An efficient protocol for the palladium-catalysed Suzuki–Miyaura cross-coupling†
Alexander N.
Marziale
a,
Dominik
Jantke
a,
Stefan H.
Faul
a,
Thomas
Reiner
b,
Eberhardt
Herdtweck
b and
Jörg
Eppinger
*a
aGreen Chemistry & Catalysis (GCC) Laboratories, KAUST Catalysis Center(KCC), 4700 King Abdullah University of Science and Technology, Thuwal, 23955-6900, Kingdom of Saudi Arabia. E-mail: jorg.eppinger@kaust.edu
bTechnischeUniversitätMünchen, DepartmentChemie, Lichtenbergstr. 4, D-85747, Garching, Germany
First published on 13th December 2010
Abstract
The palladacyclic catalyst precursor received by ortho-palladation of ([1,1′-biphenyl]-2-yloxy)diisopropyl-phosphine represents a highly active system for Suzuki–Miyaura cross-coupling reactions when used in neat water. An efficient, broadly applicable and sustainable aqueous protocol was developed using 2.5 eq. of Na2CO3 as base, allowing the reaction to be performed under air and at ambient temperature with Pd loadings of 0.04 mol%. Coupling products are obtained in high yields and excellent purity by simple filtration with no organic solvents needed throughout the whole reaction. A broad variety of functional groups are tolerated and a large number of substrates can be applied with this protocol. The crystal structure of the palladacyclic catalyst precursor is presented as well as investigations targeting the nature of catalyst activation and the active catalytic species.
Introduction
The development of sustainable and efficient reaction procedures has become a major driving force of synthetic organic chemistry in academia and industry.1 The search for environmentally acceptable concepts in chemical synthesis led to an enormous increase of research activities in the field of green chemistry.2 To judge the quality of these efforts in terms of sustainability several fundamental principles have been introduced.3 Besides the reduction of chemical waste and the number of synthetic steps, catalyst recovery and safer as well as more efficient reaction conditions, one of the most important aspects is the application of environmentally friendly solvents such as water, ionic-liquids or supercritical carbon dioxide.4Water, in contrast to common organic reaction media, is particularly favourable as it is cheap, non-flammable, non-toxic and abundantly available.5 Moreover, a broad variety of organometallic catalytic reactions have been successfully applied in water.6 Among these, the Suzuki–Miyaura cross-coupling reaction is undoubtedly the most powerful tool to selectively generate biaryls. Casalnuovo and Calabrese transferred this reaction to aqueous conditions as early as 1990.7 Research in the field has mainly focused on biphasic protocols based on water-soluble catalysts ever since.Important contributions to this field were made by Beller,8 Buchwald,9 Genet,10 Leadbeater,11 Miyaura,12 Plenio13 and Shaughnessy.14 Another approach to aqueous Suzuki coupling represents the concept of micellar catalysis, reported by Sheldon and Lipshutz.15 Further, phase transfer catalysts and the addition of water-soluble organic co-solvents such as DMF and acetonitrile, have been employed with great success in recent years.16 Modern reaction procedures like microwave heating and ultrasonic irradiation have largely contributed to reduce reaction times and make aqueous catalysis more efficient.17 Yet, most aqueous protocols for cross-coupling reactions suffer from high catalyst loadings,18 elevated temperatures,19 complex work-up procedures including column chromatography10–19 or require the addition of organic co-solvents. However, catalysis in water in principle offers the possibility of facile separation of the solid lipophilic products generated. Nevertheless, such examples remain scarce.13,20
Recently we have reported a protocol for Suzuki–Miyaura coupling in water,21 applying a newly developed Bedford-type palladacycle 2.22 The catalyst was synthesised after refluxing 2-phenylphenol with diisopropylchlorophosphine and triethylamine in toluene followed by palladation of the resulting phosphinite ligand 1 (Scheme 1). The resulting complex exhibits high thermal, air and moisture stability and shows superior catalytic activity in comparison to a variety of catalyst motifs.21
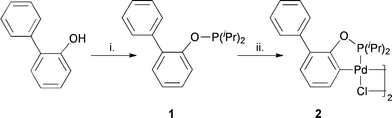
We herein present an improved protocol with broad applicability regarding the range of substrates and functional group tolerance. This protocol not only meets the criteria for green chemistry as they have been mentioned before but also the demands of “click-reactions” as defined by Sharpless and co-workers.23 Furthermore we have been examining the limitations of this protocol with respect to reaction temperature, substrate scope and product isolation. To clarify the role of the palladacyclic catalyst during the reaction, we structurally characterised the catalyst and investigated its behaviour under reaction conditions by means of kinetic studies and quantitative poisoning experiments.
Results and discussion
Suzuki cross-coupling of bromophenol with phenylbornic acid in water at room temperature was chosen as a test reaction to establish guidelines and limitations for the optimization of our protocol.21 We studied the influence of a variety of parameters on product yields such as catalyst loading, substrate concentrations and stoichiometry.Optimization of the protocol
