Glucoraphanin does not reduce plasma homocysteine in rats with sufficient Se supply via the induction of liver ARE-regulated glutathione biosynthesis enzymes
Nicole M.
Blum
a,
Kristin
Mueller
a,
Frank
Hirche
a,
Doris
Lippmann
a,
Erika
Most
b,
Josef
Pallauf
b,
Thomas
Linn
c and
Andreas S.
Mueller
*a
aInstitute of Agricultural and Nutritional Sciences, Preventive Nutrition Group, Martin Luther University Halle Wittenberg, Von Danckelmann Platz 2, D-06120, Halle (Saale), Germany. E-mail: andreas.mueller@landw.uni-halle.de
bInterdisciplinary Research Centre, Institute of Animal Nutrition and Nutritional Physiology, Justus Liebig University Giessen, Heinrich Buff Ring 26-32, D-35392, Giessen, Germany
cMedical Clinic for Internal Medicine, Endocrinology and Diabetes, Justus Liebig University Giessen, Rodthohl 6, D-35392, Giessen, Germany
First published on 29th September 2011
Abstract
Data from human and animal trials have revealed contradictory results regarding the influence of selenium (Se) status on homocysteine (HCys) metabolism. It was hypothesised that sufficient Se reduces the flux of HCys through the transsulphuration pathway by decreasing the expression of glutathione (GSH) synthesising enzymes. Glucoraphanin (GRA) is a potent inducer of genes regulated via an antioxidant response element (ARE), including those of GSH biosynthesis. We tested the hypothesis that GRA supplementation to rat diets lowers plasma HCys levels by increasing GSH synthesis. Therefore 96 weaned albino rats were assigned to 8 groups of 12 and fed diets containing four different Se levels (15, 50, 150 and 450 μg kg(diet)−1), either without GRA (groups: C15, C50, C150 and C450) or in combination with 700 μmol GRA kg(diet)−1 (groups G15, G50, G150 and G450). Rats fed the low Se diets C15 and G15 showed an impressive decrease of plasma HCys. Se supplementation increased plasma HCys and lowered GSH significantly by reducing the expression of GSH biosynthesis enzymes. As new molecular targets explaining these results, we found a significant down-regulation of the hepatic GSH exporter MRP4 and an up-regulation of the HCys exporter Slco1a4. In contrast to our hypothesis, GRA feeding did not reduce plasma HCys levels in Se supplemented rats (G50, G150 and 450) through inducing GSH biosynthesis enzymes and MRP4, but reduced their mRNA in some cases to a higher extent than Se alone. We conclude: 1. That the long-term supplementation of moderate GRA doses reduces ARE-driven gene expression in the liver by increasing the intestinal barrier against oxidative stress. 2. That the up-regulation of ARE-regulated genes in the liver largely depends on GRA cleavage to free sulforaphane and glucose by plant-derived myrosinase or bacterial β-glucosidases. As a consequence, higher dietary GRA concentrations should be used in future experiments to test if GRA or sulforaphane can be established as HCys lowering compounds.
Introduction
A large number of human and animal studies has associated a high level of total plasma homocysteine (tHCys) with an increased risk for the accelerated onset of coronary artery disease, myocardial infarction and ischemic stroke.1–5 Several mechanisms, including the generation of reactive oxygen species, an increase in LDL oxidation, the liberation of pro-inflammatory transcription factors and cytokines, and a reduction of vessel relaxation via a decreased NO availability are discussed as molecular mechanisms underlying the hypertensive and proatherogenic effects of HCys.5–9 Besides the inherited disorders cystathionine-β-synthase (CBS) deficiency and methionine synthase deficiency, a high dietary methionine intake and other factors, like age, sex and race, can trigger hyperhomocysteinemia.10–14 Whereas a number of studies has shown an effective reduction of plasma tHCys by combined vitamin B6, B9 (folic acid) and B12 supplementation, benefits of these supplements on risk reduction, recurrence of associated diseases, and on CVD- and ischemic stroke-endpoints are recently in doubt.15 Nevertheless, taking a standard multivitamin preparation to achieve the recommended vitamin B6, B9 and B12 amounts is advised to risk patients by the international professional societies.16,17Conflicting results have also been reported with regard to the influence of selenium (Se) on HCys metabolism. Studies with chicks, mice and rats have shown that Se deficiency reduced plasma tHCys concentration impressively compared to animals with adequate or slightly supranutritive Se supply.18–21 Quite in contrast, data collected from human cross-sectional observational studies in different countries uniquely demonstrated an inverse correlation between Se status and plasma tHCys concentration.22–24 Accordingly, a recent U.S. cross sectional observational study clearly confirmed the above mentioned inverse relation between Se status and plasma tHCys.25 Opposite to these studies, Se intervention in humans with 100–300 μg selenomethionine day−1 for 3 to 6 months produced no effects on plasma tHCys compared to the placebo treated controls.26,27 Nevertheless, in a rat study plasma HCys concentration in Se supplemented rats could be lowered by the addition of a high folic acid concentration to the diet.28 In a current study, no effects of high Se supplementation for 48 weeks could be measured on brachial artery occlusion parameters of healthy young men in comparison with placebo treated study participants. However, in the placebo group plasma tHCys concentration declined from 7.2 μmol L−1 to 6.4 μmol L−1, whereas in the intervention group a minimal increase from 7.1 μmol L−1 to 7.2 μmol L−1 was observed.29 These data confirm information from the rat trials and contradict data from the above mentioned human studies. In analogy to variations in Se status, another study with elderly people found a positive correlation between plasma vitamin C and plasma tHCys.30 These findings were supported by the results of a study in which participants with a high Trolox Equivalent Antioxidant Capacity (TEAC) in plasma came along with increased plasma tHCys levels.31
An in vitro study with HepG2 cells showed that HCys flux through the transsulphuration pathway and subsequent GSH biosynthesis was stimulated by prooxidants, whereas the addition of various antioxidants to the culture media reduced GSH biosynthesis due to diminished HCys utilisation.32 A similar hypothesis regarding HCys metabolism has been also postulated for Se: “The up-regulation of GSH biosynthesis in Se deficiency produces a pull on the transsulphuration pathway via an increased HCys utilization.”19 Both the GSH biosynthesis enzymes glutamate cysteine ligase (GCL) and glutathione synthase (GS), and a number of phase II enzymes, like glutathione-S-transferases (GSTs), nicotinamide-adenine-dinucleotide-(phosphate)-quinone-oxidase 1 (NQO1) and heme oxygenase 1 (HO1) share the existence of an antioxidant response element (ARE) in their DNA promoter region as a common feature.33 Increased oxidative stress, as present in Se deficiency or under conditions of high Se supply,34,35 and electrophilic isothiocyanates, like sulforaphane from cruciferous vegetables potently induce phase II-and GSH biosynthesis enzymes.36,37 When cells are sufficiently protected against oxidative stress the transcription factor NF-E2-related factor 2 (Nrf2) is bound in the cytosol to the Kelch-like ECH-associated protein 1 (KEAP1), and it is presented for ubiquitination and proteasomal degradation. Both oxidative stress and electrophilic isothiocyanates modify KEAP1 sensor-SH-residues, effecting Nrf2 liberation, its nuclear translocation and its association with the DNA-ARE sequences of target genes. As a consequence, transcription and translation of GSH biosynthesis enzymes and of phase II enzymes increases.36Sulforaphane has been demonstrated to act as a potent inducer of phase II enzymes, in particular in the intestine.36 Results of very recent studies, in which rat liver and lung slices were incubated with the sulforaphane glucosinolate precursor glucoraphanin (GRA), revealed that GRA has a comparable potential to induce ARE-regulated enzymes like pure sulforaphane.38,39
The aim of our study was to investigate if feeding a diet containing GRA can reduce the higher HCys levels as present under Se sufficiency or when Se is applied in slightly supranutritive concentrations via the induction of GSH biosynthesis enzymes (Fig. 1).
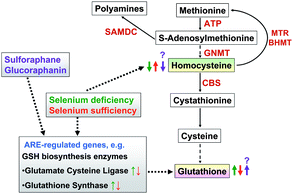 | ||
| Fig. 1 We studied the hypothesis that GRA supplementation to rat diets lowers plasma HCys, which is high under Se sufficiency due to a reduced GSH synthesis. | ||
Experimental
Animals and diets
96 healthy weaned male albino rats (mean body weight: 71.9 ± 2.26 g) from the Interdisciplinary Research Center, Department of Animal Nutrition and Nutritional Physiology's own strain HK51 were randomly assigned to eight groups of 12 (C15, C50, C150, C450, G15, G50, G150 and G450).The Se deficient basal diet of group C15 was based on Torula yeast and Se deficient wheat. Its composition has been previously described in detail.12 The analysed Se concentration of the Se deficient basal diet was approximately 15 μg kg(diet)−1. The diets of groups C50, C150 and C450 were supplemented with 50 μg Se kg(diet)−1 (one third of the recommended level), 150 μg Se kg(diet)−1 (recommended level) and 450 μg Se kg(diet)−1 (three times the recommended level) as sodium selenate. Dietary Se concentrations of groups G15, G50, G150 and G450 were identical to those in the corresponding C groups. Broccoli extract (Jarrow-Formulas®) was additionally added to the diets of the G groups at a level of 3000 mg kg−1 diet, providing 300 mg GRA kg(diet)−1 (=700 μmol GRA kg(diet)−1). With the exception of Se and GRA, the diets were composed according to the American Institute of Nutrition-93G recommendations.40 The rats were housed individually and had ad libitum access to their respective diets and water. After 8 weeks the animals were decapitated under CO2 anesthesia. Liver samples for gene expression analyses and enzymatic determinations were immediately excised, transferred into snap tubes, frozen in liquid nitrogen, and stored at −80 °C until further analysis. Blood was collected in heparinised tubes and centrifuged for 20 min at 3000g for plasma preparation. Plasma was stored at −80 °C until analysis.
