catalyst for multiple use in sustainable hydrocarbon bromination. The most important findings from this study showed that binding of the enzyme to epoxide- and amino-functionalized micrometre-sized magnetic supports was feasible in quantitative yields with up to 40% retention of the initial BPO activity. This material served for at least 14 consecutive runs as catalyst for biomimetic methylpyrrole-2-carboxylate bromination.20
Results and interpretation
1 Enzyme purification and characterization
VBrPO(AnI) was separated from a mixture of isoenzymes according to a published procedure, starting from lyophyllized A. nodosum (Fig. 1). The enzyme was stored in tris-(hydroxymethyl)-aminomethane(HCl), i.e., Tris(HCl)-buffered stock solutions (pH 9.0) at 4 °C.21Bromoperoxidase activity was retained with a half-life time of approximately 120 days under such conditions. The identity of the enzyme was verified via MALDI-TOF analysis of trypsine-digested samples and database analysis of measured tryptic peptide masses, in comparison with the known primary structure of the enzyme (PDB 1QI9). Activity of VBrPO(AnI) preparations ranged between 405 ± 43 and 526 ± 11 UT mg−1 (triiodide assay22 in phosphate buffer at pH 6.2).5,23‡ | ||
| Fig. 1 Polyacrylamide gel electrophoresis (1 h, 18 mA; non-denaturing conditions) of crude bromoperoxidase fraction (right) in comparison to VBrPO(AnI) preparation obtained from hydrophobic interaction and size exclusion chromatography (left, BPB = bromophenol blue; staining was performed in a solution prepared from phosphate buffer, H2O2, aq. KI, and 1,4-phenyldiammonium dichloride). | ||
2 Enzyme immobilization
Magnetic beads (M) were chosen as support, which were coated with a matrix of polyvinyl alcohol (PVA) and functionalized by the supplier with epoxide groups (E), or aminated with an eight atomic spacer (N) (see Experimental). The magnetite level was 50–60% (w/w), epoxide group loading 200–220 μmol, amino group loading 650–750 μmol per g of particle. Particles with a narrower (0.5–1.3 μm for E01 and N11) and a broader (0.7–4.5 μm for E02 and N12) polydisperse size distribution were used for both types of functionalized beads. The materials were sufficiently attracted by a Nd/Fe bar magnet (20 × 10 × 5 mm, N42 magnetization) to allow swift and quantitative separation from a suspension mimicking a standard reaction mixture, as evident from optical density measurements (600 nm) (Fig. 2 and ESI).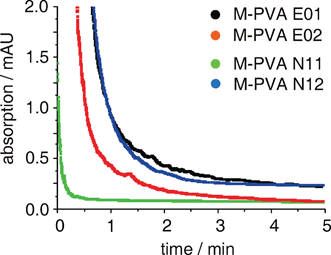 | ||
| Fig. 2 Optical density measurement (600 nm) for checking magnetic attraction of applied beads composed of polyvinyl alcohol (PVA)-coated magnetite (M) particles having epoxide (E) or amino (N) endgroups (for size distribution of the particles refer to Table 1; Nd/Fe bar magnet). | ||
VBrPO(AnI) loading onto epoxide-functionalized supports M-PVA E01 and E02 at 23 °C was feasible after adjusting pH to 6.2 with MES buffer and mixing of the components. Preparations obtained from these manipulations retained 29% (1a) and 17% (1b) of initial BPO activity as evident from triiodide tests (Table 1, entry 1–2). BPO immobilization on amino-functionalized supports was attainable viain situ-treatment of M-PVA N11 and N12 with glutardialdehyde (GA)24 in K2HPO4 buffer (20 mM, pH 8.0) for 1 h at 23 °C, which was followed by addition of the VBrPO(AnI)-stock solution in aqueous Tris(HCl) buffer (pH 9.0) (Scheme 2). The latter two reactions furnished materials showing 24% (2a) and 37–39% (2b) of the original BPO activity (Table 1, entries 3–4). The activity values were in a reproducible manner independent from the batch of enzyme used for immobilization (section 2.1 and Table 1, entries 4–5). The data also clarified that VBrPO(AnI) binding was not inhibited by the presence of an excess of Tris from the buffer.