When studying the time-dependence of coupling yields, it became evident that at reaction times shorter than 16 h product yields were not reproducible. Also varying induction periods of 2–6 h were observed for the formation of the biarylic products. Reaction times and reproducibility were improved dramatically by stirring the catalyst with the boronic acid in aqueous buffer vigorously for 30 min at rt before addition of the arene halide. This optimized protocol nearly eliminated the induction period and allows substantially shorter reaction times (Fig. 1). For the coupling of phenylboronic acid with 4-bromophenol, yields of 85% after 1 h and quantitative conversion after less than 5 h were achieved.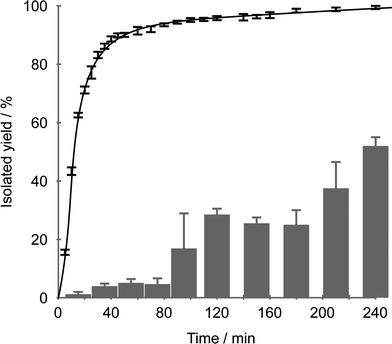 | ||
| Fig. 1 Yield of 4-phenylphenol as function of time using 0.02 mol% of complex 2 as the pre-catalyst in water at rt. Bars: average of 3 runs, co-addition of bromophenol and phenylboronic acid. Curve: average of 2 runs after sequential addition of phenylboronic acid followed by bromophenol. | ||
As illustrated by the widely accepted mechanism of Suzuki–Miyaura cross coupling catalysis, the pH of the reaction media strongly influences catalyst activity.24 Hence, we tested catalyst activity in various aqueous buffered solutions which were prepared from a 1 M NaHCO3 and 5 M NaOH solution and adjusted to the desired pH, covering a range from pH 9 to 11.
As shown in Fig. 2 yields increase for higher pH values until full conversion (5 h reaction time) is reached at pH 10.5. Instead of a 1.0 M buffer, 2.5 eq. of Na2CO3 in neat water (pH 11.7) as base are sufficient for quantitative formation of the coupling product. Thus, the concentration of salt in water could be considerably lowered to 0.25 mol L−1, which further increased the sustainability and environmental compatibility of the protocol.
 | ||
| Fig. 2 Yield of 4-phenylphenol as function of varying pH (left) and substrate concentrations (right) using 0.02 mol% of complex 2 as pre-catalyst in water at rt. | ||
During our studies we also discovered a distinct influence of substrate concentration on catalytic activity (Fig. 2). While decreasing yields at low substrate concentrations are expected due to the typical 1st order dependence in aryl halide concentration of cross-coupling catalysis, high substrate concentrations lead to the formation of a solid product phase at low conversions. We reason, that the lipophilic aryl halide and catalyst may be trapped in the product phase which leads to reduced yields due to diffusion limited availability of the hydrophilic boronic acid within this phase. As a consequence, the substrate concentration has to be chosen such that formation of the product layer, which facilitates workup by filtration, occurs close to quantitative conversion only.
Addition of the small quantities of pre-catalyst, which are required for the reaction, pose a significant experimental difficulty. Hence, we investigated the possibility of adding the catalyst from a stock solution. Interestingly, we discovered, that already the presence of small quantities of organic co-solvents dramatically reduces the observed activity.
While in neat water quantitative conversion is observed, among the eight different organic solvents that were tested in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with water, only methanol, dioxane and acetone gave reasonable yet reduced yields (Table 1). For toluene, acetonitrile and DMSO no conversion could be observed under these conditions. Surprisingly catalyst 2 shows low (methanol) to no catalytic activity (all other solvents tested) when the cross-coupling is performed in purely organic reaction media. This observation suggests a crucial role of water for the transformation of 2 into the catalytically active species. Also, a reduced solubility of the required base in organic solvents may be partially responsible for the observed loss in activity.
:1 ratio with water, only methanol, dioxane and acetone gave reasonable yet reduced yields (Table 1). For toluene, acetonitrile and DMSO no conversion could be observed under these conditions. Surprisingly catalyst 2 shows low (methanol) to no catalytic activity (all other solvents tested) when the cross-coupling is performed in purely organic reaction media. This observation suggests a crucial role of water for the transformation of 2 into the catalytically active species. Also, a reduced solubility of the required base in organic solvents may be partially responsible for the observed loss in activity.
| Entry | Solvent | Yield [%] using additive | |
|---|---|---|---|
| 50%a | 100%b | ||
| Reaction conditions: 0–1 ml water, 1–2 ml organic solvent, rt, air, [2] = 0.02 mol%, 2.5 eq. Na2CO3, reaction time 16 h, [Ar–Br] = 0.1 mol L−1.a 1.25 eq. crown ether.b 2.5 eq crown ether. | |||
| 1 | Methanol | 84 | 20 |
| 2 | Dimethylsulfoxide | 0 | 0 |
| 3 | Toluene | 0 | 0 |
| 4 | Acetonitrile | 0 | 0 |
| 5 | Dioxane | 71 | 0 |
| 6 | Tetrahydrofurane | 35 | 0 |
| 7 | Dimethylformamide | 13 | 0 |
| 8 | Acetone | 86 | 0 |
Scope of the protocol
Using catalyst 2 a broad range of substrates can be converted in the Suzuki–Miyaura reaction when the optimized protocol is applied. In total a combination of eight different aryl halides and thirteen boronic acids were coupled. Among all possible combinations yields of 52 coupling reactions are presented in Table 2.| Reaction conditions: 1.0 eq. aryl halide, 1.0 eq. boronic acid, 2 ml water, 2.5 eq. Na2CO3, rt, 6 h, air, 0.02 mol% palladacycle 2.a Yields in brackets were determined by GC analysis. | |||
|---|---|---|---|