All experiments with live animals were performed according to the German Animal Welfare Act. The protocol of this rat nutrition study was approved by the Regional Council of Giessen and by the Animal Welfare Committee of the Justus Liebig University Giessen (Germany) [record token: V54-19c20/15cGI 19/3; 39-2008A].
Se assay
Se concentrations in diets and plasma were analyzed by hydride generation atomic absorption spectrometry (Unicam PU 9400 X; PU 3960 X), as described previously. Soft winter wheat starch (No. 8438, National Institute of Standard and Technology) and control serum (Metalle S, Medichem, Steinenbronn, Germany) were used as reference materials.41Activity of liver glutathione peroxidase 1 and of plasma glutathione peroxidase 3
The activity of liver cytosolic glutathione peroxidase (GPx1) and of plasma glutathione peroxidase (GPx3) was measured spectrophotometrically (Ultrospec 3300 pro, Amersham Pharmacia Biotech, Freiburg, Germany) at 340 nm using the assay protocol coupled to glutathione reductase (GR) and NADPH.42NADPH oxidation, which is proportional to GPx-dependent peroxide reduction, was recorded for 3 min. For both enzymes H2O2 was used as substrate. One unit of GPx1 and GPx3 was defined as 1 μmol NADPH oxidized per minute and normalized to 1 mg protein.Glutathione and homocysteine concentrations in liver and plasma
The concentration of total glutathione in the liver (tGSH) was analyzed using the spectrophotometrical standard protocol coupled to 5,5′-dithiobis-(2-nitrobenzoic acid) [DTNB] and GR.43GSH concentration of samples was calculated from a standard curve prepared with oxidized glutathione in the concentration range 0–0.066 μmol oxidised glutathione mL−1. Total homocysteine concentration (tHCys) in plasma and liver and total plasma glutathione (tGSH) were analyzed using a modified standard protocol by reversed-phase HPLC, as described in detail previously.12 Liver values for tHCys and tGSH were normalized to 1 mg protein.Protein concentration of samples
Protein concentration in liver cytosol, plasma, and whole liver tissue lysates for Nrf2-immunoblotting was determined in microtitre plates according to a standard protocol using the plate reader Tecan SpectraFluor Plus (Tecan, Grödig, Austria).44Real-time RT-PCR analysis
Liver RNA was extracted with TRIzol ® reagent (Invitrogen, Karlsruhe, Germany) according to the manufacturer's protocol. Total RNA concentration and RNA purity were determined spectrophotometrically at 260 and 280 nm. Following reverse transcription of 3.0 μg total RNA using a commercial cDNA synthesis kit (RevertAid™ First strand synthesis kit, MBI Fermentas,Vilnius, Latvia), RNA of two rats per group was pooled. For use in real-time RT-PCR the cDNA solutions were diluted 2.5-fold (v/v) with sterile diethyl pyrocarbonate (DEPC) treated water. Amplification of specific sequences of the genes investigated were performed in duplicate using the Rotor-Gene 6000™ real-time detection apparatus (Corbett Research, Mortlake, Australia). Information on the genes investigated and primer data are given in Table 1. Amplification data were analyzed with the Rotor-Gene 6000™ series software using the ΔΔCt method.45 The expression of the genes investigated was normalized to β-Actin expression. Prior to this a ranking of expression stability was performed for different housekeeping genes and revealed β-Actin as being the most stable gene in the liver.46 Relative mRNA expression levels are expressed as x-fold changes relative to group C15 = 1.0.| Gene name and (abbreviation used) | Gene bank accession. number | Primer sequences (5′ → 3′) |
|---|---|---|
| for = forward; rev = reverse; TA = annealingT/°C | ||
| Betaine hydroxymethyltransferase (BHMT) | NM_030850 | for: 5′ GCACCAGCTTGCAGACAATA 3′ |
| rev: 5′ TGTGCATGTCCAAACCACTT 3′ | ||
| TA = 55 | ||
| Cystathionine beta synthase (CBS) | NM_012522 | for: 5′ ATGCTGCAGAAAGGCTTCAT 3′ |
| rev: 5′ GCGGTATTGGATCTGCTCAT 3′ | ||
| TA = 55 | ||
| Glutamate-cysteine ligase modifier subunit (GCLM) | NM_017305 | for: 5′ AGGCACCTCGGATCTAGACA 3′ |
| rev: 5′ AAATCTGGTGGCATCACACA 3′ | ||
| TA = 57 | ||
| Glycine N-methyltransferase (GNMT) | NM_017084 | for: 5′ CCACCGCAACTACGACTACA 3′ |
| rev: 5′ TCTTCTTGAGCACGTGGATG 3′ | ||
| TA = 55 | ||
| Glutathione synthase (GS) | NM_012962 | for: 5′ AGATGGCTACATGCCCAGTC 3′ |
| rev: 5′ TGTCTTTCAGCTGCTCCAGA 3′ | ||
| TA = 57 | ||
| Kelch-like ECH-associated protein1 (Keap1) | NM_057152 | for: 5′ GTGGCGGATGATTACACCAAT′ 3′ |
| rev: 5′ GAAAAGTGTGGCCATCGTAGC 3′ | ||
| TA = 57 | ||
| Multidrug resistance associated protein 4 (MRP4) | NM_133411 | for: 5′ CTGGATCCAATTTCAGTGTTG 3′ |
| rev: 5′ GGCAAACTTCTCCCGGATTT 3′ | ||
| TA = 56 | ||
| 5-methyltetrahydrofolate-homocysteine methyltransferase(Mtr) | NM_030864 | for: 5′ CCTGCTTTGGGGTTGAAGAG 3′ |
| rev: 5′ GGAGTTTGCGCAAGTCTGTG 3′ | ||
| TA = 57 | ||
| Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) | NM_031789 | for: 5′ CCAAGGAGCAATTCAACGAAG 3′ |
| rev: 5′ CTCTTGGGAACAAGGAACACG 3′ | ||
| TA = 57 | ||
| S-adenosylmethionine decarboxylase (SAMDC) | NM_031011 | for: 5′ CCCAGCAGTTATGGACCAGT 3′ |
| rev: 5′ TCCATCCGATTTCATTCCAT 3′ | ||
| TA = 55 | ||
| Solute carrier organic anion transporter family, member 1a4(Slco1a4) | NM_131906 | for: 5′ GTCATCGGGAAACTCATCTGC 3′ |
| rev: 5′ CCAAAGTAAATGGGTGCAGGA 3′ | ||
| TA = 57 | ||
| Cytochrome P450 member 1A1(Cyp1A1) | NM_012540 | for: 5′ CAGGAACTATGGGGTGATCCA 3′ |
| rev: 5′ ATATCCACCTTCTCGCCTGGT 3′ | ||
| TA = 60 | ||
| NAD(P)H dehydrogenase [quinone] 1 (NQO1) | NM_017000 | for: 5′ CGCAGAGAGGACATCATTCA 3′ |
| rev: 5′ CGCCAGAGATGACTCAACAG 3′ | ||
| TA = 57 | ||
| Heme oxygenase 1 (HO1) | NM_012580 | for: 5′ AGGCACTGCTGACAGAGGAAC 3′ |
| rev: 5′ AGCGGTGTCTGGGATGAACTA 3′ | ||
| TA = 61 | ||
| β-actin | NM_031144 | for: 5′ ATCGTGCGTGACATTAAAGAGAAG 3′ |
| rev: 5′ GGACAGTGAGGCCAGGATAGAG 3′ | ||
| TA = 60 |
Immunoblot analysis of Nrf2
For analysis of Nrf2 protein expression in whole liver cell lysate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 (w/v) liver homogenates were prepared in a non-reducing RIPA lysis buffer [50 mmol L−1 TRIS-HCl, 150 mmol L−1NaCl, 1 mM phenylmethylsulphonylfluoride (PMSF), 1 mM EDTA, 1.0% sodium desoxycholate, 0.1% sodiumdodecylsulphate (SDS) and 1% TritonX-100, pH = 7.4]. 60 μg of protein were separated according to the standard method47 under non-reducing conditions on 10% SDS-polyacrylamide gels (50 mA, 4 °C, 2 h). Separated proteins were transferred onto a PVDF membrane (PALL Biotrace 0.45 μm™) by semi-dry blotting [25 min at constant 6 V (∼60 mA)]. After blocking membranes overnight at 4 °C in TBST (20 mmol L−1 Tris-HCl, 150 mmol L−1NaCl, 0.1% Tween 20, pH = 7.6) containing 5% non-fat dry milk and 0.2% bovine serum albumin (BSA) analysis was continued by a 12 h incubation with the monoclonal Nrf2 antibody (R&D systems, MAB3925) in TBS buffer (1:1000) followed by a 1 h incubation with the secondary antibody (1:3000) linked to alkaline phosphatase (Goat Anti-Mouse IgG-h + I). Membranes were stained in reaction buffer (0.1 mol L−1 TRIS, 0.1 mol L−1NaCl, 0.05 mol L−1MgCl2) containing 0.00375% Nitro-Blue Tetrazolium (NBT)-and 0.0025% 5-bromo-4-chloro-3-indoylphosphate (BCIP). Optical density of the 67 kDA Nrf2-band was evaluated (Gene Tools, Syngene) on scanned membranes (CanoScan LiDe 500F). β-Actin protein expression was determined as control.