| Entry | Support | Particle size/μm | 1–2 |
m
0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/mg a/mg |
UT0 mg−1b |
BPO-loading/mg g−1 bead | UTloaded mg−1c |
|---|---|---|---|---|---|---|---|
| a Amount of VBrPO(AnI) prior to immobilization. b Bromoperoxidase (BPO)-activity of VBrPO(AnI) preparation (triiodide test). c BPO-activity of immobilized VBrPO(AnI) (triiodide test). | |||||||
| 1 | M-PVA E 01 | 0.5–1.9 | 1a | 0.14 | 405 ± 43 | 2.82 | 118 ± 12 |
| 2 | M-PVA E 02 | 0.7–6.0 | 1b | 0.14 | 405 ± 43 | 2.15 | 69 ± 7 |
| 3 | M-PVA N 11 | 0.5–1.9 | 2a | 0.10 | 493 ± 6 | 2.13 | 120 ± 28 |
| 4 | M-PVA N 12 | 0.7–6.0 | 2b | 0.08 | 446 ± 24 | 1.60 | 168 ± 47 |
| 5 | M-PVA N 12 | 0.7–6.0 | 2b | 0.10 | 493 ± 6 | 2.13 | 194 ± 39 |
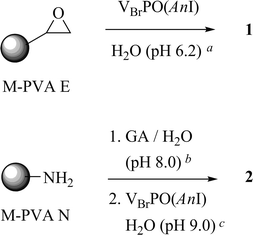 | ||
| Scheme 2 Immobilization of VBrPO(AnI) on polyvinyl alcohol (PVA)-functionalized (E or N) magnetite (M) (GA = glutardialdehyde; solid supports drawn as circles of an arbitrary radius; for further abbreviations see legend of Fig. 2, for results Table 1). aMES buffer. bPhosphate buffer. cTris(HCl) buffer. | ||
Yields of VBrPO(AnI) binding to functionalized supports were essentially quantitative, as judged on the basis of the Bradford assay.25,26 Since crystalline VBrPO(AnI) was not available in sufficient amounts, the protein concentration in solution was referenced versus bovine serum albumin (BSA). The results were independently checked viatriiodide BPO activity tests of the VBrPO(AnI) stock solution, the filtrate after separation of materials 1–2, and MES buffer washings of the latter preparations.
In view of the considerable fraction of nucleophilic endgroups at the periphery of the protein8 and the inertness of active enzyme toward leaching, attachment of VBrPO(AnI) to applied beads (Scheme 2) was assumed to occur via covalent bonds. VBrPO(AnI) immobilization to GA-activated amino-functionalized beads thereby occurred faster than attachment to epoxide-substituted particles. For practical reasons, however, the latter preparations offered the advantage of directly providing immobilized VBrPO(AnI) in MES-buffered solution, i.e. the medium the oxidation experiments were performed in. For experiments using preparations 2a and 2b, washing with MES buffer was necessary to lower pH from 9.0 to 6.2, i.e. the value required for attaining maximum BPO activity.
Long term studies indicated that storage of preparations 1a–b in MES buffer and materials 2a–b in Tris(HCl) buffer at 4 °C progressively decreased in BPO activity during the first 70 days. In solution this activity loss prevailed for the free enzyme. For immobilized VBrPO(AnI), BPO activity from this point on remained approximately constant (Fig. 3). Although triiodide BPO activity tests using suspensions of preparation 1–2 showed stronger scatter than for homogeneous solutions containing the free enzyme, presumably for the time span required for quantitative magnetic attraction prior to UV/Vis data collection, the outlined trends were diagnostic and reproducible. In view of the activity decay, oxidation experiments summarized in the following section were conducted with freshly immobilized materials 1–2.