|
|||
| Entry | Product | Isolated yield (%) | |
| X = Br | X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) I I |
||
| 1 |

|
≥99 | (≥99)a |
| 2 |

|
96 | (92)a |
| 3 |

|
≥99 | |
| 4 |

|
≥99 | ≥99 |
| 5 |

|
(79)a | |
| 6 |

|
(86)a | (81)a |
| 7 |
|
98 | (84)a |
| 8 |

|
90 | |
| 9 |

|
≥99 | |
| 10 |

|
93 | (89)a |
| 11 |

|
83 | |
| 12 |

|
(84)a | (78)a |
| 13 |

|
81 | (75)a |
| 14 |
|
(84)a | |
| 15 |
|
(68)a | |
| 16 |

|
31 | |
| 17 |
|
81 | |
| 18 |
|
49 | |
| 19 |

|
97 | (97)a |
| 20 |

|
83 | (86)a |
| 21 |

|
72 | |
| 22 |
|
(98)a | (88)a |
| 23 |

|
(≥99)a | |
| 24 |

|
(94)a | (87)a |
| 25 |

|
65 | |
| 26 |

|
(≥99)a | (95)a |
| 27 |

|
(86)a | |
| 28 |

|
(82)a | |
| 29 |
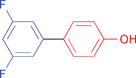
|
(82)a | (90)a |
| 30 |