:10 (w/v) liver homogenates were prepared in a non-reducing RIPA lysis buffer [50 mmol L−1 TRIS-HCl, 150 mmol L−1NaCl, 1 mM phenylmethylsulphonylfluoride (PMSF), 1 mM EDTA, 1.0% sodium desoxycholate, 0.1% sodiumdodecylsulphate (SDS) and 1% TritonX-100, pH = 7.4]. 60 μg of protein were separated according to the standard method47 under non-reducing conditions on 10% SDS-polyacrylamide gels (50 mA, 4 °C, 2 h). Separated proteins were transferred onto a PVDF membrane (PALL Biotrace 0.45 μm™) by semi-dry blotting [25 min at constant 6 V (∼60 mA)]. After blocking membranes overnight at 4 °C in TBST (20 mmol L−1 Tris-HCl, 150 mmol L−1NaCl, 0.1% Tween 20, pH = 7.6) containing 5% non-fat dry milk and 0.2% bovine serum albumin (BSA) analysis was continued by a 12 h incubation with the monoclonal Nrf2 antibody (R&D systems, MAB3925) in TBS buffer (1:1000) followed by a 1 h incubation with the secondary antibody (1:3000) linked to alkaline phosphatase (Goat Anti-Mouse IgG-h + I). Membranes were stained in reaction buffer (0.1 mol L−1 TRIS, 0.1 mol L−1NaCl, 0.05 mol L−1MgCl2) containing 0.00375% Nitro-Blue Tetrazolium (NBT)-and 0.0025% 5-bromo-4-chloro-3-indoylphosphate (BCIP). Optical density of the 67 kDA Nrf2-band was evaluated (Gene Tools, Syngene) on scanned membranes (CanoScan LiDe 500F). β-Actin protein expression was determined as control.
Statistical analysis
Data are given as means ± their standard error of the mean (S.E.M.). Statistical differences were analyzed with SPSS 19.0 for Windows (IBM, New York, USA) using one-way ANOVA after testing the normality of distribution (Kolmogorov Smirnov test and Shapiro Wilk test) and the homogeneity of variances (Levene test). If the variances were homogenous, the Least Significant Difference (LSD) test was used to analyze significant differences between means, if not the Games-Howell test was used. Differences between means were considered statistically significant at an error probability of P < 0.05.Results
Performance parameters
At the beginning of the experiment body weight did not differ among groups (Table 2). Se deficient rats of groups C15 and G15 had a lower final mean body weight than all Se-supplemented rats. This difference was significant between group C15 and groups C150, G150, C450 and G450. Despite a higher final body weight in all Se and GRA supplemented groups (G50, G150 and G450), the weight differences were not statistically significant compared to the matching Se deficient G15 rats. The differences in final body weight were accompanied by a significantly reduced feed intake in Se deficient C15- and G15 rats compared to their Se supplemented companions of groups C50, G50, C150, G150, C450 and G450. Independent of additional GRA supplementation the feed conversion ratio (g feed intake:g weight gain) did not differ between Se deficient and Se supplemented rats (Table 2).
| C15 | C50 | C150 | C450 | G15 | G50 | G150 | G450 | |
|---|---|---|---|---|---|---|---|---|
| Unlike superscripts within a line indicate significant differences between means (P < 0.05). n = 12 rats per experimental group. Se and enzymatic analyses were performed in duplicate for each individual. | ||||||||
| Performance parameters | ||||||||
| Initial body weight (g) | 71.9 ± 2.29 | 71.9 ± 2.25 | 72.0 ± 2.22 | 72.0 ± 2.19 | 71.3 ± 2.54 | 72.2 ± 2.18 | 71.9 ± 2.26 | 72.0 ± 2.21 |
| Final body weight (g) | 328 ± 6.40a | 343 ± 6.32ab | 348 ± 9.24b | 354 ± 6.41b | 336 ± 6.28ab | 342 ± 6.49ab | 350 ± 4.28b | 351 ± 6.02b |
| Total feed intake (g) | 984 ± 5.59a | 1027 ± 12.7b | 1054 ± 16.0b | 1068 ± 12.9bc | 1011 ± 7.80a | 1036 ± 13.6b | 1042 ± 7.00b | 1076 ± 8.28c |
| Feed conversion (g/g) | 3.87 ± 0.11 | 3.81 ± 0.07 | 3.86 ± 0.13 | 3.80 ± 0.07 | 3.84 ± 0.09 | 3.85 ± 0.07 | 3.81 ± 0.07 | 3.87 ± 0.08 |
| Daily feed intake (g) | 17.5 ± 0.02a | 18.3 ± 0.22ab | 18.8 ± 0.28ab | 19.1 ± 0.23ab | 18.1 ± 0.14ab | 18.5 ± 0.24ab | 18.6 ± 0.13ab | 19.2 ± 0.14b |
| Daily GRA intake per rat (μmol) | 12.3 ± 0.06a | 12.8 ± 0.16ab | 13.2 ± 0.20ab | 13.4 ± 0.16ab | 12.6 ± 0.12ab | 13.0 ± 0.17ab | 13.0 ± 0.09ab | 13.4 ± 0.10b |
| Selenium status | ||||||||
| Liver | ||||||||
| GPx1 (mU mg(prot.)−1) | 4.08 ± 0.46a | 137 ± 10.7b | 259 ± 15.6c | 282 ± 14.9c | 4.49 ± 0.26a | 168 ± 13.6b | 273 ± 20.0c | 314 ± 23.9c |
| GPx1 expression (fold of C15) | 1.00 ± 0.12a | 8.17 ± 0.86b | 9.56 ± 0.67b | 12.4 ± 1.09b | 2.07 ± 0.49a | 6.60 ± 1.36ab | 8.82 ± 1.09b | 9.63 ± 0.39b |
| GSTA3 (U mg(prot.)−1) | 0.38 ± 0.01a | 0.25 ± 0.11c | 0.22 ± 0.01c | 0.24 ± 0.01c | 0.33 ± 0.01b | 0.25 ± 0.01c | 0.24 ± 0.01c | 0.27 ± 0.02c |
| GSTA3 expression (fold of C15) | 1.00 ± 0.06a | 0.54 ± 0.03b | 0.39 ± 0.02b | 0.41 ± 0.04b | 0.45 ± 0.01b | 0.26 ± 0.01c | 0.29 ± 0.03c | 0.35 ± 0.03bc |
| Plasma | ||||||||
| Se (μg L−1) | 20.7 ± 0.62a | 415 ± 11.8c | 572 ± 8.55d | 628 ± 10.9e | 27.1 ± 1.03b | 457 ± 11.4c | 577 ± 6.88d | 630 ± 10.4e |
| GPx3 (mU mg(prot.)−1) | 0.18 ± 0.02a | 10.1 ± 0.53b | 16.0 ± 0.77c | 16.3 ± 1.19cd | 0.33 ± 0.04a | 11.7 ± 0.80bd | 15.9 ± 0.95cd | 16.7 ± 0.90c |
Selenium status
Final plasma Se concentrations in Se deficient C15 and G15 rats were 96.4 and 94.3% lower than in rats of groups C150 and G150, fed the recommended dietary Se amount (Table 2). Independent of additional GRA supplementation, plasma Se concentration increased gradually and significantly by raising the dietary Se level from 15 to 450 μg kg−1. Only in GRA supplemented Se deficient rats (G15) plasma was Se significantly (30.9%) higher than in group C15, whereas no differences in plasma Se existed between the Se supplemented G groups (G50, G150 and G450) and the matching C groups. Liver GPx1 activity was 65- to 70-fold higher in the Se-supplemented groups C150, C450, G150 and G450 than in the Se deficient groups C15 and G15 (Table 2). Rats fed one third of the recommended dietary Se level (C50 and G50) reached an intermediate liver GPx1 activity compared to their companions fed the recommended dietary Se concentration (C150 and G150). No significant differences in liver GPx1 activity were measured between GRA supplemented rats and their control littermates (C15 vs. G15, C50vs.G50, C150 vs.G150 and C450vs.G450). GPx3 activity was regulated in an analogous manner as described for liver GPx1. GSTA3 is a further sensitive indicator of oxidative stress. The highest GSTA3 expression was measured in Se deficient control rats (C15). The addition of only one third of the recommended dietary Se amount (group G50) significantly reduced both, the expression and the activity of GSTA3 compared to C15 rats. The addition of higher Se amounts to the diets (C150 and C450) caused no further changes in GSTA3 expression and activity. In Se deficient rats with a GRA supplement (G15), GSTA3 expression was reduced by 55% compared to C15 rats whereas the enzymes' activity was reduced only by 24%. GSTA3 expression in GRA- and Se-supplemented rats (G50, G150 and G450) was significantly lower than in their companions of the respective C groups. GSTA3 activity did not differ between Se supplemented C and G rats.Glutathione and homocysteine concentrations in liver and plasma
Total liver glutathione (tGSH) was significantly higher in Se deficient control rats (C15) than in Se supplemented rats of groups C50 and C150. Liver tGSH in group C450 was also distinctly lower (P = 0.22) compared to group C15. GRA addition to the Se deficient diet (G15) lowered liver tGSH significantly compared to C15 rats. The highest liver tGSH concentration within all groups was measured in group G450. Significantly higher liver tGSH levels were also analysed in rats of the Se and GRA supplemented groups G50 and G450 compared to their Se deficient companions of group G15 and their matching C groups C50 and C450 (Fig. 2A). Neither Se nor GRA significantly influenced liver tHCys concentration. However, liver tHCys tended to be higher in all Se supplemented groups compared to the Se deficient groups C15 and G15 (effect size: P = 0.15) (Fig. 2B). tGSH in plasma was reduced by approximately 30% in all Se supplemented control groups (C50, C150 and C450) compared to the Se deficient group (C15). Both Se deficient and Se supplemented rats with dietary GRA (G15, G50, G150 and G450) had significantly lower plasma tGSH concentrations than Se deficient control rats (C15). Plasma tGSH concentration of Se- and GRA supplemented rats either tended to be lower (G150, P = 0.10 and G450, P = 0.15), or it was significantly lower (G50, P = 0.045) than in rats of the respective Se deficient group G15 (Fig. 2C). C15 rats had an about 2.4-fold lower plasma tHCys concentration than their Se supplemented companions (C50, C150 and C450). GRA did not influence plasma tHCys concentration at all dietary Se levels investigated (Fig. 2D).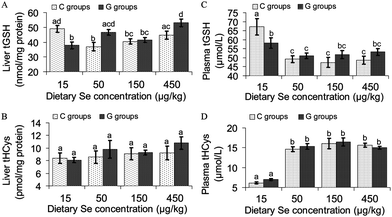 | ||