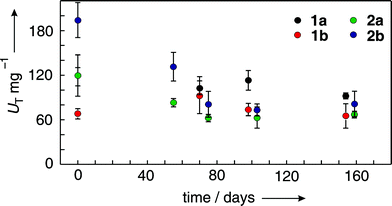 | ||
| Fig. 3 Time dependence of specific BPO-activity of immobilized VBrPO(AnI) stored at 4 °C in MES buffer (samples 1a–b) or Tris(HCl) buffer (samples 2a–b; error bars refer to standard deviations from three independent runs). | ||
3 Oxidation catalysis
Conditions suitable for organic compound bromination in VBrPO(AnI)-catalyzed oxidations were established using methyl 1H-pyrrole-2-carboxylate (3) as reporter substrate. The π-excess heteroaromatic compound undergoes substitution of bromine for hydrogen already with comparatively weakly electrophilic reagents, such as HOBr or Br2, without the necessity of adding external Lewis acids.27 It furthermore provides a diagnostic mixture of mono- and disubstitution products 4 and 5 that are sufficiently stable to allow quantitative analysis viagas chromatography and subsequent column chromatographic separation, for determining the site of aromatic substitutionviaNMR spectroscopy.27,28 Since pyrrole-2-carboxylic acid in nature is formed from the amino acid metabolism,29 the experiments were considered to be relevant for providing insight into genesis of naturally occurring, brominated pyrroles.|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | n 3 /μmol | Conv. 3/% | Time/h | BPO | UT | UTfinal mg−1a |
4/% (4/5)b |
| a Initial enzyme activity 526 UT0 mg−1 for entries 1–4; for entries 5–6 refer to Table 1; active refers to residual enzyme activity as judged by qualitative triiodide tests. b Ratio of 4/5-regioisomers of bromopyrrole 4. c c 3 0 = 7.2 mM, H2O2 addition in 3 batches every 30 min; total amount of substrates: 1.0 equiv. of H2O2 and 3.0 equiv. of NaBr. d H2O2 added in a single batch; total amount of substrates: 1.0 equiv. of H2O2 and 1.0 equiv. of NaBr. | |||||||
| 1 | 36 | quant.c | 24 | VBrPO(AnI) | 1.3 | 117 | 94 (93/7) |
| 2 | 36 | 92d | 5 | VBrPO(AnI) | 1.3 | 99 | 73 (89/11) |
| 3 | 36 | 77d | 3 | VBrPO(AnI) | 1.3 | 239 | 60 (82/18) |
| 4 | 36 | 27d | 1 | VBrPO(AnI) | 1.3 | 231 | 26 (81/19) |
| 5 | 200 | 54d | 3 | 1a | 6.9 | active | 40 (90/10) |
| 6 | 200 | 75d | 3 | 2b | 6.9 | active | 32 (93/8) |
![Combination of d-glucoseoxidase (GO)-catalyzed H2O2 generation and VBrPO(AnI)-catalyzed bromide oxidation for sustainable pyrrole bromination [d-Glc = d-glucose; 6.2 UT for VBrPO(AnI); 1.67 U for GO; pH 5.8 (MES); tBuOH/H2O; c30 = 7.2 mM; for stoichiometry of the BPO-catalyzed reaction see Scheme 1].](/image/article/2011/GC/c0gc00499e/c0gc00499e-s3.gif) | ||
| Scheme 3 Combination of D-glucoseoxidase (GO)-catalyzed H2O2 generation and VBrPO(AnI)-catalyzed bromide oxidation for sustainable pyrrole bromination [D-Glc = D-glucose; 6.2 UT for VBrPO(AnI); 1.67 U for GO; pH 5.8 (MES); tBuOH/H2O; c30 = 7.2 mM; for stoichiometry of the BPO-catalyzed reaction see Scheme 1]. | ||
A survey of the time dependence of bromopyrrole formation showed that substrate conversion reached a level of approximately 70% after 3 h. Since changes in terms of reactivity and selectivity originating from the support would be most evident at this conversion level, a reaction period of 3 h and thus incomplete substrate turnover was chosen as standard for validating catalytic properties of immobilized VBrPO(AnI). Comparison of product formation and substrate consumption clarified that the use of the enzyme in homogeneous solutions and in immobilized form (i.e.1a and 2b) afforded almost equivalent turnover rates. A separate study for maximizing yields therefore was not performed for validating highest performance of solid phase bound VBrPO(AnI)-preparations, since those data were indirectly available from experiments conducted in homogeneous solution. Additional products for explaining deviation between turnover of 3 and yield of bromopyrrole 4 in the analytical scale experiments using immobilized VBrPO(AnI) 1a and 2b (Table 2, entries 5–6) were not detected (GC, NMR).
