Microwave-promoted synthesis of polyol esters for lubrication oil using a composite catalyst in a solvent-free procedure†
Fengxiu
Zhang‡
*a and
Guangxian
Zhang‡
b
aCollege of Chemistry and Chemical Engineering, Southwest University, 2 Tiansheng Street, Chongqing, 400715, P. R. China. E-mail: zhangfx656472@sina.com.cn; Fax: +86 023 68251228; Tel: +86 023 68251681
bCollege of Textile and Garment, Southwest University, 2 Tiansheng Street, Chongqing, 400715, P. R. China. Fax: +86 023 68251228; Tel: +86 023 68251681
First published on 22nd November 2010
Abstract
A rapid, highly efficient and green synthetic approach to polyol esters for lubrication oils is proposed. Using sulfuric acid and p-toluene sulfonic acid as a composite catalyst and C5–C9 straight-chain monocarboxylic acids, with pentaerythritol (PE) and dipentaerythritol (di-PE) as starting materials, a series of lubrication oil polyol esters were synthesized in the absence of solvent under microwave irradiation. Compared with the conventional synthetic method, our proposed microwave method exhibits advantages, including higher yields, shorter reaction times and lower reaction temperatures. The viscosity coefficients of the products at 40 °C and their refractive indices at 25 °C were investigated. In addition, the thermal behavior of the starting materials under microwave irradiation was also investigated. The results reveal that the microwave absorbance of n-pentanoic acid is stronger than that of n-hexanoic acid and that of n-heptanoic acid is stronger than that of n-octanoic acid. The microwave absorbance of di-PE is also stronger than that of PE.
Introduction
Many polyol esters are commonly used as bases for lubrication oils because they have excellent properties,1,2 such as high flash point, high viscosity index, low pour point, low volatility and excellent thermal stability. Because they are physiologically harmless and readily biodegrade in the natural environment, polyol ester-based lubrication oils are regarded as eco-friendly.2,3,4Two major synthetic methods have been adopted for the preparation of lubrication oil esters. One is direct esterification, where monobasic straight-chain fatty acids are reacted with a polyalcohol in the presence of acidic catalysts, and organic solvents with low boiling points (such as benzene, toluene and dimethylbenzene) are used as water-carrying reagents.5,6,7 However, a disadvantage of above method is that the products are commonly dark in color.7,8 The other involves a two-step reaction. Firstly, fatty acid methyl esters are obtained by the methanol alcoholysis of triglycerides. Secondly, they are transesterified with a polyhydric alcohol to produce polyol esters.9 Gryglewiecz and co-workers9 have employed the transesterification method to synthesize fatty acid polyol esters of neopentyl glycol (NPG) and trimethylol propane (TMP). They used vegetable and animal fats as sources of fatty acids and calcium methoxide as a catalyst. The second step yield was about 85–90%, with isooctane as an azeotropic solvent, but the first step was generally low-yielding. Thus, the total yield was not satisfactory. Furthermore, the reaction time was very long (20 h).
In the synthesis of lubrication oil esters, typically used catalysts include sulfuric acid, p-toluene sulfonic acid, phosphoric acid, phosphate, tributyl phosphate, sodium methoxide, sodium ethoxide, tetraoctyl zirconate, tetrabutyl titanate and sodium hydroxide.6,7,8,10 Recently, various lipases have also been employed as catalysts for the synthesis of lubrication oil esters. For example, Uosukainen et al.11 used TMP and rapeseed oil methyl ester as starting materials and various lipases as catalysts to synthesize trisubstituted TMP ester by a transesterification reaction. The highest yield of trisubstituted TMP ester was 85.05% and the lowest was 4.87% after 48 h at 50 °C. Linko et al.12 also applied the transesterification method to prepare the TMP triester of rapeseed oil fatty acids with lipase immobilized on Duolite ES-561; the highest yield of about 70% was reached after 78 h at 47 °C. With the commercial immobilized R. mieheilipase Lipozyme IM 20, the yield of TMP triester was about 75% in 24 h at 58 °C.13 Although lipase-catalyzed esterification may offer many significant advantages, such as milder reaction conditions, simplicity of product isolation, enzyme reuse and lower environmental pollution, the reaction efficiency is low in terms of long reaction time and unsatisfactory yield.