| Fig. 2 Concentration of total glutathione (tGSH) and total homocysteine (tHCys) in the liver and the plasma of growing rats fed diets supplemented with increasing dietary selenium concentrations (15 μg kg−1, 50 μg kg−1, 150 μg kg−1 or 450 μg kg−1) either without (C15, C50, C150, C450) or with the addition of 700 μmol glucoraphanin/kg diet (G15, G50, G150, G450). Unlike small letters in a figure indicate significant differences between means (P < 0.05). n = 12 rats per experimental group. Analyses were performed in duplicate for each individual. | ||
mRNA expression of key enzymes and transporters of homocysteine and glutathione metabolism
In order to study the molecular mechanisms underlying the measured GSH and HCys concentrations in plasma and liver, the mRNA expression of key enzymes and of transport proteins involved in GSH and HCys metabolism was analyzed in the liver as the main organ of GSH and HCys synthesis and metabolism (Table 3). GCLM and GS, two important key enzymes of GSH biosynthesis, revealed the highest mRNA expression in the Se deficient groups C15 and G15. In the C groups C50, C150 and C450 Se supply at all levels investigated decreased GCLM mRNA to levels of 61 to 78% of that in group C15. GRA addition to the Se deficient diet (G15) had no influence on GCLM mRNA compared to C15 rats. GRA plus Se (G50, G150, G450) reduced GCLM expression by 24 to76% in comparison with the respective Se deficient rats (G15). GS showed a different regulation profile. Se deficient rats with or without GRA supply (C15 and G15) had the highest GS mRNA. Independent of GRA, Se supplementation at all levels investigated reduced GS expression by 23 to 45% of that in Se deficient C15 rats. Independent of GRA, rats receiving the highest Se supplementation (C450 and G450), had the highest GS expression levels within all Se supplemented groups. GNMT, the first enzyme in HCys metabolism, converting S-adenosyl-methionine to S-adenosyl-homocysteine, was down-regulated in tendency by Se supplementation at the recommended (C150, P = 0.10) and at the supranutritive level (C450, P = 0.08) compared to Se deficient control rats (C15). GRA effected a general down-regulation of GNMT expression compared to C15 rats. The combination of Se and GRA (G50, G150, G450) reduced GNMT mRNA to a higher extent than Se alone. The mRNA of SAMDC, passing S-adenosyl-methionine into the polyamine pathway, was reduced by Se at all levels investigated by about 50% compared to Se deficiency (C15). Independent of the dietary Se level, GRA decreased SAMDC expression by about 60% compared to C15 rats. The mRNA level of Mtr, the key enzyme of folate and vitamin B12 dependent HCys remethylation, was neither affected by Se nor by GRA. In contrast, Se supplementation at all levels, reduced expression of BHMT, the second HCys remethylating enzyme, by 23% to 32% compared to Se deficiency (C15). GRAper se decreased BHMT expression by 36% to 47% compared to Se deficient controls (C15). BHMT mRNA was already reduced by GRA supply to Se deficient rats (G15) compared to Se deficient controls without GRA (C15). In relation to Se deficient controls (C15), the combined Se- and GRA-supplementation (G50, G15, G450) reduced BHMT mRNA to a comparable extent than Se alone (C50, C150, C450). The mRNA of CBS, the key enzyme of HCys utilization for cysteine production, was not influenced by variations in the dietary Se concentration in all C groups. At all dietary Se levels GRA supply reduced CBS mRNA compared to the C groups. This reduction was significant in Se- and GRA-supplemented rats of groups G50, G150 and G450 compared to Se deficient controls (C15). mRNA levels of MRP 4, the main GSH exporter into plasma, were 50 to 60% lower in all Se supplemented controls (C50, C150, C450) than in their Se deficient companion (C15). GRA supply reduced MRP4 mRNA already in Se deficiency by 34% compared to Se deficient controls (C15). MRP4 mRNA levels of GRA- and Se-supplemented rats (G50, G150 and G450) were comparable to those of their respective controls without GRA (G50. G15, G450). mRNA expression of Slco1a4, releasing HCys into the plasma, was higher with Se supplementation (C50, C150, C450, G50, G150 and G450) than in Se deficiency (C15 and G15). This raise was only significant in C450 and G150 rats compared to their respective Se deficient companions (C15 and G15).| C15 | C50 | C150 | C450 | G15 | G50 | G150 | G450 | |
|---|---|---|---|---|---|---|---|---|
| Unlike superscripts within a line indicate significant differences between means (P < 0.05). n = 6 cDNA pools of 2 rats per experimental group. Analyses were performed in duplicate for each pool. | ||||||||
| Glutathione biosynthesis | ||||||||
| GCLM (fold of C15) | 1.00 ± 0.06a | 0.64 ± 0.04bd | 0.61 ± 0.03bd | 0.78 ± 0.03bc | 0.99 ± 0.13ac | 0.76 ± 0.06b | 0.53 ± 0.01d | 0.24 ± 0.09e |
| GS (fold of C15) | 1.00 ± 0.04a | 0.60 ± 0.05bc | 0.69 ± 0.7bc | 0.73 ± 0.05b | 0.94 ± 0.06a | 0.55 ± 0.04c | 0.57 ± 0.05bc | 0.67 ± 0.04bc |
| S-adenosyl-methionine utilisation | ||||||||
| GNMT (fold of C15) | 1.00 ± 0.08a | 0.96 ± 0.09ab | 0.78 ± 0.08abc | 0.86 ± 0.07abc | 0.74 ± 0.09abc | 0.72 ± 0.11bc | 0.69 ± 0.05bc | 0.66 ± 0.09c |
| SAMDC (fold of C15) | 1.00 ± 0.14a | 0.59 ± 0.07b | 0.47 ± 0.07b | 0.48 ± 0.05b | 0.38 ± 0.05b | 0.42 ± 0.09b | 0.39 ± 0.08b | 0.40 ± 0.06b |
| HCys remethylation and utilisation of HCys for cysteine production | ||||||||
| Mtr (fold of C15) | 1.00 ± 0.08a | 1.12 ± 0.12a | 1.17 ± 0.24a | 0.91 ± 0.05 a | 0.91 ± 0.09 a | 0.99 ± 0.10a | 0.87 ± 0.08a | 0.97 ± 0.08a |
| BHMT (fold of C15) | 1.00 ± 0.03a | 0.77 ± 0.09b | 0.66 ± 0.09bc | 0.68 ± 0.05bc | 0.64 ± 0.04bc | 0.56 ± 0.08bc | 0.53 ± 0.06c | 0.58 ± 0.07bc |
| CBS (fold of C15) | 1.00 ± 0.09a | 0.96 ± 0.08a | 1.00 ± 0.19ab | 1.05 ± 0.09a | 0.83 ± 0.07ab | 0.69 ± 0.01c | 0.77 ± 0.03bc | 0.71 ± 0.04c |
| Glutathione and homocysteine exporters | ||||||||
| MRP4 (fold of C15) | 1.00 ± 0.06a | 0.45 ± 0.03b | 0.49 ± 0.06b | 0.48 ± 0.01b | 0.76 ± 0.10c | 0.45 ± 0.03b | 0.39 ± 0.01b | 0.48 ± 0.04b |
| Slco1a4 (fold of C15) | 1.00 ± 0.09ac | 1.37 ± 0.18ad | 1.10 ± 0.10acd | 1.77 ± 0.11b | 0.93 ± 0.07c | 1.30 ± 0.06acd | 1.52 ± 0.20bd | 1.26 ± 0.10cd |
mRNA expression of the phase I enzyme CYP1A1 and of the antioxidant phase II enzymes NQO1 and HO1
Se deficient control rats (C15) had an about 3-fold higher CYP1A1 expression than all their Se supplemented companions (C50, C150 and C450) (Fig. 3A). Independent of the Se effects, GRA supply reduced CYP1A1 mRNA by about 60% of that in Se deficient controls (C15). The mRNA of NQO1, preventing free radical formation by one electron reductions, was the highest in Se deficient control rats (C15). In Se deficiency GRA supply (G15) reduced NQO1 expression by 20%. Se supply alone (C50, C150, C450) or in combination with GRA (G50, G150, G450) further reduced NQO1 expression by 66 to 74% compared to the respective Se deficient groups (C15 and G15) (Fig. 3B). The antioxidant HO1 had the highest expression in the liver of Se deficient controls (C15). Se addition to the diets reduced HO1 mRNA dose dependently (C50, −10%), (C150, −37%), (C450, −49%). Irrespective of the dietary Se level, GRA reduced HO1 mRNA by 42 to 55% compared to Se deficient controls (Fig. 3C).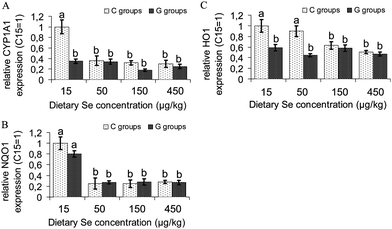 | ||
| Fig. 3 Expression of the phase I cytochrome P450 oxidase CYP1A1, and of the phase II antioxidant enzymes NAD(P)H Quinone Reductase 1 (NQO1) and Heme Oxygenase 1 (HO1) in the liver of growing rats fed diets supplemented with increasing dietary selenium concentrations (15 μg kg−1, 50 μg kg−1, 150 μg kg−1 or 450 μg kg−1) either without (C15, C50, C150, C450) or with the addition of 700 μmol glucoraphanin kg(diet)−1 (G15, G50, G150, G450). Unlike superscripts in a figure indicate significant differences between means (P < 0.05). n = 6 cDNA pools of 2 rats per experimental group. Analyses were performed in duplicate for each pool. | ||
mRNA expression of KEAP 1 and Nrf2 and KEAP 1:Nrf2-ratio