|
65 | |
| 31 |

|
92 | |
| 32 |
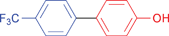
|
≥99 | |
| 33 |

|
72 | |
| 34 |

|
≥99 | |
| 35 |

|
≥99 | |
| 36 |

|
93 | |
| 37 |
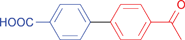
|
88 | 88 |
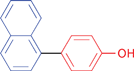
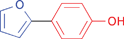
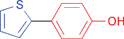
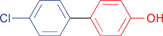
A variety of functionalities are tolerated for the aryl bromide including aldehyde, ketone, carboxylic and hydroxylic groups. Furthermore, a diverse choice of arylboronic acids bearing methyl, chlorine, fluorine, carboxylic, methoxy, and 3,4-methylendioxy substituents were successfully coupled in the Suzuki–Miyaura reaction. Notably, also sterically more demanding substrates such as naphthylboronic acid could be applied. The optimized protocol is suited to couple electron-deficient as well as electron-rich aryl iodides or bromides with arylboronic acids. Among the heteroaromatic boronic acids (Table 2, entries 16, 17 and 18) only the combination of 2-furanylboronic acid and iodophenol was coupled in good yields.
Aryl chlorides in general and non-polar aryl bromides, such as bromobenzene or bromoanisol, could not be converted at 30 °C. Using the standard substrate concentration of 0.1 mol L−1, 27 coupling products can be isolated in good to excellent yields and high purity by simple filtration. In order to extend this isolation protocol to a broader range of products, the reaction parameters may need to be optimized for each case.
So far, we have presented an efficient and simple protocol for Suzuki–Miyaura cross coupling reactions under mild and environmentally benign conditions, which was developed based on current knowledge of the reaction and optimized by screening procedures.
Catalyst structure and active species
To further improve the method, establish a catalyst recycling procedure and to transfer the tempting simplicity of the approach to other reactions, a better understanding of the structure of the catalytically active species is required. Hence, we first turned our attention to the structural characterization of palladacycle 2. Crystals suitable for X-ray diffraction were grown from a THF/pentane mixture. In contrast to similar Bedford-type palladacycles,22 both the trans- and the cis-isomers of 2 are present in the unit cell (Fig. 3a and 3b). All bond length and angles are within the expected range and in good agreement with literature values of similar compounds.22 | ||
| Fig. 3 (a) Molecular structure of complex cis-2. Selected bond lengths (Å) and angles (°): Pd1–C1 2.010(4), Pd1–P1 2.1800(9), Pd1–Cl1 2.4388(9); P1–Pd1–C1 80.21(10), C1–Pd1–Cl1 98.47(10), P1–Pd1–Cl1 178.25(4). (b) Molecular structure of complex trans-2. Selected bond lengths (Å) and angles (°): Pd3–C37 2.005(4), Pd3–P3 2.1926(10), Pd3–Cl4 2.4392(9); P3–Pd3–C37 79.63(11), C37–Pd3–Cl4 97.34(11), P3–Pd3–Cl4 176.66(3). | ||
A distinct feature of both isomers, however, is the sterically crowded environment of the oxygen atom, which is bridging the phosphite and the arene moiety of the ligand. This is induced by the phenyl substituent in the 2-position of the palladated arene ring as well as the bulky iso-propyl groups at the phosphorous atom. The particular steric and electronic environment of the P–O bond may explain, why palladacycle 2 excels in comparison to all other catalyst precursors tested. The phosphorous centre is rich in electron density favouring oxidative addition of the haloarene and a well-balanced steric bulk stabilizes the palladacyclic motive. On the one hand, a further increase in steric bulk induces strain destabilizing the palladacyclic structure, which might explain why either introduction of tert-butyl substituents or replacement of the oxygen atom by a methylene moiety significantly reduces the activity observed.21 On the other hand, the phosphorous centre is embedded in-between the 4-phenyl and the isopropyl substituent and thus well shielded against cleavage by nucleophilic attack. Hence, hydrolysis of the phosphite moiety is very slow and the catalytically active palladacyclic moiety is highly stable under reaction conditions (see below). Correspondingly, decreased steric shielding increases hydrolysis rates of 2 and decreased activities are observed, since the hydroxyl diisopropylphosphine ligand 3 resulting from hydrolysis is much less effective in stabilizing the active species. This is confirmed by low yields when a mixture of 3 and PdCl2 is used to catalyse aqueous the Suzuki–Miyaura cross coupling reaction.21 Also, when preformed with complex 425 only conversions below 50% were reached.
In solution both isomers of 2 are present and can be identified by NMR spectroscopy. E.g. two signals are evident in the 31P NMR spectrum at 202.8 and 201.7 ppm with a ratio of 1:1.5.