Microwave irradiation (MW) can enormously shorten reaction times and remarkably increase reaction yields in most cases compared to traditional synthesis.14 The combination of solvent-free reaction conditions and microwave irradiation is considered to be an eco-friendly approach.15 Here, we report a MW method for the direct synthesis of polyol esters in the absence of solvent, using C5–C9 straight-chain monocarboxylic acids and a series of polyols as starting materials in the presence of a composite catalyst. The synthetic regime is shown in Scheme 1:
 | ||
| Scheme 1 Reaction of polyols and fatty acids. | ||
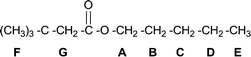
The properties of polyol esters are related to the structures of the fatty acids and alcohols, i.e. the length of the aliphatic chain, the number of hydroxyl groups in the alcohol and the presence or absence of a β-carbon–hydrogen (C–H) bond relative to the hydroxyl group (Fig. 1).16 The sensitivity to thermal oxidation of the C–H bonds at positions F, G, A and E of Fig. 1 is comparatively low. In contrast, the C–H bonds at positions B, C and D (Fig. 1) are easily oxidized to unsaturated compounds at high temperature (Fig. 2).9
 | ||
| Fig. 1 The various carbon–hydrogen bonds (C–H) of an ester. | ||
 | ||
| Fig. 2 Mechanism of the thermal destruction of esters with a β-hydrogen. | ||
Polyol esters without a β-H are more thermally stable than those with a β-H. Therefore, pentaerythritol (PE) and dipentaerythritol (di-PE) are commonly used as starting materials in industry.17
Results and discussion
Microwave absorbance of starting materials
Molecules with different polarities showed different microwave absorbances. Fig. 3 shows the increases in temperature for the various materials as a function of irradiation time. n-Pentanoic acid and n-heptanoic acid exhibited greater microwave absorbances than n-hexanoic acid and n-octanoic acid.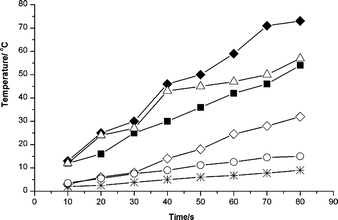 | ||
| Fig. 3 Changes of temperature with time: (◆) n-pentanoic acid, (■) n-hexanoic acid, (△) n-heptanoic acid, (◇) n-octanoic acid, (*) pentaerythritol and (○) dipentaerythritol. | ||
In the odd-numbered carbon fatty acids (n-pentanoic acid and n-heptanoic acid), the carboxyl and chain-end methyl groups in their structures are located on the same side of the carbon chain. These asymmetric molecular structures lead to greater intermolecular distances, and hence weaker intermolecular forces, than for the even-numbered carbon fatty acids (n-hexanoic and n-octanoic acid). This also implies that the dipoles in the odd-numbered carbon fatty acids align themselves more readily with the applied microwave field by rotation and re-orientation than the dipoles in the even-numbered carbon fatty acids. While the dipoles change their mutual orientation, the applied field is also changing, resulting in a phase difference between the orientation of the field and that of the dipole. This phase difference leads to energy loss from the dipole by molecular collisions and friction, thus giving rise to dielectric heating.15 Therefore, in odd-numbered carbon fatty acids, heat generation will be faster than in even-numbered carbon fatty acids. In the case of low odd-numbered carbon fatty acids, molecules with shorter carbon chains will also be more easily rotated and re-oriented, and hence produce more heat as a result of agitation and the intermolecular friction of molecules.15 Moreover, the abilities of molecules of different polarity to absorb microwave energy and to convert that absorbed energy are related to their dielectric constants (ε′) and their loss factors (ε′′). These factors are usually considered using the loss angle (δ), which is expressed as its tangent (eqn (1)): 15
| tan δ = ε′′/ε′ | (1) |
For molecules of comparable dielectric constant, higher loss tangent values imply the better absorption of MW irradiation, resulting in a more rapid temperature increase. The dipole moments of n-pentanoic acid, n-hexanoic acid, n-heptanoic acid and n-octanoic acid reduce gradually over the small range 1.679–2.0,17c and their dielectric constants are similar. Hence, the above results for the temperature increase of different monocarboxylic acids (Fig. 3) indicate that the loss tangent value of n-pentanoic acid is higher than that of n-heptanoic acid, n-hexanoic and n-octanoic acid. Because the re-orientation of dipole moments in the applied field brings about a phase difference between the orientation of the field and the dipoles, the energy absorbed from the electric field is converted into heat as the dielectric loss, described here by the loss tangent values. Thus, it can be seen that the tangent values and dipole moments correspond. This is why n-pentanoic acid gives the highest microwave absorbance of the molecules studied.
PE and di-PE have melting points beyond 220 °C and thus remain solid for the irradiation times used. Fig. 3 shows that PE and di-PE have a very weak microwave absorbance compared with that of the liquid fatty acids. There are several possible reasons for the stronger microwave absorbance of di-PE relative to PE: the structure of di-PE is more asymmetric and its dipole moment is stronger than that of PE; in addition, PE is a granular solid, while di-PE is a powder.
Choice of catalyst
The choice of catalyst is crucial to the synthesis of polyol esters. On the one hand, a weakly polar catalyst absorbs the microwave energy poorly, leading to a lower reaction temperature, which is not beneficial to the esterification reaction. On the other hand, a strongly polar catalyst leads to a sharp rise in the reaction temperature, causing undesirable carbonization of the reactants. Therefore, a suitable catalyst and the dosage of the catalyst were very important in our study. We investigated four kinds of acidic catalysts: phosphoric acid, p-toluene sulfonic acid, sulfuric acid and a composite catalyst made up of p-toluene sulfonic acid and sulfuric acid. Activated charcoal was also added into the reaction system to increase the microwave absorbance and also to decolor the product. Catalysts were screened (Table 1) using the example synthesis of di-PE hexa-hexanoate (4b) at 320 W MW power for 10 min and conventional heating for 10 or 110 min. All esterification reactions were carried out in the absence of solvent. The catalyst loading was 3% (w/w) for trials 1–5. For trials 4 and 5, the catalyst comprised 0.1876 g sulfuric acid and 0.5253 g p-toluene sulfonic acid (i.e., a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.8). As can be seen from Table 1, phosphoric acid (trial 1) had a strong microwave absorption and a correspondingly high reaction temperature, but the resultant yield was low, only 42.6%. p-Toluene sulfonic acid (trial 2) had a weak microwave absorption, leading to a lower reaction temperature and also a relatively low yield (67.6%). Sulfuric acid (trial 3) was the better catalyst, but it made the reactants and product char easily and darken under MW irradiation. Thus, we designed a composite catalyst consisting of sulfuric acid and p-toluene sulfonic acid. As expected, the composite catalyst with a 1:2.8 mass ratio of sulfuric acid to p-toluene sulfonic acid gave both a high percentage yield and a light-colored product (trial 4). Subsequently, the composite catalyst was used for the synthesis of polyol esters under MW. The results of reactions with conventional heating are shown in trial 5 for comparison. When the reaction time was 10 min, the same as the MW reaction, the yield was very low, only 25.0%. Meanwhile, an 83.5% yield was obtained after 110 min. Although the reaction temperature and catalyst were the same as for the MW reaction (trial 4), the conventional yields were 70.2 and 11.7% lower after 10 and 110 min, respectively, than those with MW irradiation. The reaction time with MW irradiation was 11 times shorter than that for conventional heating. Moreover, the product color from the MW reaction was more satisfactory than that from conventional heating for 110 min. Hence, the conventional heating method produced a poorer yield and product color, and required a longer reaction time. Therefore, there are very significant advantages to applying a MW method to synthesize polyol esters for lubrication oils.
:2.8). As can be seen from Table 1, phosphoric acid (trial 1) had a strong microwave absorption and a correspondingly high reaction temperature, but the resultant yield was low, only 42.6%. p-Toluene sulfonic acid (trial 2) had a weak microwave absorption, leading to a lower reaction temperature and also a relatively low yield (67.6%). Sulfuric acid (trial 3) was the better catalyst, but it made the reactants and product char easily and darken under MW irradiation. Thus, we designed a composite catalyst consisting of sulfuric acid and p-toluene sulfonic acid. As expected, the composite catalyst with a 1:2.8 mass ratio of sulfuric acid to p-toluene sulfonic acid gave both a high percentage yield and a light-colored product (trial 4). Subsequently, the composite catalyst was used for the synthesis of polyol esters under MW. The results of reactions with conventional heating are shown in trial 5 for comparison. When the reaction time was 10 min, the same as the MW reaction, the yield was very low, only 25.0%. Meanwhile, an 83.5% yield was obtained after 110 min. Although the reaction temperature and catalyst were the same as for the MW reaction (trial 4), the conventional yields were 70.2 and 11.7% lower after 10 and 110 min, respectively, than those with MW irradiation. The reaction time with MW irradiation was 11 times shorter than that for conventional heating. Moreover, the product color from the MW reaction was more satisfactory than that from conventional heating for 110 min. Hence, the conventional heating method produced a poorer yield and product color, and required a longer reaction time. Therefore, there are very significant advantages to applying a MW method to synthesize polyol esters for lubrication oils.
| Trial | Catalyst | T/°C | Yield (%) | Color |
|---|---|---|---|---|
| a Conventional heating. b Reaction for 10 min. c Reaction for 110 min. | ||||
| 1 | Phosphoric acid | 134 | 42.6 | Brown |
| 2 | p-Toluene sulfonic acid | 96 | 67.6 | Colorless |
| 3 | Sulfuric acid | 120 | 93.0 | Brown |
| 4 | Sulfuric acid and p-toluene sulfonic acid | 128 | 95.2 | Yellowish |
| 5a | Sulfuric acid and p-toluene sulfonic acid | 128 | 25.0b | Colorless |
| 83.5c | Sandy beige |
Because sulfuric acid has a stronger microwave absorbance than p-toluene sulfonic acid, different mass ratios of sulfuric acidvs. p-toluene sulfonic acid were expected to influence both the yield and the product color. The example synthesis of 4b under MW irradiation was used to investigate the effect of the ratio of sulfuric acid to p-toluene sulfonic acid, and the results are given in Table 2. The reaction time was the time taken at 320 W MW power for the esterification reaction to cease (as determined by no further emission of the water). Ratios of sulfuric acid to p-toluene sulfonic acid of 1:2.0 or 1:2.4 gave lower yields (79.6 and 85.2%, respectively) and poor product colors. A ratio of 1:3.2 produced a colorless product. However, the yield was only 68.6%. Hence, a ratio of 1:2.8 for the composite catalyst was chosen to give a high yield with a satisfactory product color at 128 °C.
|
Sulfuric acid:p-toluene sulfonic acid |
Time/min | T/°C | Yield (%) | Color |
|---|---|---|---|---|
| a Reaction conditions: 0.73 g composite catalyst, 320 W MW power. | ||||
| 1:2.0 |
15.9 | 140 | 79.6 | Brown |
| 1:2.4 |
12 | 134 | 85.2 | Yellow |
| 1:2.8 |
8.5 | 128 | 97.0 | Yellowish |
| 1:3.2 |
6.0 | 108 | 68.6 | Colorless |
Optimization of microwave power
MW power has a remarkable effect on the esterification reaction. The example synthesis of 4b under MW irradiation was used to investigate the effect of MW power, and the results are presented in Table 3. As previously described, the reaction time is the time taken for the esterification reaction to cease, as evaluated by no further water being produced. Table 3 shows that an increase in MW power leads to higher reaction temperatures and a gradual increase in yield from 230 to 320 W. When the MW power was 230–260 W, the reaction temperature and yield were both low. When the MW power was increased to 290–320 W, the yield observably increased from 73.5 to 97.0% and the reaction time was only 1–2 min longer than that with 230–260 W MW power. When a MW power of 340–360 W was used, the yield decreased and the product became yellow or brown in color. Considering the various factors, such as product color, yield and reaction time, the optimal MW power was 320 W for 4b. Furthermore, the results of conventional heating at a similar temperature to that obtained during the MW reaction are given in Table 3. When the reaction temperature was 75–83 °C, the yield was very low. From 83 to 98 and 98 to 128 °C, the yields were enhanced by 46.9 and 27.7%, respectively. A further small increase in yield was found from 128 to 148 °C. The conventional heating reactions displayed a consistent trend of increasing yield with increasing reaction temperature. In contrast, for the MW reactions the yields decreased from 128 to 148 °C. The results indicate that MW power can directly influence the esterification reaction, and hence selecting the optimal MW power is very important.| Trial | MW power/W | T/°C | Time/min | Color | Yield (%) |
|---|---|---|---|---|---|
|
a Reaction conditions: sulfuric acid:p-toluene sulfonic acid ration was 1:2.8.
b Conventional heating.
|
|||||
| 1 | 230 | 75 | 6.5 | Colorless | 53.5 |
| — | 110b | Yellowishb | 2.37b | ||
| 2 | 260 | 83 | 7.0 | Colorless | 64.3 |
| — | 110b | Yellowishb | 8.9b | ||
| 3 | 290 | 98 | 7.5 | Colorless | 73.5 |
| — | 110b | Buffb | 55.8b | ||
| 4 | 320 | 128 | 8.5 | Yellowish | 97.0 |
| — | 110b | Yellowb | 83.5b | ||
| 5 | 340 | 136 | 10 | Yellow | 87.6 |
| — | 110b | Brownb | 85.1b | ||
| 6 | 360 | 148 | 15 | Brown | 76.6 |
| — | 110b | Brownb | 88.5b |
Optimization of the synthesis parameters for 4a–d
The parameters for the MW synthesis of polyol esters, including the mass ratio of the composite catalyst, MW power, reaction time and reaction temperature, were optimized for each ester, and the optimal conditions are reported in Table 4. The polyol esters included di-PE hexa-pentanoate (4a), di-PE hexa-hexanoate (4b), di-PE hexa-heptanoate (4c) and di-PE hexa-octanoate (4d). The esterification temperatures of compounds 4a, 4b, 4c and 4d were 115, 128, 135 and 142 °C, respectively, indicating that the esterification temperature gradually increased with increasing carbon chain length. The yields of 4a–d were 90–98% within 9–11 min (Table 4).The advantage of using microwave heating
Compounds 5a–e (PE tetra-pentanoate (5a), PE tetra-hexanoate (5b), PE tetra-heptanoate (5c), PE tetra-octanoate (5d) and PE tetra-nonanoate (5e)) were synthesized from PE and C5–C9straight-chain fatty acids. The esterification reactions of 5a–e were carried out in the presence of 0.67 g of the composite catalyst and 0.13 g of activated charcoal. For the MW method at a power of 300–320 W, the optimal synthesis parameters (mass ratio of the composite catalyst, reaction time and reaction temperature) were investigated, and the results are shown in Table 5. We also compared the MW method and the conventional heating method in terms of reaction time, reaction temperature, yield and color of the product—these results are also shown in Table 5. Compared to conventional heating, the MW method achieved an excellent yield within 9–10 min (a reduction by a factor of 6 to 17.8). The yields from 5a–e were 14, 10, 18, 14 and 12% higher than the conventional heating yield, respectively. In addition, the reaction temperatures decreased by 15–53 °C, and the products were colorless. Therefore, the MW synthesis method exhibited several advantages over the conventional synthesis method: enhanced reaction yield, reduced undesired side products, dramatically shortened reaction times, decreased reaction temperatures and better color of the products.| Compound | Mass ratio of composite catalyst/g | Time/min | T/°C | Yield (%) | Color |
|---|---|---|---|---|---|
| a Conventional heating. | |||||
| 5a | 1:3.0 |
9 | 103 | 97 | Colorless |
| 120a | 118a | 83a | Buffa | ||
| 5b | 1:2.8 |
9 | 105 | 96 | Colorless |
| 160a | 158a | 86a | Yellowisha | ||
| 5c | 1:3.0 |
10 | 110 | 93 | Colorless |
| 120a | 130a | 75a | Buff a | ||
| 5d | 1:2.5 |
10 | 114 | 95 | Colorless |
| 105a | 138a | 81a | Buff a | ||
| 5e | 1:3.0 |
10 | 118 | 88 | Colorless |
| 60a | 154a | 76a | Buffa |
Physical properties of the polyol esters
The physical properties, including viscosity coefficient, refractive index and color, of polyol esters 4a–d and 5a–e were investigated, and the results are shown in Table 6. When the chroma of a compound is below 175 Hazen units, it appears as a colorless and clear liquid. From 200 to 300 Hazen units, the compound is a yellowish liquid. From 325 to 500 Hazen units, the compound’s color varies from buff to yellow. Over 500 Hazen units, the compound gradually darkens in color. Table 6 shows that products 5a–e were colorless clear liquids, and that compounds 4a–d were mostly yellowish to fuscous liquids. This may be because di-PE and fatty acids, in particular, n-octanoic acid, are easily charred at high temperatures. Alternatively, it may be a consequence of the higher temperatures reached during the preparation of hexa-esters (4a–d) compared with tetra-esters (5a–e), because the esterification reactions to make polyol esters occur gradually rather than in a single step. The products produced by conventional heating were darker in color than the corresponding products from MW irradiation.| Trial | Compound | Viscosity/mm2s−1a |
n 25 D | Color/Hazen | ||||
|---|---|---|---|---|---|---|---|---|
| MWc | Cd | MWc | Cd | MWc | Cd | |||
| a Determined at 40 °C. b Literature data at 40 °C. c MW method. d Conventional heating. | ||||||||
| 1 | 4a | 45.2 | — | 1.4521 | — | 425 | — | |
| 2 | 4b | 49.4 | — | 1.4556 | — | 300 | — | |
| 3 | 4c | 54.5 | — | 1.4571 | — | 450 | — | |
| 4 | 4d | 60.1 | — | 1.4576 | — | >500 | — | |
| 5 | 5a | 14.3 | 13.51 | 1.4446 | 1.4445 | 150 | 200 | |
| 6 | 5b | 16.5 | 15.68 | 1.4491 | 1.4490 | 70 | 95 | |
| 7 | 5c | 23.46 | 22.35 | (b Lit.4 23.65) | 1.4521 | 1.4506 | 175 | 225 |
| 8 | 5d | 26.18 | 25.22 | (b Lit.4 26.45) | 1.4531 | 1.4516 | 125 | 200 |
| 9 | 5e | 27.60 | 26.33 | (b Lit.4 27.40) | 1.4577 | 1.4576 | 120 | 225 |
Refractive index is a widely used physical property for the classification of lubricating oils. In industry, compounds 4a–d and 5a–e are synthesized from PE or di-PE and the appropriate mole ratio of C5–C9straight-chain fatty acids. Thus far, there are only a few reported values of viscosity and refractive indices for these and similar compounds. Here, we present the refractive indices of 4a–d and 5a–e for the first time. The refractive indices of 5a, 5b and 5e produced using MW irradiation are almost the same as the corresponding products using conventional heating. For compounds 5c and 5d, the refractive indices of the MW products are higher than those from conventional heating, but the difference is not sufficient to affect the grade of lubricating oil. A possible reason for this difference is that the fraction of hydroxyl groups converted during the MW reaction is higher than during conventional heating.
The viscosity coefficients of polyol esters are important property parameters as a criterion of lubricating oil quality and are also the main parameters governing the choice of lubricating oil for various applications in equipment, engines and aviation.1,2 They also affect lubrication performance, including abrasion and surface rubbing prevention, rubbing power loss and the work efficiency of equipment.18 As can be seen from Table 6, the viscosity coefficients of 4a–d and 5a–e increase gradually with increasing carbon chain length of the fatty acid, and the trend in viscosity with chain length is as expected from theory.19 The results for products 5c–e from MW reactions are in agreement with the available literature data.4 The viscosity coefficients of the MW reaction products are significantly higher than those produced by conventional heating. These results show that the two different synthetic methods markedly affect the viscosity coefficient and color of polyol esters, and that the MW method produced materials with better viscosity coefficients than those from the conventional heating method.
Experimental
Materials
Pentaerythritol (PE) was purchased from the Shanghai Chemical Company (Shanghai, China). Dipentaerythritol (di-PE, 85% purity) was purchased from the Shandong Plant Protection Drug Factory (China), purified by recrystallizing twice from re-distilled water and then dried at 60–70 °C until of constant weight. All other chemicals were of analytical grade and used without further purification. PE, p-toluene sulfonic acid and activated charcoal were dried at 60 °C until of constant weight before use. C5–C9 straight-chain monocarboxylic acids were dehydrated by 4 Å molecular sieves, and sulfuric acid was stored under vacuum for some time before use.Analytical methods
IR spectra were recorded on a Perkin-Elmer GX FT-IR spectrophotometer in KBr pellets. NMR spectra were measured on a Bruker AV 300 spectrometer (300 MHz) in CDCl3. MS spectra were obtained using a Bruker HCT Agilent Technologies 1200 Series instrument. The colors of the prepared esters were determined by platinum-cobaltous colorimetry (GB/T3143-82).20 Refractive indices were measured on a ZWA-J refractometer at 25 °C. Product viscosity was measured at 40 ± 0.05 °C using an Ubbelohde viscometer in a temperature-equilibrated bath. Re-distilled water was used as the calibrant.General procedure for evaluating the microwave absorbance of different materials
The MW reactor used (WP-650, Nanjing Lingjiang Science and Technology Corporation) could process in distillation or reflux modes, and the MW power was adjustable from 0 to 650 W. Temperatures were measured by placing a thermocouple into the sample immediately before, and again at the end of, each irradiation.C5–C8carboxylic acids 1 (20 ml) or solid samples of 2 or 3 (5.0 g) were added into a 50 ml beaker. Then, the beaker was placed into the MW oven. The samples were heated at 300 W for 1 or 320 W for 2 and 3 for 10, 20, 30, 40, 50, 60, 70 or 80 s, and the final temperatures were then measured. ΔT, the change in temperature, was defined as in eqn (2):
| ΔT = T2 − T1 | (2) |
General procedure for MW-activated and conventional heating reactions of 4a–d and 5a–e
di-PE 2 (10 mmol) or PE 3 (22 mmol), C5–C9carboxylic acids 1 (80 mmol for di-PE 2 and 96 mmol for PE 3), composite catalyst and activated charcoal (0.13 g) were added into a 100 ml round-bottomed flask with two anti-bumping zeolite granules. The above mixture was MW-irradiated at a power of 300–320 W and the water produced was continually distilled out under a reduced pressure (46–63 KPa) until no further water was distillable and the mixture ceased to boil. At this point, the reaction was considered “ceased”, and the reaction time was determined using a stopwatch by counting from the beginning to the ceasing of boiling of the reaction mixture. The reaction temperature was also promptly determined by placing a thermocouple into the reaction mixture when the reaction had ceased. In the conventional heating procedure, the same reactants, composite catalyst and activated charcoal as the MW reaction were added into a 100 ml round-bottomed flask with two anti-bumping zeolite granules. Then, the reaction mixture was heated by an immersion heater for a set time at a set temperature, and the water produced was continually distilled out under a reduced pressure (46–63 KPa) until no further water could be removed. The water produced was collected in a recipient vessel.After cooling down to ambient temperature, the reaction mixture was filtered. The filtrate was washed with 15 ml distilled water, followed separately 2 or 3 times by 15 ml of a 10% sodium hydroxide solution. The final repeated washing was with distilled water until the organic layer was neutral. The organic layer was dried over anhydrous sodium sulfate. The crude product was purified by silica gel column chromatography using diethyl ether as the eluent. The product structures were characterized by IR, 1H NMR, 13C NMR and mass spectroscopy.
Due to the molecular weights of products 4a–d and 5a–e being higher, both their boiling points and viscosities are also higher.18 Therefore, the products cannot be measured on a gas chromatogram, and are also easily lost during separation and purification. Therefore, the yield (Y) was calculated from the ratio of the experimental (We) and theoretical (Wt) values of the water produced (eqn (3)): 21
| Y(%) = 100 × We/Wt | (3) |
| We = W2 − W1 | (4) |
Conclusions
This study presents the thermal behavior of starting materials of different polarities under microwave irradiation. An efficient composite catalyst consisting of sulfuric acid and p-toluene sulfonic acid was screened. Optimization of the reaction conditions was performed for di-PE and PE. The physical properties of the polyol esters were investigated. From a green chemical standpoint, the present method is a rapid, highly efficient and eco-friendly synthetic approach for the preparation of polyol esters for lubrication oils.Acknowledgements
The authors gratefully acknowledge financial support from the Chongqing Grand Natural Science Department in China (Grant no. 6683). F. Z. thanks Dr Shi-Hong Chen for language revision of the paper.References
- (a) C. S. Li, Sci. Technol. Chem. Ind., 2000, 8, 76 Search PubMed; (b) B. Z. Zhang, Synth. Lubricants, 1997, 24, 26 Search PubMed; (c) Y. K. Luo and D. H. Hu, Synth. Lubricants, 1994, 21, 36 Search PubMed.
- T. G. Zhong, Synth. Lubricants, 2000, 27, 12 Search PubMed.
- (a) L. Jin, X. J. Wang, Z. L Gu, D. Z. Zhou and S. Q. Xie, Pedosphere, 2006, 16, 540 CrossRef CAS; (b) P. Mercurio, K. A. Burns and A. Negri, Environ. Pollut., 2004, 129, 165 CrossRef CAS; (c) P. Mercurio, K. A. Burns and J. Cavanagh, Environ. Pollut., 2004, 129, 175 CrossRef CAS; (d) P. Mercurio, A. P. Negri, K. A. Burns and A. J. Heyward, Environ. Pollut., 2004, 129, 183 CrossRef CAS; (e) N. S. Battersby, Chemosphere, 2000, 41, 1011 CrossRef CAS.
- J. Y. Cheng, Q. Zhu, Y. S. Yang, L. L. Shi, Z. Q. Yao and Z. H. Lu, Acta Scientiae Circumstantiae, 2001, 21, 120 Search PubMed.
- L. B. Wang, Lubes Fuels, 2003, 13, 5 Search PubMed.
- (a) X. G. Chen and B. Zhou, Chem. Bioeng., 2005, 22, 16 Search PubMed; (b) J. Q. Fei and W. Zhao, Lubricating Oil, 2009, 24, 42 Search PubMed.
- Y. H. Gao, Z. S. Lin and M. Y. Huang, Guangzhou Chem., 2000, 25, 24 Search PubMed.
- S. L. Guan, J. F. Chen, W. Li and J. B. Wang, Guizhou Chem. Ind., 2004, 29, 17 Search PubMed.
- S. Gryglewicz, W. Piechocki and G. Gryglewicz, Bioresour. Technol., 2003, 87, 35 CrossRef CAS.
- (a) M. I. D. Sairre, S. B. U. Érika and P. M. Donate, Tetrahedron Lett., 2005, 46, 2705 CrossRef; (b) H. Wagner, R. Luther and T. Mang, Appl. Catal., A, 2001, 221, 429 CrossRef CAS.
- E. Uosukainen, M. Lämsä, Y. Y. Linko, P. Linko and M. Leisola, Enzyme Microb. Technol., 1999, 25, 236 CrossRef CAS.
- Y. Y. Linko, M. Lämsä, X. Wu, E. Uosukainen, J. Seppälä and P. Linko, J. Biotechnol., 1998, 66, 41 CrossRef CAS.
- Y. Y. Linko, T. Tervakangas, M. Lämsä and P. Linko, Biotechnol. Tech., 1997, 11, 889 CrossRef CAS.
- (a) A. Kulkarni and B. Török, Green Chem., 2010, 12, 875 RSC; (b) S. Herrero, J. A. Reyes, J. Perles, J. L. Priego and F. A. Urbanos, Green Chem., 2010, 12, 965 RSC; (c) D. H. Yu, L. Tian, D. X. Ma, H. Wu, Z. Wang, L. Wang and X. X. Fang, Green Chem., 2010, 12, 844 RSC.
- (a) P. Lidström, J. Tierney, B. Wathey and J. Westman, Tetrahedron, 2001, 57, 9225 CrossRef CAS; (b) L. Perreux and A. Loupy, Tetrahedron, 2001, 57, 9199 CrossRef CAS.
- Y. K. Luo and D. H. Hu, Synth. Lubricants, 1994, 21, 35 Search PubMed.
- (a) D. F. Zhao, L. B. Chen, M. Z. Yao and J. R. Gao, Synthetical Chemistry of Refined Chemical and Application, Chemical Industry Press, Beijing, 2001, pp. 280–281 Search PubMed; (b) X. H. Qian and S. C. Mo, Contemporaneity Technique Encyclopaedia on Refined Chemistry and the Product of Chemical Technology, Science Press, Beijing, 2001, pp. 563–565 Search PubMed; (c) C. L. Yaws and P. K. Narasimhan, Thermophysical Properties of Chemicals and Hydrocarbons, William Andrew Inc., 13 Eaton Avenue, 2009, ch. 19, pp. 672–682 Search PubMed.
- X. H. Li and X. J. Zhao, Lubricating Oil, 2009, 24, 59 Search PubMed.
- M. Xu, Lubricating Oil, 1999, 14, 56 Search PubMed.
- Compiling groups, Manual of Analyzing Way on General Chemistry Industry Production, Chemical Industry Press, Beijing, 1999, pp. 974–975 Search PubMed.
- (a) J. Q. Fei and W. Zhao, Lubricating Oil, 2009, 24, 42 Search PubMed; (b) D. Q. Li, B. Li and H. B. Lou, Chemical Propellants & Polymeric Materials, 2000, 4, 28 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: IR, 1H NMR, 13C NMR and MS analyses of compounds 4a–d and 5a–e. See DOI: 10.1039/c0gc00351d |
| ‡ The authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2011 |