In C rats Se supply reduced KEAP1 mRNA expression dose-dependently (relative expression: C50 = 0.74, C150 = 0.62, C450 = 0.54) compared to Se deficient controls (C15, relative expression =1.00) (Fig. 4A). GRAper se significantly reduced KEAP1 mRNA already in Se deficiency (relative expression: G15 = 0.61). The lowest KEAP1 mRNA levels were measured in the Se supplemented G groups (relative expression: G50 = 0.36, G150 = 0.52, G450 = 0.42). Irrespective of GRA supply, Nrf2 expression was higher in Se supplemented rats (relative expression: C50 = 1.18, C150 = 1.15, C450 = 1.32, G50 = 1.14, G150 = 1.11, G450 = 1.25) than in the respective Se deficient rats (C15 and G15, relative expression: C15 = 1.00, G15 = 0.98). The results for slightly increased Nrf2 expression in Se supplemented rats could be confirmed by immunoblotting (Fig. 4B). As a consequence of these results the KEAP1:Nrf2-ratio decreased dose-dependently in Se supplemented C rats (C15 = 1.00, C50 = 0.62, C150 = 0.47, C450 = 0.41). Moreover the KEAP 1: Nrf2-ratio of GRA supplemented Se deficient rats (G15, 0.59) was significantly lower than that of their Se deficient companions without GRA (C15). Se in combination with GRA (G50, G150, G150) reduced the KEAP 1: Nrf2-ratio (Se treatment effect: P = 0.041) compared to their companions with Se deficiency (G15). The KEAP 1:Nrf2-ratio of rats receiving diets with combined GRA- and Se-supply (G50 = 0.31, G150 = 0.47 and G450 = 0.31) tended to be lower than in their companions of the respective C groups (C50, C150, C450, GRA treatment effect: P = 0.10).
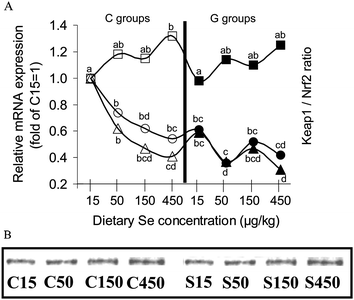 | ||
| Fig. 4 (A) Expression of Keap1 and Nrf2 and Keap1:Nrf2-ratio in the liver of growing rats fed diets supplemented with increasing dietary selenium concentrations (15 μg kg−1, 50 μg kg−1, 150 μg kg−1 or 450 μg kg−1) either without (C15, C50, C150, C450) or with the addition of 700 μmol glucoraphanin kg(diet)−1 (G15, G50, G150, G450). Circles show the expression of Keap1 relative to group C15 = 1, in the left side of the figure empty circles (○) represent Keap1 mRNA expression of the C groups, in the right side of the figure black-filled circles (●) represent the Keap1 mRNA expression of the G groups. Squares show the expression of Nrf2 relative to group C15 = 1, in the left side of the figure empty squares (□) represent Nrf2 mRNA expression of the C groups, in the right side of the figure black-filled squares (■) represent the Nrf2 mRNA expression of the G groups. Triangles show the expression ratio of Keap1:Nrf2 relative to group C15 = 1, in the left side of the figure empty triangles (△) represent the ratio of Keap1:Nrf2 in the C groups, in the right side of the figure black-filled triangles (▲) represent the ratio of Keap1: Nrf2 in the C groups. Unlike superscripts in the figure indicate significant differences between means (P < 0.05). n = 6 cDNA pools of 2 rats per experimental group. Analyses were performed in duplicate for each pool. (B) Protein expression of Nrf2 in whole liver lysate of growing rats fed diets supplemented with increasing dietary selenium concentrations (15 μg kg−1, 50 μg kg−1, 150 μg kg−1 or 450 μg kg−1) either without (C15, C50, C150, C450) or with the addition of 700 μmol glucoraphanin kg(diet)−1 (G15, G50, G150, G450). The figure shows a representative immunoblot prepared from one rat per group, representing the mean body weight of its group. | ||
Discussion
Performance parameters
In accordance with prior research from our group and with other rat studies we could confirm that marginal Se supply (C15, G15) reduces feed intake and weight gain of growing rats.48–50 Although final body weights of Se deficient C -and G-rats did not differ significantly, GRA influenced feed intake and weight gain slightly positively. Irrespective of GRA, no significant differences with regard to performance parameters could be observed between the Se supplemented groups (C50, C150, C450, G50, G150 and G450) (Table 2).Se status
Se status, assayed by means of plasma Se concentration, plasma GPx3 activity, liver GPx1 activity- and expression, indicated a marked Se deficiency in groups C15 and G15 and a dose dependent increase in plasma Se concentration by raising the dietary Se level. The achievement of a plateau of GPx3 activity and of liver GPx1 activity- and expression by feeding the recommended dietary Se amount confirmed the well established regulation-hierarchy of functional selenoproteins, which was also the topic of numerous studies dealing with the determination of rats' Se requirement.51,52 That the ARE-regulated GSTA3 was up-regulated under conditions of oxidative stress and in particular of Se deficiency was also observed in two earlier mouse studies and could be confirmed by our data.53,54 That GSTA3 expression- and activity were drastically reduced by supplementing only one third of the recommended Se amount (group C50) indicate a massive decrease of oxidative stress already with this low Se dose. The reduction of GSTA3 expression and activity by GRA supply to Se deficient rats (G15) compared to C15 rats indicated a reduction of oxidative stress by GRA. Decreased oxidative stress by GRA supplementation was also reflected by distinctly lower GSTA3 mRNA levels in Se supplemented G rats (G50, G150, G450) compared to their corresponding C companions (C50, C150, C450) (Table 2).Influence of various dietary Se levels on GSH and HCys metabolism
Studies with rats and humans have reported conflicting results with regard to the influence of Se status on plasma HCys level. Whereas rat studies clearly have demonstrated that a low Se status reduces plasma HCys and increases plasma GSH, a human study with subjects having a low Se status (plasma Se concentration range: 54–70 μg L−1)23 found a significantly inverse relationship between Se status and plasma HCys. Contrariwise in populations with a higher baseline Se status (plasma Se concentration range: 80–100 μg L−1), Se supplements (100 to 300 μg Se per day in form of Se enriched yeast) remained without an effect on plasma tHCys. In these trials participants of the intervention groups achieved final plasma Se levels of 150–250 μg L−1 after 2.5 and 6 months.26,27 In our rat study plasma Se levels in the Se deficient groups C15 and G15 (Table 2) were even lower compared to the human study with a low basal Se status.23 An increase in plasma tHCys by adding graded Se amounts to our rat diets could be observed only between the Se deficient groups C15 and G15 and the groups supplemented with the lowest Se concentration of 50 μg kg−1 diet (C50 and G50). Additional Se supply, up to slightly supranutritive levels (450 μg Se kg(diet)−1), produced no further effects (Table 3). Our data rather support the results of the human supplementation trials, which have demonstrated the ineffectiveness of Se as a HCys lowering agent.26,27Moreover our data confirm the hypothesis of prior investigations that only severe Se deficiency lowers plasma tHCys concentration by increasing GSH biosynthesis.12,19–21 In addition to this generally accepted information our study revealed some interesting new results as to how a low Se status contributes to a decrease of plasma tHCys and an increase in plasma tGSH. The up-regulation of HCys remethylation by BHMT in Se deficiency (C15, Table 3) in combination with decreased HCys export (Slco1a4) and increased GSH export (MRP4) into the plasma provide additional information for the particular changes in liver and plasma tHCys-and tGSH levels. This new information is supported by the knowledge that both GSH biosynthesis enzymes (GCLM and GS) and MRP 4 are Nrf2 targets with ARE-containing promoters.53–57
Finally, it should be mentioned that positive effects of an adequate or high Se status may derive from the distinct down-regulation of SAMDC, involved in spermidine sythesis. Since spermidine is discussed as a cancer-promoting compound, the reduction of SAMDC expression by Se may represent an additional and novel mechanism explaining its cancer protective potential.58
Influence of GRA in combination with various dietary Se levels on GSH and HCys metabolism
The main hypothesis of our rat study was to investigate if feeding GRA could lower plasma tHCys concentration in Se sufficiency by increasing the expression of the ARE-regulated GSH biosynthesis enzymes. The up-regulation of ARE-regulated genes in the liver thereby was expected to deriving from GRA cleavage by bacterial β-glucosidases in the caecum followed by the release of sulforaphane into the blood.59In contrast to our expectations at all Se levels investigated, GRA feeding had no mentionable influence on liver mRNA levels of GSH biosynthesis enzymes (Table 3).