An ongoing discussion has developed regarding the nature of the active species in cross-coupling reactions catalyzed by palladacycles. Particularly the ‘classical’ Pd(0)/Pd(II) pathway has been challenged by some indications for a catalytic cycle involving Pd(IV).26 However, most experimental evidences point towards the conventional Pd(0)/Pd(II) mechanism, based on a highly active, zero valent and under-coordinated palladium species formed in situ by reductive processes.27 Reductive elimination of the palladated ligand aryl group and an aryl moiety originating from transmetalation of a boronic acid nucleophile has been discussed as a possible mechanism for the formation of a Pd(0) species from a palladacyclic catalyst precursor.22
In order to establish whether and how complex 2 is transformed into an active Pd(0) catalyst under these reaction conditions, we investigated the stability of this palladacycle in water and its reactivity with components of the Suzuki reaction using 31P NMR and GC-MS analysis. Addition of aqueous buffer to the catalyst does not result in significant decomposition at room temperature, and after 64 h less than 6% was hydrolysed. However, addition of phenylboronic acid to compound 2 and stirring at reflux for 72 h in aqueous buffer resulted only in the formation of trace amounts (<1%) of the 2,6-arylated ligand. These observations imply that the palladacyclic structure remains mostly unaltered under reaction conditions. However, considerable amounts of biphenyl were formed even at room temperature. Hence, we suggest that addition of phenylboronic acid induces first a double transmetallation of phenyl groups, followed by a reductive elimination of biphenyl resulting in an under-coordinated, palladacyclic, anionic Pd(0) species as illustrated in Scheme 2. This is in agreement with the observation, that pre-treatment of the catalyst with arylboronic acid eliminates the induction period of the reaction and leads to more reproducible results.
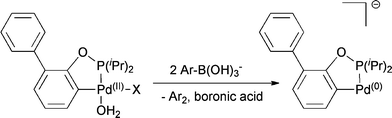 | ||
| Scheme 2 Suggested pathway for the formation of an active, palladacyclic, anionic Pd(0) species. | ||
When the stability of 2 in water was examined in the presence of phenylboronic acid, addition of small amounts of THF resulted in the immediate formation of Pd-black. We assume, that addition of the organic co-solvent solubilizes the catalyst (the pre-catalyst 2 is virtually insoluble in water) and thus increases the concentration of the under-coordinated Pd(0) species in solution. Formation of Pd-black is 2nd order in Pd(0) and therefore sensitive to Pd(0) concentration. We reason, that the solubilising effect of organic co-solvents might therefore to some extent be responsible for the activity reduction observed, if the active catalyst consists of a palladacycle (as suggested in Scheme 2) rather than Pd- nanoparticles or Pd-black.
To further support this conclusion, we performed a mercury drop test, which is widely used to exclude catalysis by nanoparticles. Amalgamation should only deactivate heterogeneous metal particle catalyst, yet is not expected to occur for ligated homogenous Pd(II) species.28 Despite addition of 400 eq. of mercury to the catalytic reaction yields remained as high as 72%. The loss in activity can be attributed to the elimination of temporarily emerging Pd(0) species within the catalytic cycle of the Suzuki coupling. Another suitable experiment to examine the formation of palladium nanoparticles involves poisoning by thiophene addition. A substantial decrease in yields would be expected if metallic palladium contributes to the catalytic activity. Nevertheless, addition of 400 eq. of thiophene did not affect the catalysts performance and quantitative yields for the Suzuki coupling product were observed. Accordingly, these poisoning experiments present a strong evidence for a homogenous nature of the catalytically active species, presumably a palladacycle and no indication of formation of palladium nanoparticles was found.
Catalyst recycling
Concluding that the palladacyclic structure of 2 remains intact under reaction conditions of our optimized protocol we assumed that it should be possible to recycle the catalyst from the aqueous reaction filtrate. Recycling experiments were performed using the standard bromophenol/phenylboronic coupling test reaction and simple filtration of the reaction mixture for isolation of the coupling product. Substrates were added again to the reused aqueous filtrate, which was stirred for another 6 h (Table 3).| Entry | Catalytic run | Yield [%] |
|---|---|---|
| Reaction conditions: 1.0 eq. bromophenol, 1.0 eq. phenyl boronic acid, 2.5 eq. Na2CO3, 0.02 mol% palladacycle 2, rt, air, reaction time 6 h. | ||
| 1 | 1 | ≥99 |
| 2 | 2 | 99 |
| 3 | 3 | 94 |
| 4 | 4 | 88 |
| 5 | 5 | 71 |
After the first recycling step full catalytic activity is preserved and a considerable loss in activity is observed for only after the 4th run. Correspondingly several cycles of catalyst use and reuse are possible, despite the fact that salt by-products accumulate in the solution. This finding is not only a further indication for the homogenous nature of the active catalyst, reuse is also most desirable in terms of environmental sustainability.
Since catalyst recycling is possible, the majority of the palladium added should be found in the aqueous filtrate rather than in the isolated products. Pd-concentrations of the biarylic coupling products support this assumption. ICP-OES analysis revealed Pd contaminations between 1.1 and 28.6 ppm depending on the nature of the product and the filtration protocol used. Most effective for reduction of Pd contaminations proved to be a thorough squeezing of the product between two filter papers until all remaining moisture was removed. In the reaction mixture, the initial concentration of Pd relative to the substrates is roughly 200 pm. Therefore less than 15% of the catalyst remains in the product.
Conclusions
In conclusion we report an environmentally friendly reaction protocol for aqueous Suzuki–Miyaura cross-coupling catalysis under air at rt, which is applicable to a broad range of substrates. Since no organic co-solvents or other additives are required, isolation of substrates in excellent yields and purity can be achieved in most cases using simple filtration. Pre-treatment of the catalyst with aryl boronic acid significantly shortens the reaction times required. The palladacyclic structure of the pre-catalyst was found to be stable under reaction conditions. Correspondingly, recycling of the active catalytic species can be performed several times and the palladium contaminations of the isolated products are in the range of levels tolerated for drug substances (5 ppm).29 Moreover, it has been shown that the addition of organic co-solvents partially or fully deactivates the catalyst. This shines a new light on many aqueous protocols, which require both, organic co-solvents and elevated temperature. It may well be that the elevated temperature is very often only required since the catalyst activity is reduced by the addition of an organic solvent. Correspondingly, pure water might be the solvent of choice not only for Suzuki–Miyaura cross-coupling reactions.Experimental section
Materials and methods
All experiments were carried out under an atmosphere of argon using standard Schlenk and glove-box techniques. Toluene was dried over Na/benzophenone, distilled under argon and deoxygenated prior to use. Ethanol was dried over Na/diethylphtalate, distilled under argon and deoxygenated prior to use. Pentane was dried and deoxygenized by passing through columns packed with activated alumina and Q5. Deuterated solvents were dried by distillation from CaH2 (CD2Cl2 and CDCl3) and deoxygenated by three freeze-pump-thaw cycles. NEt3 was purchased from Merck and dried over CaH2, distilled under argon and deoxygenated prior to use. PdCl2 (ABCR) was used as purchased. Compounds 1 and 2 were synthesized according to the procedure published previously.21Analytical methods
Determination of palladium concentrations was performed by ICP-OES, Dr. Dennis Jurkin, Lehrstuhl für Radiochemie, Technische Universität München. Calibration against palladium standards and a blank provided a linear relation of signal to concentration. Weighed samples of the coupling products were digested in concentrated sulfuric and nitric acid in a microwave prior to injection into the ICP-OES instrument. NMR spectra were recorded on Jeol Lambda 400 spectrometer. GC-yields of the catalytic reactions were determined using a Varian CP 3800 (1200 L MS (single quad.)) and a Varian CP3800 (1200 L MS/MS (triple quad.)).Procedures
Single-crystal X-ray structure determination
Acknowledgements
We are grateful to the Elitenetzwerk Bayern (graduate fellowship for A.N.M.), KAUST (graduate fellowship for A.N.M. and D.J.) and the IDK NanoCat for funding of this project. Experimental support by Markus Scheibel is gratefully acknowledged. Finally we are particularly grateful to the Aramco PMT team, KAUST HSE and KAUST Procurement, who all worked very hard for more than a year to finishing our research laboratories.Notes and references
- (a) Green Chemistry: Theory and Practice ed. P. T. Anastas and J. C. Warner, Oxfordy University Press, Oxford, UK, 2000 Search PubMed; (b) V. Polshettiwar and R. S. Varma, Curr. Opin. Drug Discovery Dev., 2007, 10, 723–737 CAS; (c) I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2167–2168 CrossRef CAS.
- (a) I. T. Horváth, Green Chem., 2008, 10, 1024–1028 RSC; (b) C.-J. Li, Acc. Chem. Res., 2002, 35, 533–538 CrossRef CAS.
- V. Polshettiwar and R. Varma, Chem. Soc. Rev., 2008, 37, 1546–1557 RSC; V. Polshettiwar, A. Decottignies, C. Len and A. Fihri, ChemSusChem, 2010, 3, 502–522 CrossRef CAS.
- I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 107, 2169–2173 CrossRef CAS.
- (a) Organic Reactions in Aqueous Media, ed. C.-J. Li and T.-H. Chan, Wiley, New York, 1997 Search PubMed; (b) Organic Synthesis in Water ed. P. A. Grieco, Academic Press, Dordrecht, The Netherlands, 1997 Search PubMed; (c) Aqueous-Phase Organometallic Catalysis, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, Weinheim, 2nd edn, 2004 Search PubMed; (d) K. H. Shaughnessy and R. B. DeVasher, Curr. Org. Chem., 2005, 9, 585 CrossRef CAS.
- C.-J. Li, Chem. Rev., 2005, 105, 3095–3165 CrossRef CAS.
- A. L. Casalnuovo and J. C. Calabrese, J. Am. Chem. Soc., 1990, 112, 4324 CrossRef.
- M. Beller, J. G. E. Krauter, A. Zapf and S. Bogdanovic, Catal. Today, 1999, 48, 279–290 CrossRef CAS; M. Beller, J. G. E. Krauter and A. Zapf, Angew. Chem., Int. Ed. Engl., 1997, 36, 772–774 CrossRef CAS.
- K. W. Anderson and L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS.
- J. P. Genêt, A. Linquist, E. Blart, V. Mouriès and M. Savignac, Tetrahedron Lett., 1995, 36, 1443–1446 CrossRef CAS; C. Dupuis, K. Adiey, L. Charruault, V. Michelet, M. Savignac and J. P. Genêt, Tetrahedron Lett., 2001, 42, 6523–6526 CrossRef CAS.
- N. E. Leadbeater, Chem. Commun., 2005, 2881 RSC.
- M. Ueda, M. Nishimura and N. Miyaura, Synlett, 2000, 6, 856–858.
- C. A. Fleckenstein and H. Plenio, Green Chem., 2007, 9, 1287–1291 RSC; C. A. Fleckenstein, S. Roy, S. Leuthäußer and H. Plenio, Chem. Commun., 2007, 2870–2872 RSC; C. A. Fleckenstein and H. Plenio, Chem.–Eur. J., 2007, 13, 2701–2716 CrossRef CAS; C. A. Fleckenstein and H. Plenio, J. Org. Chem., 2008, 73, 3236–3244 CrossRef CAS; C. A. Fleckenstein and H. Plenio, Chem.–Eur. J., 2008, 14, 4267–4279 CrossRef CAS.
- L. R. Moore and K. H. Shaughnessy, Org. Lett., 2001, 3, 2757–2759 CrossRef CAS; L. R. Moore and K. H. Shaughnessy, Org. Lett., 2004, 6, 225–228 CrossRef CAS; R. Huang and K. H. Shaughnessy, Organometallics, 2006, 25, 4105–4112 CrossRef; L. R. Moore, E. C. Western, R. Craciun, J. M. Spruell, D. A. Dixon, K. P. O'Halloran and K. H. Shaughnessy, Organometallics, 2008, 27, 576–593 CrossRef CAS.