VBrPO(AnI)-activity that was conserved in methylpyrrole-2-carboxylate bromination in homogeneous solutions was applicable for further experiments after addition of aliquots of substrate 3, H2O2, NaBr, and MES buffer. BPO-activity after the fourth run, however, fell below a synthetically relevant level (Fig. 4).
![Efficiency of product formation in VBrPO(AnI)-catalyzed reactions in sequential runs [4.96 UT corresponding to 0.05 mmol % VBrPO(AnI); 144 μmol of 3, 1 equiv. of H2O2 and NaBr, H2O/tBuOH = 75/25 (v/v); additional product: 10% of methyl 3,4-dibromopyrrole-2-carboxylate (5) in experiment number 1 and <1% in experiment number 2].](/image/article/2011/GC/c0gc00499e/c0gc00499e-f4.gif) | ||
| Fig. 4 Efficiency of product formation in VBrPO(AnI)-catalyzed reactions in sequential runs [4.96 UT corresponding to 0.05 mmol % VBrPO(AnI); 144 μmol of 3, 1 equiv. of H2O2 and NaBr, H2O/tBuOH = 75/25 (v/v); additional product: 10% of methyl 3,4-dibromopyrrole-2-carboxylate (5) in experiment number 1 and <1% in experiment number 2]. | ||
Addition of catalyst suspension to aqueous tBuOH containing reporter substrate 3 was followed by addition of NaBr and H2O2. At fixed reaction times of 3 h, catalyst particles were separated from the solution with a magnet that was held against the wall of the reaction flask (vide supra). The reaction medium was decanted off to afford a clear solution that was worked up for clarifying product identity, yields, and substrate conversion. Routine analysis showed weak BPO activity in this solution during the first 3 runs but not beyond this point. Additional standard checks indicated no detectable BPO activity in solutions obtained from catalyst washings from runs 1–15.
The next cycle was started by addition of fresh buffer and substrates in standard concentrations into the flask, which was followed by removal of the magnet from the exterior of the reaction vessel. With increasing number of reaction cycles, the yield of bromopyrrole 4 increased and reached a maximum at approximately the 5th cycle (Fig. 5). High catalyst performance prevailed until the 9th cycle. The following runs (10–15), in a reproducible manner, led to progressive decrease in turnover and yield. These changes were paralleled by a gradual decrease of terminal pH of the reaction mixture from 7.8 (1st run) via 7.6 (5th cycle), 6.8 (9th cycle) to 6.2 (14th cycle), by the time the reaction was stopped (3 h). The results obtained from oxidations using catalyst 2b (ESI) were nearly identical in terms of yields and activity changes upon multiple use, compared to those described for experiments performed with preparation 1a (Fig. 5).
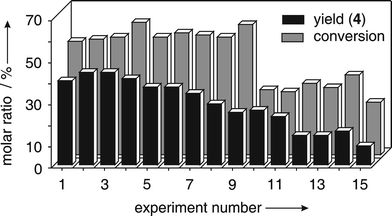 | ||
| Fig. 5 Conversion of substrate 3 (c0 = 35 mM; gray columns) and efficiency of methylpyrrole-2-carboxylate bromination (black columns) upon multiple use of VBrPO(AnI)-preparation 1a (6.9 UT) for catalyzing oxidative bromination in aqueous tBuOH (pH 6.2, 23 °C) using 1.0 equiv. each of H2O2 and NaBr (3 h, see text). | ||
The origin of irreversible loss of catalyst activity remained unclear. Leaching of active enzyme was excluded on the basis of the above-mentioned results from routine triiodide tests of reaction media and catalyst washings. Possible vanadate loss was checked by soaking the immobilized catalyst in an orthovanadate solution between consecutive oxidation experiments. This modification, however, had no effect on performance of the material and activity loss after the 8–10th cycle.
Concluding remarks
Immobilization of VBrPO(AnI) to solid supports provided three distinctive results relevant for future development of sustainable oxidative brominations.(i) VBrPO(AnI) binding either to epoxide-functionalized or to glutardialdehyde-activated amino-substituted magnetic beads was feasible in quantitative yields, with almost 40% of the original catalyst activity being conserved.
(ii) Immobilized VBrPO(AnI) showed similar turnover characteristics for methyl pyrrole-2-carboxylate oxidative bromination as the free enzyme in homogeneous solutions. The unexpectedly high performance of immobilized enzyme was explained with substrate bromination that occurred in bulk solution and not in proximity to the active site. In this model, diffusion of bromide to the histidine-bound vanadate(V) co-factor would be rate determining and not migration of the organic substrate to a specific binding site.
(iii) Supported bromoperoxidase was applicable for 8–10 consecutive cycles without notable activity loss.
The most significant achievement compared to other techniques used for instance for immobilization of the iron-dependent chloroperoxidase from Caldariomyces fumago [FeClPO(CfI)] on mesoporous supports31 was the ease and the efficiency of magnetic separation of immobilized VBrPO(AnI) from reaction mixtures. Chemicals originating from a preceding transformation thus were removable from the catalyst by simply washing with the buffer needed for the subsequent run. This aspect has been noted previously for application of immobilized lipases in biofuel preparation32 but has not yet been applied to haloperoxidase chemistry. With this material in hand, application of VBrPO(AnI) in oxidative bromination has the potential to become an interesting alternative in instances where substitution of molecular bromine is desired for environmental reasons by the reagent combination of H2O2, possibly generated in situ from aerobic D-Glc oxidation catalyzed by GO, NaBr and an adequate source of protons.
Experimental
1 General
For instrumentation, comments on laboratory standards, and details of compound preparations, see the ESI. VBrPO(AnI) was isolated from A. nodosum collected in April 2004 (France, 48° 43′ N, 3° 58′ W) according to a published procedure.21 Magnetic beads were obtained from chemagen Biopolymer-Technologie AG, Germany.2 Immobilization of VBrPO(AnI)
3 Bromination of methyl pyrrole-(1H)-2-carboxylate (3)
Acknowledgements
This work was supported by the Deutsche Bundesstiftung Umwelt (Scholarship for D.W.) and the State of Rheinland-Pfalz (NanoKat). We also express our gratitude to chemagen for generous donations of M-PVA E0x and M-PVA N1x particles used in this study, and to Dr Thomas Sommer (chemagen) for helpful discussions. This work is part of the Ph.D. thesis of D.W.Notes and References
- H. Vilter, Phytochemistry, 1984, 23, 1387–1390 CrossRef CAS.
- A. Butler and J. V. Walker, Chem. Rev., 1993, 93, 1937–1944 CrossRef CAS.
- G. E. Meister and A. Butler, Inorg. Chem., 1994, 33, 3269–3275 CrossRef CAS.
- E. de Boer, H. Plat and R. Wever, in Biocatalysis in Organic Media, ed. C. Laane, J. Tramper and M. D. Lilly, Elsevier, Amsterdam, 1987, pp. 317–322 Search PubMed.
- J. Hartung, Y. Dumont, M. Greb, D. Hach, F. Köhler, H. Schulz, M. Časný, D. Rehder and H. Vilter, Pure Appl. Chem., 2009, 81, 1251–1264 CrossRef CAS.
- H. Vilter, in Vanadium and its Role in Life, Metal Ions in Biological Systems, ed. H. Sigel and A. Sigel, Dekker, New York, 1995, vol. 31, pp. 325–362 Search PubMed.
- E. E. Coupe, M. G. Smyth, A. Fosberry, R. M. Hall and J. A. Littlechild, Protein Expression Purif., 2007, 52, 265–272 CrossRef CAS.
- M. Weyand, H. J. Hecht, M. Kiess, M. F. Liaud, H. Vilter and D. Schomburg, J. Mol. Biol., 1999, 293, 595–611 CrossRef CAS.
- O. Bortolini, M. Carraro, V. Conte and S. Moro, Eur. J. Inorg. Chem., 2003, 42–46 CrossRef.
- A. Butler and J. N. Carter-Franklin, Nat. Prod. Rep., 2004, 21, 180–188 RSC.
- F. H. Vaillancourt, E. Yey, D. A. Vosburg, S. Garneau-Tsodikova and C. T. Walsh, Chem. Rev., 2006, 106, 3364–3378 CrossRef.
- G. J. Gribble, Environ. Sci. Pollut. Res., 2000, 7, 37–49 CrossRef CAS.
- G. Rothenberg and J. H. Clark, Green Chem., 2000, 2, 248–251 RSC.
- A. Podgoršek, M. Zupan and J. Iskra, Angew. Chem., Int. Ed., 2009, 48, 8424–8450 CrossRef CAS.
- C. W. Jones, in Applications of Hydrogen Peroxide and Derivatives, RSC Clean Technology Monographs, series ed. J. H. Clark, RSC, Cambridge, 1999, pp. 156–162 Search PubMed.
- For guidelines of sustainable synthesis see: F. M. Kerton in Introduction from Chemicals from Biomass, ed. J. Clark and F. Deswarte, Wiley, 2008, ch. 3, pp. 47–76 Search PubMed.
- H. Vilter, Methods Enzymol., 1994, 228, 665–672 CAS.
- L. S. Wong, F. Khan and J. Micklefield, Chem. Rev., 2009, 109, 4025–4053 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- S. Forenza, L. Minale and R. Riccio, J. Chem. Soc., Chem. Commun., 1971, 1129–1130 RSC.
- J. Hartung, O. Brücher, D. Hach, H. Schulz, H. Vilter and G. Ruick, Phytochemistry, 2008, 69, 2826–2830 CrossRef CAS.
- F. Björkstén, Eur. J. Biochem., 1968, 5, 133–142 CrossRef CAS.
- International Union of Biochemistry (IUB), Report of the Commission on Enzymes, Pergamon Press, Oxford, 1961 Search PubMed.
- S. S. Wong, in Chemistry of Protein Conjugation and Cross-Linking, CRL Press, Boca Raton, 1993, ch. 4, pp. 74–103 Search PubMed.
- M. H. Bradford, Anal. Biochem., 1976, 205, 22–26.
- N. J. Krüger, Meth. Mol. Biol., 1994, 32, 9–15 Search PubMed.
- C. Schroif-Gregoire, N. Travert, A. Zaparucha and A. Al-Mourabit, Org. Lett., 2006, 8, 2961–2964 CrossRef CAS.
- B. M. Trost and G. Dong, J. Am. Chem. Soc., 2006, 128, 6054–6055 CrossRef CAS.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23, 517–531 RSC.
- D. D. Keilin and E. F. Hartree, Biochem. J., 1952, 50, 331–341 CAS.
- M. Hartmann and C. Streb, J. Porous Mater., 2006, 13, 347–352 CrossRef CAS.
- X. Wang, P. Dou, P. Zaho, C. Zhao, Y. Ding and P. Xu, ChemSusChem, 2009, 2, 947–950 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Standard instrumentation, BPO-analysis, determination of BPO-activity, synthesis of methyl 1H-pyrrole-2-carboxylate (3), details for the use of D-Glc/GO for in situ H2O2 generation in BPO-catalyzed oxidations, details for the use of VBrPO(AnI)-preparations 1a and 2b in multiple runs. See DOI: 10.1039/c0gc00499e |
| ‡ Notation for enzymatic activity: 1 unit (U) refers to the amount of enzyme required for turning over 1 μmol of substrate per minute. BPO-activity determined with the aid of the triiodide assay (T) was abbreviated as UT. Specific BPO-activity referred to the number of units per mg of enzyme preparation, i.e.UT mg−1. |
| This journal is © The Royal Society of Chemistry 2011 |