In the Se deficient group G15 the GSH exporter MRP 456,57 was even down-regulated by additional GRA feeding, whereas no expression differences existed between the Se supplemented groups. This result was directly reflected by a significantly lower plasma tGSH concentration in GRA treated Se deficient rats (G15) compared to the Se deficient controls (C15) (Fig. 2C).
Another interesting result of GRA feeding could be made with regard to CBS expression. Whereas CBS mRNA was not influenced by Se (C groups), its expression was markedly down-regulated by GRA supplementation (G groups) (Table 3). This particular result may base on a more pronounced down-regulation of the antioxidant HO1 in the G groups (Fig. 3C). Beside sufficient vitamin B6, heme is a second major factor responsible for full CBS activity.60 With GRA supply CBS activity may have reached a maximum activity followed by the reduction of its expression due to reduced heme degradation by HO1.
A further difference between C and G groups could be observed with regard to BHMT expression.61Se deficient C rats (C15) had the highest BHMT expression. Se supplementation and, in particular, additional GRA supply distinctly reduced BHMT mRNA.
The combination of reduced BHMT- and CBS expression by GRA feeding thus could be expected to cause an accumulation of liver HCys and higher plasma HCys values. However, liver and plasma tHCys concentration in G rats only tended to be higher than in C rats.
As observed for the Se supplemented C rats (C50, C150 and C450), additional GRA supply reduced SAMDC mRNA to a somewhat greater extent than Se alone and therefore its function as a cancer protective compound is supported.58
Finally, a conflicting result of our study should be addressed. We have measured the highest GNMT mRNA concentration in Se deficient control rats. Se supply, in particular in combination with GRA, led to a dose-dependent decline of GNMT mRNA (Table 3). This result would imply higher liver and plasma HCys concentrations in C15 rats than in Se supplemented groups. An explanation for the lacking effect of the high GNMT expression on liver and plasma HCys in C15 rats may be based on the dual function of the enzyme. GNMT acts as both a methylase and as the 4S Polycyclic Aromatic Hydrocarbon Receptor (4S-PAHR), which in turn is one important factor for CYP1A1 induction.62 Concordant with these facts, C15 rats had the highest GNMT and the highest CYP1A1 expression. This implicates that GNMT under conditions of increased oxidative stress (Se deficiency) acts as both a methylase and as the 4S-PAHR. When oxidative stress is reduced by Se- and/or GRA-supply it works just as a methylase, which implies that changes in GNMT do not influence the particular changes of liver and plasma HCys.
We could not establish GRA as a HCys lowering dietary supplement under the conditions tested. Presumably the dietary GRA amount was too low for sufficient sulforaphane release, which induces ARE-regulated GSH biosynthesis enzymes in peripheral organs. This topic will be discussed in the following section.
Discussion of the lacking effect of GRA on the induction of ARE-regulated genes in the liver
In contrast to earlier trials, which have studied the induction of phase II enzymes in cell culture models and in rats using pure sulforaphane, the purpose of our study was to investigate the effects of a broccoli extract with a guaranteed GRA concentration of 10% (w/w). Another intention of our study was to adapt the GRA supply of the rats to a realistic uptake of 500–1000 μmol GRA per day in humans. These concentrations can be realised by eating 250–400 g cooked broccoli with almost inactivated myrosinase or taking a dietary supplement.
The latest results from a rat study have shown that in the absence of plant-derived myrosinase, 10 to 15% of dietary GRA are cleaved by β-glucosidases from ceacal bacteria and that a peak value of free sulforaphane in mesenterial blood is reached 2–3 h after GRA ingestion. In this study a single dose of 150 μmol of GRA was applied to the rats for one time only.65 In contrast, in our study the average daily GRA intake of G group rats was only about 13 μmol (∼730 μmol during the whole experiment). That way the GRA intake in our trial was distinctly lower than in the above mentioned study and may provide one explanation for the lacking response on the induction of ARE-regulated liver enzymes. Whereas prior studies with tissue cultures and laboratory animals have postulated that only pure isothiocyanates (e.g.sulforaphane) induce ARE-regulated enzymes, the latest investigations have shown that GRA also potently up-regulates these enzymes.38,39 Effective GRA concentrations for a maximum phase II enzyme induction varied between 1 μM and 25 μM, depending on the enzyme.38,39 However, it must be remarked that the mentioned studies were carried out in vitro by incubating tissue slices with GRA. In our study we have measured a distinct induction of a broad panel of ARE-regulated genes, including different subclasses of GSTs, NQO1, epoxide hydrolase1, HO1, GPx2, and TrxR1, in the small and in the large intestine of the GRA fed rats (data are not shown and will be published separately). Transferring the in vitro data38,39 to our study, it can be concluded that GRA, in the concentration used, rather has produced a local response in the gastrointestinal tract than systemic effects. Local concentrations in the intestine of our rats, having an average daily GRA intake of ∼13 μmol per rat, correspond well to the mentioned in vitro incubation studies.38,39 and to results from another trial in which 9 μmol pure sulforaphane potently induced ARE-regulated genes in the intestine of rats.63
Another explanation for the lacking response of GRA on ARE gene induction in the liver may consist of an increased intestinal barrier against oxidative stress,66,67 which reduces oxidative stress in the organism and may produce quite contrary effects on ARE enzymes in peripheral organs.
This hypothesis is confirmed by data of a rat study. In this trial a distinct increase in the expression of a large number of liver phase II enzymes was induced by the application of 50 μmol pure sulforaphane per rat for two times.64 In this study a 2.2-fold increase in liver CBS expression was also achieved by sulforaphane application. Potentially in this study tHCys values would have responded to the high sulforaphane dose.
A very recent study with human lung cells confirmed our results for the liver. The incubation of these cells with low doses of pure sulforaphane (1 and 2 μmol L−1) caused a distinct up-regulation of NQO1 mRNA expression for 2 days. Interestingly, after 6 days NQO1 mRNA levels drastically dropped even below that in the DMSO treated control group, whereas the protein level was kept up-regulated.68 In our study we could observe a similar effect for GSTA3 expression and activity in group G15. In accordance with the above mentioned study,68 in our trial the mRNA levels of many ARE genes were also lower in GRA rats than in the respective C groups. These particular changes in ARE genes were most obvious when Se deficient C15 rats were compared to their GRA supplemented companions of group G15. Nevertheless, gene expression levels of some ARE-regulated genes (e.g.GCLM, GS, HO1) were also lower in the Se- and GRA-supplemented groups (G50, G150 and G450) than in the respective C groups.