- (a) Green Chemistry and Catalysis ed. R. A. Sheldon, I. Arden and U. Hanefeld, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (b) B. H. Lipshutz and A. R. Abela, Org. Lett., 2008, 10, 5329–5332 CrossRef CAS; B. H. Lipshutz, T. B. Petersen and A. R. Abela, Org. Lett., 2008, 10, 1333–1336 CrossRef CAS.
- (a) C. Nájera, J. Gil-Moltó, S. Karlström and L. R. Falvello, Org. Lett., 2003, 5, 1451–1454 CrossRef CAS; C. Nájera, J. Gil-Moltó and S. Karlström, Adv. Synth. Catal., 2004, 346, 1798–1811 CrossRef CAS; L. Botella and C. Nájera, Angew. Chem., Int. Ed., 2002, 41, 179–181 CrossRef CAS; K. Botella and C. Nájera, Angew. Chem., 2002, 114, 187–189 CrossRef; D. A. Alonso, C. Nájera and M. C. Pacheco, J. Org. Chem., 2002, 67, 5588–5594 CrossRef CAS; L. Botella and C. Nájera, J. Organomet. Chem., 2002, 663, 46–57 CrossRef CAS; D. A. Alonso, L. Botella, C. Nájera and M. C. Pacheco, Synthesis, 2004, 10, 1713–1718; (b) R. B. DeVasher, L. R. Moore and K. H. Shaughnessy, J. Org. Chem., 2004, 69, 7919–7927 CrossRef CAS; R. B. DeVasher, J. M. Spruell, D. A. Dixon, G. A. Broker, R. D. Rogers and K. H. Shaughnessy, Organometallics, 2005, 24, 962–971 CrossRef CAS.
- (a) R. K. Arvela and N. E. Leadbeater, Org. Lett., 2005, 7, 2101–2104 CrossRef; (b) K. M. Dawood, Tetrahedron, 2007, 63, 9642–9651 CrossRef CAS; (c) V. Polackova and S. Toma, Chem. Pap., 2007, 61, 41–45 Search PubMed; (d) A. Cohen, M. D. Cozet, P. Rathelot and P. Vanelle, Green Chem., 2009, 11, 1736–1742 RSC; (e) V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629–639 CrossRef CAS; (f) V. Polackova, M. Hutka and S. Toma, Ultrason. Sonochem., 2005, 12, 99–102 CrossRef CAS; (g) J. Zhang, F. Yang, G. Ren, T. C. W. Mak, M. Song and Y. Wu, Ultrason. Sonochem., 2008, 15, 115–118 CrossRef CAS.
- L. Liu, Y. Zhang and B. Xin, J. Org. Chem., 2006, 71, 3994–3997 CrossRef CAS; Y. Han, H. V. Huynh and G. K. Tan, Organometallics, 2007, 26, 6581–6585 CrossRef CAS; T. Brendgen, M. Frank and J. Schatz, Eur. J. Org. Chem., 2006, 2378–2383 CrossRef CAS.
- A. M. Sheloumov, P. Tundo, F. M. Dolgushin and A. A. Koridze, Eur. J. Inorg. Chem., 2008, 572–576 CrossRef CAS.
- B. Inés, R. SanMartin, F. Churruca, E. Domínguez, M. K. Urtiaga and M. I. Arriortua, Organometallics, 2008, 27, 2833–2839 CrossRef CAS; B. Inés, I. Moreno, R. SanMartin and E. Domínguez, J. Org. Chem., 2008, 73, 8448–8451 CrossRef CAS.
- A. N. Marziale, Stefan H. Faul, T. Reiner, S. Schneider and J. Eppinger, Green Chem., 2010, 12, 35–38 RSC.
- R. B. Bedford and S. L. Welch, Chem. Commun., 2001, 129–130 RSC; R. B. Bedford, S. L. Hazelwood (née Welch) and M. E. Limmert, Chem. Commun., 2002, 2610–2612 RSC; R. B. Bedford, C. S. J. Cazin and S. L. Hazelwood (née Welch), Angew. Chem., Int. Ed., 2002, 41, 4120–4122 CrossRef CAS; R. B. Bedford, S. L. Hazelwood (née Welch), P. N. Horton and M. B. Hursthouse, Dalton Trans., 2003, 4164–4174 RSC; R. B. Bedford and M. E. Blake, Adv. Synth. Catal., 2003, 345, 1107–1110 CrossRef CAS; R. B. Bedford, Chem. Commun., 2003, 1787–1796 RSC.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS; S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275–3279 CrossRef CAS; E. J. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249–1262 RSC.
- S. Ogo, Y. Takebe, K. Uehara, T. Yamazaki, H. Nakai, Y. Watanabe and S. Fukuzumi, Organometallics, 2006, 25, 331–338 CrossRef CAS.
- A. J. Carty, S. E. Jacobson, R. T. Simpson and N. J. Taylor, J. Am. Chem. Soc., 1975, 97, 7254–7262 CrossRef CAS.
- W. A. Herrman, V. P. W. Böhm and C.-P. Reisinger, J. Organomet. Chem., 1999, 576, 23 CrossRef CAS.
- J. Louie and J. F. Hartwig, Angew. Chem., Int. Ed. Engl., 1996, 35, 2359 CrossRef CAS.
- B. Inés, R. SanMartin, M. J. Moure and E. Domínguez, Adv. Synth. Catal., 2009, 351, 2124–2132 CrossRef CAS.
- C. E. Garrett and K. Prasad, Adv. Synth. Catal., 2004, 346, 889–900 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic data or references for all products. CCDC reference numbers: 788105 (2), 788104 (4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0gc00522c |
| This journal is © The Royal Society of Chemistry 2011 |