The observed changes in ARE gene expression were also reflected by KEAP1 (Fig. 4A). Both Se supplementation and in particular GRA supplementation reduced KEAP1 expression. KEAP1 modification at sensitive –SH groups by oxidative stress or electrophilic compounds like GRA or sulforaphane36,38,39 leads to Nrf2 liberation, its nuclear translocation and the induction of phase II enzymes.37,56 Latest results have revealed that KEAP1per se is regulated via an ARE.55 Thus the reduced expression of KEAP1 by Se and long-term GRA may indicate that GRA treated rats (in particular C15 vs.G15) had reduced oxidative stress in their livers. Interestingly, Nrf2 expression responded with a nearly constant or a slightly increased mRNA- and protein-expression due to Se and GRA supplementation (Fig. 4A, B). This would implicate a higher nuclear Nrf2 translocation and contradicts the down-regulation of ARE genes by Se and GRA. Potentially, the up-regulation of Nrf2 due to reduced KEAP1 expression represents a counter regulatory mechanism. However, this aspect needs further intensive investigation with regard to nuclear Nrf2 translocation by immunoblotting or EMSA.
In our study we could show that CYP1A1 was down-regulated by Se supplementation and even more by additional long-term GRA supplementation (Fig. 3A). As mentioned above, potentially the down-regulation of GNMT contributes to this effect.62 Moreover we could measure a reduction of phase II and also of phase III enzymes (MRP4) by the applied dietary regimes. In our study CYP1A1 was down-regulated to a higher extent than some phase II and III enzymes (GCLM, GS, HO1,MRP4) (Table 3 and Fig. 3C) or down-regulation was nearly comparable (GSTA3, NQO1) (Table 2 and Fig. 3B). Therefore our results suggest that Se supplementation, in particular with combined long-term low dose GRA supply, reduces oxidative stress in the liver of rats, resulting in a balanced down-regulation of phase I, II and III enzymes.
Conclusions and perspectives
Conclusions
We have hypothesised that long-term GRA supplementation at a level, corresponding to a realistic uptake in humans, lowers plasma tHCys, which is higher in Se supplemented rats than in Se deficient. Moreover, we assumed that GRA reduces plasma tHCys by a pull on the transsulphuration pathway via the induction of ARE-regulated enzymes of GSH biosynthesis. As expected, Se impressively reduced the expression of GSH biosynthesis enzymes and that of other ARE-regulated genes. As a consequence, plasma tHCy levels were higher in Se supplemented rats than in Se deficient.In contrast to our hypothesis GRA did not counteract the Se dependent mRNA decrease of ARE genes, but reduced their mRNA in some cases to a higher extent than Se alone. Under the conditions tested we could not establish GRA as a HCys lowering compound.
We conclude:
• That GRA at the dietary level tested (∼13 μmol per rat and day) reduces oxidative stress in the liver via increasing the intestinal barrier against oxidative stress. As a consequence the organism responds rather with a down-regulation of ARE genes than with an up-regulation.
• That an up-regulation of ARE-regulated genes in peripheral organs, like the liver, largely seems to depend on GRA cleavage to free sulforaphane and glucose by plant-derived myrosinase or bacterial β-glucosidases.
• That the GRA amount tested in our study was too low for the production of a sulforaphane amount (∼50 μmol per rat and day) sufficient for the up-regulation of ARE genes also in the liver. As a consequence the reduction of plasma tHCys failed.
Perspectives
• In future studies dose-response experiments starting with distinctly higher GRA concentrations should be carried out to examine if these higher GRA concentrations influence the mRNA of ARE genes also in the liver.• In addition to the above mentioned dose-response, experiments with animal models with defects in HCys metabolism or with dietary-induced hyperhomocyteinemia should be used to test if GRA or sulforaphane can be established as complementary HCys lowering compounds, supporting the conventional therapy with vitamins B6, B9 and B12.
Abbreviations
| BHMT | Betaine homocysteine methyl transferase |
| CBS | Cystathionine beta synthase |
| tGSH | total glutathione |
| tHCys | total homocysteine |
| GNMT | Glycine-N-methyl transferase |
| GPx1 | Glutathione peroxidase 1 |
| GPx3 | Glutathione peroxidase 3 |
| GCLM | Glutamate cysteine ligase (modifier subunit) |
| GS | Glutathione synthetase |
| KEAP1 | Kelch-like ECH-associated protein 1 |
| MRP4 = ABCG4 | Multidrug resistance protein 4 = ATP-binding cassette transporter 4 |
| MTR | 5-methyltatrahydrofolate-homocysteine methyltransferase |
| Nrf2 | Nuclear factor (erythroid-derived 2)-like 2 |
| Slco1a4 | solute carrier organic anion transporter family (member 1a4) |
| Cyp1A1 | Cytochrome P450 member 1A1 |
| NQO1 | NAD(P)H dehydrogenase [quinone] 1 |
| HO1 | Heme oxygenase 1 |
Acknowledgements
We thank the DANONE FOUNDATION FOR HEALTH, D-85540 Haar, Germany for supporting this experiment by a grant dedicated to study consequences of GRA supplementation on metabolic processes (Project number 2009/6). Further thank is addressed to our diploma students Stefanie Weber, Claudia Beck and Eva Maria Steinke, helping with the laboratory analyses within the scope of their diploma theses. We thank Mrs. Kumari Hiller (Jarrow Formulas Germany, Berlin) and Mr. Jarrow L. Rogovin (founder of Jarrow Formulas, Los Angeles, USA) for providing us with the broccoli extract, produced for the European market by FRUTAROM, Belgium.References
- M. Boucelma, F. Haddoum, H. Chaudet, G. Kaplanski, N. Mazouni-Brahimi, A. Rezig-Ladjouze, M. Brouri and A. Berrah, Int. Angiol., 2011, 30, 18–24 CAS.
- S. Singh, K. R. Bailey and I. J. Kullo, Int. J. Cardiol., 2011 DOI:10.1016/j.ijcard.2011.05.068.
- S. J. Kim, Y. H. Choe, O. Y. Bang and Chaos-Biomarker Collaborators, Stroke, 2011, 42, 1464–1468 CrossRef CAS.
- R. P. Kloppenborg, P. J. Nederkoorn, Y. van der Graaf and M. I. Geerlings, Atherosclerosis, 2011, 216, 461–466 CrossRef CAS.
- J. Zhou and R. C. Austin, BioFactors, 2009, 35, 120–129 CrossRef CAS.
- A. Kassab, T. Ajmi, M. Issaoui, L. Chaeib, A. Miled and M. Hammami, Ann. Clin. Biochem., 2008, 45, 476–480 CrossRef CAS.
- C. P. Lin, Y. H. Chen, J. W. Chen, H. B. Leu, T. Z. Liu, P. L. Liu and S. L. Huang, J. Biomed. Sci., 2008, 15, 183–196 CrossRef CAS.
- C. Séguin, M. R. Abid, K. C. Spokes, I. G. Schoots, A. Brkovic, M. G. Sirois and W. C. Aird, Biomed. Pharmacother., 2008, 62, 395–400 CrossRef.
- H. R. Griffiths, S. Aldred, C. Dale, E. Nakano, G. D. Kitas, M. G. Grant, D. Nugent, F. A. Taiwo, L. Li and H. J. Powers, Free Radical Biol. Med., 2006, 40, 488–500 CrossRef CAS.
- I. Barić, J. Inherited Metab. Dis., 2009, 32, 459–471 CrossRef.
- A. M. Troen, E. Lutgens, D. E. Smith, I. H. Rosenberg and J. Selhub, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 15089–15094 CrossRef CAS.
- N. M. Wolf, K. Mueller, F. Hirche, E. Most, J. Pallauf and A. S. Mueller, Br. J. Nutr., 2010, 104, 520–532 CrossRef CAS.
- M. N. Di Minno, S. Pezzullo, V. Palmieri, A. Coppola, A. D'Angelo, F. Sampietro, V. Cavalca, E. Tremoli and G. Di Minno, Thromb. Res., 2011 DOI:10.1016/j.thromres.2011.05.017.
- X. Liu, M. Liu, D. Tsilimingras and E. L. Schiffrin, J. Am. Soc. Hypertens., 2011, 5, 239–248 CrossRef.
- Y. M. Smulders and H. J. Blom, J. Inherited Metab. Dis., 2011, 34, 93–99 CrossRef CAS.
- O. Dary, Nutr. Rev., 2009, 67, 235–244 CrossRef.
- O. Stanger, W. Herrmann, K. Pietrzik, B. Fowler, J. Geisel, J. Dierkes, M. Weger and Dach-Liga Homocystein e.V., Clin. Chem. Lab. Med., 41, pp. 1392–1403 Search PubMed.
- K. M. Halpin and D. H. Baker, J. Nutr., 1984, 114, 606–612 CAS.
- E. O. Uthus, K. Yokoi and C. D. Davis, J. Nutr., 2002, 132, 1122–1128 CAS.
- E. O. Uthus and S. A. Ross, J. Nutr., 2007, 137, 1132–1136 CAS.
- E. O. Uthus and S. Ross, Biol. Trace Elem. Res., 2009, 129, 213–220 CrossRef CAS.
- S. González, J. M. Huerta, J. Alvarez-Uría, S. Fernández, A. M. Patterson and C. Lasheras, J. Nutr., 2004, 134, 1736–1740 Search PubMed.
- B. Klapcińska, S. Poprzęcki, A. Danch, A. Sobczak and K. Kempa, Biol. Trace Elem. Res., 2005, 108, 1–15 CrossRef.
- C. J. Bates, C. W. Thane, A. Prentice and H. T. Delves, J. Trace Elem. Med. Biol., 2002, 16, 1–18 CAS.
- A. Floegel, S. J. Chung, A. von Ruesten, M. Yang, C. E. Chung, W. O. Song, S. I. Koo, T. Pischon and O. K. Chun, Public Health Nutr., 2011, 18, 1–10 CrossRef.
- B. J. Venn, A. M. Grant, C. D. Thomson and T. J. Green, J. Nutr., 2003, 133, 418–420 CAS.
- B. Bekaert, M. L. Cooper, F. R. Green, H. McNulty, K. Pentieva, J. M. Scott, A. M. Molloy and M. P. Rayman, Mol. Nutr. Food Res., 2008, 52, 1324–1333 CAS.
- E. O. Uthus, S. A. Ross and C. D. Davis, Biol. Trace Elem. Res., 2006, 109, 201–214 CrossRef CAS.
- W. C. Hawkes and L. J. Laslett, Am. J. Physiol.: Heart Circ. Physiol., 2009, 296, H256–262 CrossRef CAS.
- J. Breilmann, J. Pons-Kühnemann, C. Brunner, M. Richter and M. Neuhäuser-Berthold, Ann. Nutr. Metab., 2010, 57, 177–182 CrossRef CAS.
- B. Puchau, M. A. Zulet, A. G. de Echávarri, H. H. Hermsdorff and J. A. Martínez, Nutrition, 2010, 26, 534–541 CrossRef CAS.
- V. Vitvitsky, E. Mosharov, M. Tritt, F. Ataullakhanov and R. Banerjee, Redox Rep., 2003, 8, 57–63 CrossRef CAS.
- K. Mizuno, T. Kume, C. Muto, Y. Takada-Takatori, Y. Izumi, H. Sugimoto and A. Akaike, J. Pharmacol. Sci., 2011, 115, 320–328 CrossRef CAS.
- J. J. Bark and A. S. Chung, J. Biochem., 1989, 22, 61–67 Search PubMed.
- A. M. Raines and R. A. Sunde, BMC Genomics, 2011, 12, 26 CrossRef CAS.
- C. E. Guerrero-Beltrán, M. Calderón-Oliver, J. Pedraza-Chaverri and Y. I. Chirino, Exp. Toxicol. Pathol., 2010 DOI:10.1016/j.etp.2010.11.005.
- T. W. Kensler, N. Wakabayashi and S. Biswal, Annu. Rev. Pharmacol., 2007, 47, 89–116 CrossRef CAS.
- A. F. Abdull Razis, M. Bagatta, G. R. De Nicola, R. Iori and C. Ioannides, Toxicology, 2010, 277, 74–85 CrossRef CAS.
- A. F. Abdull Razis, M. Bagatta, G. R. De Nicola, R. Iori and C. Ioannides, Lung Cancer, 2011, 71, 298–305 CrossRef.
- P. G. Reeves, J. Nutr., 1997, 127, S838–841 Search PubMed.
- A. S. Mueller, J. Pallauf and E. Most, J. Trace Elem. Med. Biol., 2002, 16, 47–55 Search PubMed.
- R. A. Lawrence and R. F. Burk, Biochem. Biophys. Res. Commun., 1976, 71, 952–958 CrossRef CAS.
- O. W. Griffith, Anal. Biochem., 1982, 106, 207–212 CrossRef.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- M. W. Pfaffl, Nucleic Acids Res., 2001, 29, e45 CrossRef CAS.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, Clin. Chem., 2009, 55, 611–622 CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- A. S. Mueller, S. D. Klomann, N. M. Wolf, S. Schneider, R. Schmidt, J. Spielmann, G. Stangl, K. Eder and J. Pallauf, J. Nutr., 2008, 138, 2328–2336 CrossRef CAS.
- A. S. Mueller, A. C. Bosse, E. Most, S. D. Klomann, S. Schneider and J. Pallauf, J. Nutr. Biochem., 2009, 20, 235–247 CrossRef CAS.
- K. Schäfer, A. Kyriakopoulos, H. Gessner, T. Grune and D. Behne, J. Trace Elem. Med. Biol., 2004, 18, 89–97 Search PubMed.
- R. A. Sunde, Exp. Biol. Med., 2010, 235, 1046–1052 CrossRef CAS.
- R. A. Sunde, J. K. Evenson, K. M. Thompson and S. W. Sachdev, J. Nutr., 2005, 135, 2144–2150 CAS.
- R. F. Burk, K. E. Hill, A. Nakayama, V. Mostert, X. A. Levander, A. K. Motley, D. A. Johnson, J. A. Johnson, M. L. Freeman and L. M. Austin, Free Radical Biol. Med., 2008, 44, 1617–1623 CrossRef CAS.
- M. Müller, A. Banning, R. Brigelius-Flohé and A. Kipp, Genes Nutr., 2010, 5, 297–307 CrossRef.
- X. Wang, D. J. Tomso, B. N. Chorley, H. Y. Cho, V. G. Cheung, S. R. Kleeberger and D. A. Bell, Hum. Mol. Genet., 2007, 16, 1188–1200 CrossRef CAS.
- Q. Cheng, K. Taguchi, L. M. Aleksunes, J. E. Manautou, N. J. Cherrington, M. Yamamoto and A. L. Slitt, J. Biochem. Mol. Toxicol., 2011 DOI:10.1002/jbt.20392.
- C. D. Klaassen and L. M. Aleksunes, Pharmacol. Rev., 2010, 62, 1–96 CrossRef CAS.
- S. Tsujinaka, K. Soda, Y. Kano and F. Konishi, Int. J. Oncol., 2011, 38, 305–312 CAS.
- A. T. Dinkova-Kostova and P. Talalay, Mol Nutr. Food Res., 2008, 52, S128–238 Search PubMed.
- R. Banerjee and C. G. Zou, Arch. Biochem. Biophys., 2005, 433, 144–156 CrossRef CAS.
- K. M. Sterling, 4S polycyclic aromatic hydrocarbon receptor (glycine N-methyltransferase) and the aryl hydrocarbon receptor nuclear translocator (hypoxia inducible factor-1β) interaction in Chinese hamster ovary and rat hepatoma cells: 4S PAH-R/ARNT hetero-oligomers?, J. Cell. Biochem., 2011, 112, 2015–2018 CrossRef CAS.
- S. K. Kim and Y. C. Kim, J. Hepatol., 2005, 42, 907–913 CrossRef CAS.
- R. K. Thimmulappa, K. H. Mai, S. Srisuma, T. W. Kensler, M. Yamamoto and S. Biswal, Cancer Res., 2002, 62, 5196–5203 CAS.
- R. Hu, V. Hebbar, B. R. Kim, C. Chen, B. Winnik, B. Buckley, P. Soteropoulos, P. Tolias, R. P. Hart and A. N. Kong, J. Pharmacol. Exp. Ther., 2004, 310, 263–271 CrossRef CAS.
- R. H. Lai, M. J. Miller and E. Jeffery, Food Funct., 2010, 1, 161–166 CAS.
- R. Brigelius-Flohé, C. Müller, J. Menard, S. Florian, K. Schmehl and K. Wingler, BioFactors, 2001, 14, 101–106 CrossRef.
- A. Banning, S. Deubel, D. Kluth, Z. Zhou and R. Brigelius-Flohé, Mol. Cell. Biol., 2005, 25, 4914–4923 CrossRef CAS.
- X. L. Tan, M. Shi, H. Tang, W. Han and S. D. Spivack, J. Nutr., 2010, 140, 1404–1410 CrossRef CAS.
- S. Barcelo, J. M. Gardiner, A. Gescher and J. K. Chipman, Carcinogenesis, 1996, 17, 277–282 CrossRef CAS.
- K. Mahéo, F. Morel, S. Langouët, H. Kramer, E. Le Ferrec, B. Ketterer and A. Guillouzo, Cancer Res., 1997, 57, 3649–5362 Search PubMed.
- K. Skupinska, I. Misiewicz-Krzeminska, R. Stypulkowski, K. Lubelska and T. Kasprzycka-Guttman, J. Biochem. Mol. Toxicol., 2009, 23, 18–28 CrossRef CAS.
- P. Perocco, G. Bronzetti, D. Canistro, L. Valgimigli, A. Sapone, A. Affatato, G. F. Pedulli, L. Pozzetti, M. Broccoli, R. Iori, J. Barillari, V. Sblendorio, M. S. Legator, M. Paolini and S. Z. Abdel-Rahman, Mutat. Res, 2006, 595, 125–136 CrossRef CAS.
- M. G. Robbins, J. Hauder, V. Somoza, B. D. Eshelman, D. M. Barnes and P. R. Hanlon, J. Food Sci., 2010, 75, H190–199 CrossRef CAS.
- V. Yoxall, P. Kentish, N. Coldham, N. Kuhnert, M. J. Sauer and C. Ioannides, Int. J. Cancer, 2005, 117, 356–362 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |