Dietary chromones as antioxidant agents—the structural variable
M. M.
Dias
a,
N. F. L.
Machado
a and
M. P. M.
Marques
*ab
aResearch Unit “Molecular Physical Chemistry”, University of Coimbra, Portugal
bDepartment of Life Sciences, Faculty of Science and Technology, University of Coimbra, Apartado 3046, 3001-401, Portugal. E-mail: pmc@ci.uc.pt; Fax: +351 239854448; Tel: +351 239826541
First published on 7th September 2011
Abstract
This study reports an evaluation of the free radical scavenging ability of a series of chromone derivatives, in the light of their structural features and conformational behaviour. The 2,2-diphenyl-1-picrylhydrazyl radical (DPPH˙) test for the assessment of radical scavenging properties was applied, and the interpretation of the experimental results was assisted by ab initio theoretical approaches that allowed relevant parameters, such as the enthalpy of formation of the radical species, to be predicted. From the eighteen tested compounds, three—fisetin, luteolin and quercetin—are shown to act as effective antiradicals. Consistent structure–activity relationships (SARs) were established regarding the antioxidant role of this type of chromone-based system.
1. Introduction
Oxidative stress conditions—disruption of the homeostatic balance between free radical generation and the production of naturally occurring antioxidants (e.g.glutathione and regulatory enzymes such as superoxide dismutase, catalase and peroxidases)—are recognized to be directly linked to damage in numerous cell targets (DNA, lipids, proteins), which can finally result in severe diseases, namely liver toxicity, cardiovascular and neurodegenerative disorders, and cancer. Dietary habits play a key role in the prevention of these pathologies (along with lifestyle and environmental conditions)1 by controlling or restoring the homeostatic oxidative balance.Chemicals produced by plants (phytochemicals), although not considered as essential nutrients, are known to possess health-promoting properties due to their activity as chain-breaking antioxidants by radical scavenging or the reduction of free radical formation.2,3 The phenolic constituents are the largest group and comprise, among others, phenolic acids, anthocyanins, coumarins, tannins, chromones and flavones.4 Research on novel antioxidants from natural sources has been growing in the last decade within the nutritional, pharmacological and medicinal chemistry fields,1,5–17 with particular emphasis on the prevention of cancer and cardiovascular disorders through dietary intervention (nutraceutical agents), mainly when it became clear that therapy does not always succeed (e.g. due to lack of specificity or drug resistance mechanisms).18–22
Chromones and their structural analogues (e.g.flavonoids), in particular, are known to play an important protective role against oxidation processes, either from deleterious radical species or from UV radiation, therefore displaying pharmacologically relevant functions such as antibacterial, antifungal, antiviral, anti-spasmolytic, anti-inflammatory, anti-HIV or anticancer.23–29 They have therefore motivated great interest within the medicinal chemistry field, the chromone moiety supposedly being the essential component of pharmacophores of a large number of bioactive molecules.30
Structure and conformation are key factors that determine a compound's behaviour from its acid–base profile or lipophilic vs. hydrophilic character to its interaction with biochemical receptors within the cell. Therefore, the beneficial activity of phytochemicals relies on their structural preferences, namely the number and location of the phenolic OH groups.21,31,32 In the present study, a series of chromone-based compounds with different ring substitution patterns (Fig. 1) are assessed as to their free radical scavenging ability in order to ascertain the optimal molecular features (e.g.hydroxylation profile) associated with this antiradical activity.21 The experimental data was interpreted in light of relevant calculated parameters, such as the enthalpy of formation of the corresponding radical species and the spin density distribution. The results thus obtained will hopefully pave the way for the development of novel chemoprotective agents of natural origin, particularly against cancer and cardiovascular disorders.
 | ||
| Fig. 1 A: Basic chromone and flavonoid structures (the atom numbering and ring labeling is included). B: The chromone derivatives under study. | ||
2. Results and discussion
A series of chromones with different substitution patterns (Fig. 1) was assessed as to their free radical scavenging activity by examining their ability to capture the 2,2-diphenyl-1-picrylhydrazyl (DPPH˙) stable radical in solution, in order to ascertain the molecular features related to a high antiradical activity in this type of system.For chromone-based antioxidants (A), the commonly accepted mechanism for this process is represented by:
| AOH + DPPH˙ → AO˙ + DPPH2, | (1) |
 | (2) |
DPPH˙ displays a typical absorption maximum at 515 nm, which disappears upon reduction to the corresponding hydrazine (DPPH2). This colorimetric assay is commonly used for establishing structure–activity relationships (SARs).21
First, the stabilization time for a complete reduction of the DPPH˙ free radical was determined. The steady-state discoloration time was verified to be 20 min for all the compounds tested. The percentage of remaining DPPH˙ radicals in solution was then calculated for each compound and for each concentration. Table 1 comprises the effective dosage required to scavenge 50% DPPH˙ (EC50) for all the derivatives studied. The corresponding dose–response curves for the most effective antiradical compounds are shown in Fig. 2. In all cases, antioxidant activity showed a time- and concentration-dependent profile.
| Molecular core | Compounda | EC50/μMb |
|---|---|---|
| a Atom numbering according to Fig. 1. b See Materials and methods section; the asterisk refers to intergroup comparison: * non-significant; among groupsp < 0.0001. c Ref. 32. d Ref. 33. e Positive control. f Ref. 34. N. A. – experimental data did not yield convergent results. | ||
| Chromone | ||

|
Chromone | >400 |
| 7-Hydroxy-4-chromone (7-OH) | >400 | |
| Chromone 3-acid carboxylic (3-COOH) | >400 | |
| 3-Formylchromone (3-CHO) | >400 | |
| 2-Amino-3-formylchromone (2-NH2, 3-CHO) | >400 | |
| Chromone 2-acid carboxylic (2-COOH) | N. A. | |
| Flavone | ||

|
Luteolin (5,7,3′,4′-OH) | 17.64 ± 2.33* (11.04 ± 0.38c; 41.92d) |
| Apigenin (5,7,4′-OH) | N. A. (463.40 ± 22.28c) | |
| Chrysin (5,7-OH) | N. A. (492.57 ± 23.94c) | |
| 3-Methylflavone-8-carboxylic acid (3-Me, 8-COOH) | N. A. | |
| Flavone | >400 (>1000) | |
| Isoflavone | ||

|
Genistein (5,7,4′-OH) | >400 |
| Daidzein (7,4′-OH) | >400 | |
| Flavon-3-ol | ||

|
Quercetin (3,5,7,3′,4′-OH) | 16.42 ± 1.67* (10.89 ± 0.03c) |
| Fisetin (3,7,3′,4′-OH) | 21.53 ± 3.89* (14.06 ± 0.21c) | |
| Kaempferol (3,5,7,4′-OH) | 32.02 ± 1.36* (28.05 ± 0.28c) | |
| Galangin (3,5,7-OH) | 126.10 ± 17.92** (71.64 ± 1.07c) | |
| 3,6-Dihydroxyflavone (3,6-OH) | 349.50 ± 48.58*** | |
| Trolox™e | 38.90 ± 10.06* (50.08f; 99.88d) | |
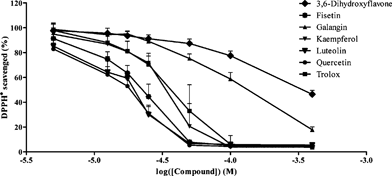 | ||
| Fig. 2 Dose–response curves for the DPPH˙ scavenging ability of the most effective compounds tested. The EC50 values were calculated from each curve as the effective concentration able to scavenge 50% DPPH˙ (the mean ± SD values are represented for each compound and each concentration, obtained from two independent experiments carried out in triplicate. Trolox™ was used as a reference). | ||
The chromone-based compounds with the simplest structure sharing the same molecular core—chromone, chromones 2- and 3-carboxylic acid, 7-hydroxy-4-chromone, 3-formyl-chromone and 2-amino-3-formylchromone (Fig. 1)—showed not to be able to reduce DPPH˙ radicals in solution (even at high concentrations), which seems to indicate that although the chromone center is important for stabilizing the semiquinone species formed upon radical reduction34 (see eqn (1)), it is not the solely responsible for the scavenging ability of the compound. In fact, for this series of chromones, corresponding to minor structural changes of the chromone parent compound, no significant antioxidant capacity is found. Even the presence of the hydroxyl group at the C-7 position was not enough to provide antiradical activity.
Three of the tested derivatives, comprising a C-2 catechol substitution—fisetin, luteolin and quercetin (Fig. 1)—were found to act as effective antiradicals (Tab. 1), reacting quickly with DPPH˙ and attaining steady state conditions almost immediately. Galangin and 3,6-dihydroxyflavone also evidenced a certain degree of radical scavenging capacity, although only at high dosages (126.10 ± 17.92 and 349.50 ± 48.58 μM, respectively).
As evidenced in Table 1, hydroxylation of the inactive flavone structure can greatly improve its scavenging ability (e.g.luteolinvs.flavone), since the hydroxyl substituents have long been established to be essential components to promote this activity.35 Furthermore, the in vitroantioxidant capacity of flavonoids is also highly dependent on the arrangement and relative orientation—structural features and conformational preferences—of the different functional entities about their core structure, apart from the number of hydroxyl ring substituent groups.32,36
Daidzein and genistein differ from the rest of the flavonoids investigated here, since they are isoflavones, displaying a phenyl group at the C-3 position (Fig. 1). For the range of concentrations tested, neither of them showed significant antioxidant activity, and even though genistein has an extra OH group at C-5, this does not lead to an enhancement of its antioxidant ability (Tab. 1). In fact, recent studies allowed the conclusion that compounds having a 4′-monohydroxylated B ring (Fig. 1) behave as weak antioxidants.37
On the other hand, when comparing genistein (an isoflavone) with luteolin (a flavone), differing in the location of the B ring—at C-3 and C-2, respectively (Fig. 1)—and in the presence of a second OH group at 5′ in luteolin, the former does not display any antioxidant activity whereas luteolin acts as a strong reducing agent (Tab. 1). Regarding the flavones chrysin and luteolin, in turn, it is verified that chrysin, lacking the catechol moiety, does not exhibit any antioxidant capacity, as opposed to luteolin (Tab. 1).
These experimental observations allow clear relationships to be established between the derivatives' structural features and their radical scavenging ability (Table 1): (i) an ortho-dihydroxy substitution (the presence of a catechol group) and its location at the C-2 position is of paramount importance, as previously suggested by Lopez-Lazaro,28 (ii) this catechol moiety at C-2 is more relevant than the presence of hydroxyl groups at positions C-5 and C-7 (ring A), (iii) in the presence of a phenol or phenyl group instead of catechol, OH substitution at C-3 is determinant (e.g.galangin (planar) vs.apigenin (non-planar) or chrysinvs.3,6-dihydroxyflavone).
Quercetin, a flavonol displaying an additional hydroxyl group at position C-3 from ring C, and luteolin (a flavone) yielded similar dose–response curves and EC50 values (Tab. 1), although the flavonol exhibited a slightly higher activity due to the presence of the C-3 hydroxyl. In addition, when comparing the activities of quercetin and fisetin, two flavonols varying solely in the number of OH groups (Fig. 1), it was verified that quercetin acted as a slightly stronger antioxidant (EC50 = 16.42 ± 1.67 vs. 21.53 ± 3.89 μM, respectively; Table 1) owing to the additional OH at position C-5 of ring A. In turn, fisetin and luteolin, differing in the position of an OH group (at C-3 and C-5, respectively), display different reducing activities, the latter being a more effective free radical scavenger (21.53 ± 3.89 vs. 17.64 ± 2.33 μM, respectively; Table 1). Therefore, despite the identical number of hydroxyl groups in these two compounds, the one at C-5 (A ring) provides a more favorable free radical capture reaction than the C-3 OH substitution. A higher activity has been reported for flavonols (quercetin and fisetin) as compared to flavones (luteolin),13,22 based solely on the planar structure of the former relative to the slightly twisted conformation characteristic of the latter. In fact, it appears that both the planarity and conjugation within the molecule enable a more effective electronic delocalization, thereby increasing the stability of the phenoxyl radical that results from the first reduction step (see eqn (1)).13,22 However, knowing that the hydroxyl substituents are essential for antioxidant ability, it is not surprising that luteolin (comprising an OH at C-5) is found to act as a more efficient antiradical agent than fisetin. In addition, calculations show that this C-5 radical displays a planar, favoured geometry.
Also quite relevant is the fact that 3,6-dihydroxyflavone (flavonol) and chrysin (flavone) present a distinct behaviour towards DPPH˙ (Table 1). While the former was found to be able to act as a radical scavenger, though at very high dosages (349.50 ± 48.58 μM), chrysin showed no antioxidant activity for any of the concentrations tested. In fact, the presence of a 7-OH substitution at the A ring in chrysin seems to be responsible for an electronic delocalisation from C-6 to C-7 relative to the dihydroxy derivative (also displaying a 3-OH group), thus disfavouring radical formation. Additionally, all the derivatives comprising this OH substitution at C-3 were found to have a planar structure, which leads to a further stabilization of the corresponding radicals. Also interesting to note is that the energy difference between the 3,6-dihydroxyflavone radicals at positions C-3 and C-6 is only 1 kJ mol−1 (Table 2), evidencing that these two sites are almost equally favoured for hydrogen abstraction in this molecule. The same occurs for kaempferol regarding its C-3 and C-4′ radical species (Table 2).
| Radical species | BDE/kJ mol−1 | Δ(BDE) a/kJ mol−1 |
|---|---|---|
| a Δ(BDE) relative to the most stable radical. b Ref. 38. c Ref. 39. d Ref. 40. e Ref. 41. f Ref. 42. g Ref. 43. | ||
| Chromones | ||
| 7-Hydroxy-4-chromone | ||
| 7-O˙ | 377.50 | — |
| Chromone 2-acid carboxylic | ||
| 2-COO˙ | 445.27 | — |
| Chromone 3-acid carboxylic | ||
| 3-COO˙ | 465.85 | — |
| Flavones | ||
| 3-Methyl-8-carboxylic acid flavone | ||
| 8-COO˙ | 437.00 | — |
| Apigenin | ||
| 4′-O˙ | 352.34 (343.92b) | 0.00 |
| 7-O˙ | 371.92 (365.63b) | 19.58 (21.71b) |
| 5-O˙ | 421.26 (443.67b) | 68.92 (99.75b) |
| Chrysin | ||
| 7-O˙ | 372.88 (384.30c; 357.10d) | 0.00 |
| 5-O˙ | 421.17 | 48.29 |
| Luteolin | ||
| 4′-O˙ | 317.96 (311.88b; 342.70d) | 0.00 |
| 3′-O˙ | 325.10 (321.42b) | 7.14 (9.54b) |
| 7-O˙ | 371.66 (365.69b) | 53.70 (53.81b) |
| 5-O˙ | 488.90 (443.47b) | 170.94 (131.59b) |
| Isoflavones | ||
| Daidzein | ||
| 4′-O˙ | 345.35 (341.03e) | 0.00 |
| 7-O˙ | 363.61 (360.28e) | 18.26 (19.25e) |
| Genistein | ||
| 4′-O˙ | 347.32 (340.37e) | 0.00 |
| 7-O˙ | 374.43 (370.33e) | 27.11 (29.96e) |
| 5-O˙ | 484.57 (381.83e) | 110.25 (41.46e) |
| Flavonols | ||
| 3,6-Dihydroxyflavone | ||
| 6-O˙ | 353.67 (316.77c) | 0.00 |
| 3-O˙ | 355.30 | 1.63 |
| Fisetin | ||
| 4′-O˙ | 308.91 (294.22c; 346.30d) | 0.00 |
| 3′-O˙ | 319.77 | 10.86 |
| 3-O˙ | 350.26 | 41.35 |
| 7-O˙ | 359.09 | 50.18 |
| Galangin | ||
| 3-O˙ | 349.21 (318.07c; 363.10d) | 0.00 |
| 7-O˙ | 370.38 | 21.17 |
| 5-O˙ | 403.30 | 54.09 |
| Kaempferol | ||
| 3-O˙ | 327.12 (339.48f; 348.90d) | 0.00 (0.83f) |
| 4′-O˙ | 328.79 (338.65f) | 1.67 (0.00f) |
| 7-O˙ | 367.89 (362.50f) | 40.77 (23.85f) |
| 5-O˙ | 387.65 (395.13f) | 60.53 (56.48f) |
| Quercetin | ||
| 4′-O˙ | 311.40 (302.71g; 343.00d) | 0.00 |
| 3′-O˙ | 322.05 (313.17g) | 10.65 (10.46g) |
| 3-O˙ | 344.70 (337.86g) | 33.30 (35.15g) |
| 7-O˙ | 367.44 (362.54g) | 56.04 (59.83g) |
| 5-O˙ | 403.78 (399.78g) | 92.38 (97.07g) |
The radical scavenging ability of flavonoids and other chromone derivatives is due to their high reactivity as hydrogen or electron donors, thus being mainly ruled by their O–H bond dissociation enthalpy (BDE), which corresponds to formation of the radical species. This hydrogen-donating capacity was therefore predicted for each compound investigated by calculating their respective BDE values (Table 2). The lower the BDE the greater is the ability to donate a H-atom from a hydroxyl group, giving rise to a stable radical (eqn (1) and eqn (2)), thus favouring the free radical scavenging process (Fig. 3).
 | ||
| Fig. 3 A graphical plot of the experimental free radical scavenging ability (EC50) as a function of the calculated BDEmin for the most effective compounds tested (BDEmin refers to the most stable radical). | ||
The catechol-comprising chromones behave in a slightly different manner during this process, as their most stable radical does not necessarily correspond to the favoured conformation of the neutral molecule after hydrogen loss. Actually, in the presence of a carbonyl, formed by H ablation from the original hydroxyl, a rearrangement of the catechol group takes place through rotation of the second group OH, leading to the formation of a stabilizing hydrogen bond between this hydroxyl and the neighbouring carbonyl (Scheme 1).
 | ||
| Scheme 1 | ||
The spin density distribution in the radical species was also theoretically predicted, a good correlation having been verified with both the BDE values and the radical scavenging experimental results, namely for the most promising compounds (quercetin, luteolin and fisetin). In fact, the regions of the molecule with the higher probability of finding the unpaired electron—positive spin density (represented in red, Fig. 4(A))—were found to correspond to the site(s) where H abstraction occurs preferentially, yielding the most stable radical species (Table 2). Furthermore, the distinct behaviour observed for 3,6-dihydroxyflavone and chrysin (Table 1) is foreseen by the calculations, which yield a lower enthalpy of formation for the 3,6-dihydroxyflavone radical at position C-6 (Table 2), corresponding to an efficient radical scavenging activity as well as to a higher spin density at this site (Fig. 4(B)) compared to chrysin.
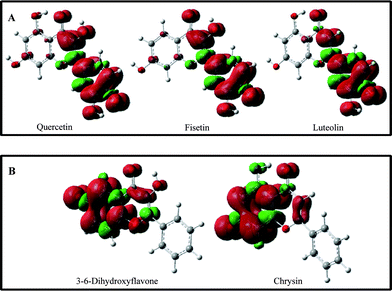 | ||
| Fig. 4 3D maps of the calculated spin density of the optimised radical species for the chromone derivatives showing a higher radical scavenging activity (red: positive spin density and green: negative spin density; calculated at the B3LYP/EPR-ii level and represented for an isovalue of 0.001). | ||
Finally, the fact that 7-hydroxy-4-chromone, largely reported as an efficient antioxidant,44 was experimentally verified to be unable to reduce DPPH˙ (Table 1) and to have a reasonably high BDE value (Table 2) is explained by the fact that this compound's antioxidant capacity is mainly due to its interference with specific signalling pathways45,46 rather than through radical scavenging processes.
3. Conclusions
This work aims to be a pioneer in the evaluation of the antioxidant properties of simple molecules such as chromone, chromones 2- and 3-carboxylic acid or 7-hydroxy-4-chromone, comparing them with more complex flavones and flavonols. The experimental procedure currently followed ensures a reproducible and reliable assessment of compounds, since the conditions are kept constant for each radical scavenging evaluation and the results are interpreted in the light of calculated parameters relevant for antioxidant capacity. Furthermore, an ab initio quantum mechanical theoretical approach was used, as opposed to the semi-empirical methodologies reported in most studies to date which, although much less demanding in terms of computing requirements, cannot accurately represent these kinds of unsaturated systems and radical formation reactions.The results gathered here allow clear structure–activity relationships (SARs) for the chromone derivatives investigated to be established, particularly regarding their radical scavenging capacity. The presence of a catechol group located at the C-2 position, as well as the number of hydroxyl substituents and their location in the molecule (preferably at C-3, C-5 and/or C-7), are determinant structural factors for their ability to scavenge free radicals (Fig. 5). For those compounds able to reduce DPPH˙ to DPPH2, the antioxidant activity was found to decrease according to the order: quercetin > luteolin > fisetin > kaempferol (> Trolox™) > galangin > 3,6-dihydroxyflavone.
 | ||
| Fig. 5 The main structure–activity relationships (SARs; highlighted by the shadowed areas) presently established for the chromone derivatives in this study regarding their radical scavenging activity in the presence (A) or absence (B) of a catechol group. | ||
In general, the chromone core, reported to be essential for a stable flavonoid structure,34 does not by itself ensure radical scavenging activity. The present study validates the theory that the substitution of this central nucleus at specific sites will lead to a tailored antioxidant capacity, thus paving the way for a rational design of new and more efficient antioxidant agents from natural sources.
4. Materials and methods
4.1. Reagents
Apigenin (97%), daidzein (97%), galangin (97%), genistein (97%), luteolin (97%) and 3-methylflavone-8-carboxylic acid (98%) were purchased from Alfa Aesar (Lancashire, UK). 2-Amino-3-formychromone (97%), chromone (99%), chromone 2-carboxylic acid (97%), chromone 3-carboxylic acid (97%), chrysin (97%), 3,6-dihydroxyflavone (98%), dimethylsulphoxide (DMSO, ≥99.9%), 2,2-diphenyl-1-picrylhydrazyl radical (DPPH˙, 95% pure), fisetin (≥99.0%), flavone, 3-formylchromone (97%), 7-hydroxy-4-chromone (97%), standard antioxidant 6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid (Trolox™, 97%), kaempferol (>97%), methanol and quercetin (≥98%) were purchased from Sigma-Aldrich Química S. A. (Sintra, Portugal). All other chemicals were of analytical grade.4.2. Free radical scavenging activity
The free radical scavenging activity was determined for chromone and the derivatives under study through the DPPH˙ test, following the procedure of Brand-Williams et al.,47 as altered by Samee et al.,34 with minor modifications, using Trolox™ (6-hydroxy-2,5,7,8-tetramethylchroman-2-carboxylic acid) as a standard. This method is based on the reduction of methanolic DPPH˙ in the presence of a hydrogen-donating antioxidant. Upon reduction, the violet DPPH˙ (absorption maximum at 515 nm) yields a yellow solution of DPPH2.A methanolic solution (100 μL) of the sample at different concentrations (10 to 800 μM) was added (in 96-well microplates) to 100 μL of a DPPH˙ methanolic solution (prepared from a 3 mg/15 mL methanol stock, diluted to obtain an A515 value between 0.9–1.0). The mixture was homogenized and left to stand for 20 min in the dark at room temperature. The A515 value was then measured in a μQuant MQX200 microplate reader (BioTek, USA) and converted to the DPPH˙ percentage in solution through the equation:34
 | (3) |
The percentage of DPPH˙ radicals in solution was plotted against the logarithmic concentration of the tested compounds in order to obtain the corresponding EC50 values (effective concentration leading to a 50% loss of DPPH˙ activity).
Methanol was used as the solvent for all compounds tested, except for apigenin, which was solubilized in a methanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMSO (l/l, v/v) mixture. In this case, since a change from a protic to a non-protic solvent has been shown to have a non-negligible effect on the measured antioxidant activity,48 the EC50 value was scaled according to Seyoum et al.:32EC50methanol = EC50methanol:DMSO/2.17 in order to compare it with the values obtained for the other compounds. A DPPH˙ methanolic solution was taken as the negative control, while Trolox™ was the positive control.
:DMSO (l/l, v/v) mixture. In this case, since a change from a protic to a non-protic solvent has been shown to have a non-negligible effect on the measured antioxidant activity,48 the EC50 value was scaled according to Seyoum et al.:32EC50methanol = EC50methanol:DMSO/2.17 in order to compare it with the values obtained for the other compounds. A DPPH˙ methanolic solution was taken as the negative control, while Trolox™ was the positive control.
Unlike several reported studies on the antioxidant capacity of phytochemicals, the screening presented here was carried out for identical experimental conditions and equal concentrations of both the tested compounds and controls. This ensures the reproducibility and accuracy of the resulting data, and allows a reliable comparison among compounds.
4.3. Statistical analysis
The experimental values were fitted with non-linear regression functions, and the results were compared to those observed with the reference antioxidant Trolox™. All measurements were performed in triplicate and repeated at least twice. The results are expressed as mean ± standard deviation (SD). Analysis of the variance was conducted, and divergence between variables was tested for significance by one-way ANOVA with the Tukey test—differences with p < 0.0001 were considered statistically significant.4.4. Calculation of bond dissociation enthalpies and spin density distribution
The quantum mechanical calculations were performed using the GAUSSIAN 03W program49 within the density functional theory (DFT) approach in order to properly account for the electron correlation effects (particularly important in these kinds of conjugated system). The widely employed hybrid method, denoted B3LYP, which includes a mixture of HF and DFT exchange terms and the gradient-corrected correlation functional of Lee, Yang and Parr,50,51 as proposed and parameterized by Becke,52,53 was used, along with the double-zeta split valence basis set 6-31G**.54 A full conformational analysis was undertaken through geometry optimisation and evaluation of the relative energy of every possible conformation of the neutral chromone derivatives using the Berny algorithm and redundant internal coordinates;55 the bond lengths were optimised to within ca. 0.1 pm and the bond angles to within ca. 0.1°. The final root-mean-square (rms) gradients were always less than 3 × 10−4 Hartree Bohr−1 or Hartree radian−1. No geometrical constraints were imposed on the molecules under study.Radicals were obtained for each neutral molecule by deletion of a hydrogen atom from the minimum energy geometry and optimization with a spin multiplicity of 2 at the same level of theory. This procedure was repeated for all the OH groups in the molecule in order to determine the most stable radical species.
The O–H bond dissociation enthalpies (BDE), associated with radical formation, were calculated according to the following equation:
| BDE = Hf(A–O˙) + Hf(H) − Hf(A–OH), | (4) |
These enthalpy values were obtained by calculating the single point energy for the most stable conformation of both the neutral molecule and its radical with the extended basis set 6-311++G**57 (including diffuse functions58 and thus yielding more reliable energy values). A thermal correction to the enthalpy was performed, as proposed by Zhang and co-workers41 (using the appropriate factor obtained from the zero-point energy (ZPVE) calculation).
The spin density (SD) values correspond to the probability of localization of the unpaired electron in the molecule. They were calculated for all possible radicals using the B3LYP functional and the EPR-ii double zeta basis set developed by Barone and co-workers,59 comprising a single set of polarization functions and an enhanced s part, optimized for the computation of hyperfine coupling constants by DFT methods. The SD maps were drawn using GaussView 3.0 and plotted for an isovalue of 0.001.
Since the calculations regarding radicals with unpaired electrons and a spin multiplicity equal to 2 require the use of the unrestricted spin option (UB3LYP), this was applied for the entire theoretical procedure (both for radicals and neutral molecules) in order to ensure consistency and a precise comparison between the conformational energies of each molecule and its radical species.
Acknowledgements
The authors acknowledge financial support from the Portuguese Foundation for Science and Technology—PEst-OE/QUI/UI0070/2011 and PhD fellowship SFRH/BD/40235/2007 (NFLM).References
- E. Riboli and T. Norat, Am. J. Clin. Nutr., 2003, 78, 559S–569S CAS.
- C. Kaur and H. C. Kapoor, Int. J. Food Sci. Technol., 2001, 36, 703–725 CrossRef CAS.
- O. Blokhina, E. Virolainen and K. V. Fagerstedt, Ann. Bot., 2003, 91, 179–194 CrossRef CAS.
- E. Haslam, Practical Polyphenolics: From Structure to Molecular Recognition and Physiological Action, Cambridge University Press, Cambridge, 1998 Search PubMed.
- L. Bravo, Nutr. Rev., 2009, 56, 317–333 CrossRef.
- P. Fresco, F. Borges, C. Diniz and M. P. Marques, Med. Res. Rev., 2006, 26, 747–766 CrossRef CAS.
- P. Fresco, F. Borges, M. Marques and C. Diniz, Curr. Pharm. Des., 2010, 16, 114–134 CrossRef CAS.
- C. A. Gomes, T. G. da Cruz, J. L. Andrade, N. Milhazes, F. Borges and M. P. Marques, J. Med. Chem., 2003, 46, 5395–5401 CrossRef CAS.
- B. A. Graf, P. E. Milbury and J. B. Blumberg, J. Med. Food, 2005, 8, 281–290 CrossRef CAS.
- C. G. Heijnen, G. R. Haenen, F. A. van Acker, W. J. van der Vijgh and A. Bast, Toxicol. in Vitro, 2001, 15, 3–6 CrossRef CAS.
- T. J. Key, A. Schatzkin, W. C. Willett, N. E. Allen, E. A. Spencer and R. C. Travis, Public Health Nutr., 2004, 7, 187–200 CrossRef.
- G. Mandalari, R. M. Faulks, C. Bisignano, K. W. Waldron, A. Narbad and M. S. Wickham, FEMS Microbiol. Lett., 2010, 304, 116–122 CrossRef CAS.
- P. G. Pietta, J. Nat. Prod., 2000, 63, 1035–1042 CrossRef CAS.
- W. Ren, Z. Qiao, H. Wang, L. Zhu and L. Zhang, Med. Res. Rev., 2003, 23, 519–534 CrossRef CAS.
- Y. J. Surh, Nat. Rev. Cancer, 2003, 3, 768–780 CrossRef CAS.
- B. Swinburn, Public Health Nutr., 2009, 12, 877–878 CrossRef.
- P. van't Veer, M. C. Jansen, M. Klerk and F. J. Kok, Public Health Nutr., 2000, 3, 103–107 CrossRef CAS.
- C. J. Dillard and J. B. German, J. Sci. Food Agric., 2000, 80, 1744–1756 CrossRef CAS.
- E. Middleton, Jr, C. Kandaswami and T. C. Theoharides, Pharmacol. Rev., 2000, 52, 673–751 Search PubMed.
- B. H. Havsteen, Pharmacol. Ther., 2002, 96, 67–202 CrossRef CAS.
- D. Amic, D. Davidovic-Amic, D. Beslo and N. Trinajstic, Croat. Chem. Acta, 2003, 76, 55–61 CAS.
- M. A. Soobrattee, V. S. Neergheen, A. Luximon-Ramma, O. I. Aruoma and T. Bahorun, Mutat. Res., Fundam. Mol. Mech. Mutagen., 2005, 579, 200–213 CrossRef CAS.
- D. Bennardi, G. Romanelli, J. Jios, J. Autino, G. Baronetti and H. Thomas, ARKIVOC, 2008, 123–130 CAS.
- M. Grazul and E. Budzisz, Coord. Chem. Rev., 2009, 253, 2588–2598 CrossRef CAS.
- Y. Li, H. Fang, W. Xu, Y. Li, H. Fang and W. Xu, Mini-Rev. Med. Chem., 2007, 7, 663–678 CrossRef CAS.
- K. N. Prasad, J. Hao, C. Yi, D. Zhang, S. Qiu, Y. Jiang, M. Zhang and F. Chen, J. Biomed. Biotechnol., 2009, 2009, 612805 CrossRef.
- M. Recanatini, A. Bisi, A. Cavalli, F. Belluti, S. Gobbi, A. Rampa, P. Valenti, M. Palzer, A. Palusczak and R. W. Hartmann, J. Med. Chem., 2001, 44, 672–680 CrossRef CAS.
- M. Lopez-Lazaro, Mini-Rev. Med. Chem., 2009, 9, 31–59 CrossRef CAS.
- F. Casetti, W. Jung, U. Wölfle, J. Reuter, K. Neumann, B. Gilb, A. Wähling, S. Wagner, I. Merfort and C. M. Schempp, J. Photochem. Photobiol., B, 2009, 96, 260–265 CrossRef CAS.
- N. Machado and M. Marques, Curr. Bioact. Compd., 2010, 6, 76–89 CrossRef CAS.
- D. Prochazkova, I. Bousova and N. Wilhelmova, Fitoterapia, 2011, 82, 513–523 CrossRef CAS.
- A. Seyoum, K. Asres and F. K. El-Fiky, Phytochemistry, 2006, 67, 2058–2070 CrossRef CAS.
- U. Wölfle, P. R. Esser, B. Simon-Haarhaus, S. F. Martin, J. Lademann and C. M. Schempp, Free Radical Biol. Med., 2011, 50, 1081–1093 CrossRef.
- W. Samee, N. Sae-Lee and J. Ungwitayatorn, SWU J. Pharm. Sci., 2004, 9, 36–42 Search PubMed.
- N. Cotelle, Curr. Top. Med. Chem., 2001, 1, 569–590 CrossRef CAS.
- K. E. Heim, A. R. Tagliaferro and D. J. Bobilya, J. Nutr. Biochem., 2002, 13, 572–584 CrossRef CAS.
- A. S. Pannala, T. S. Chan, P. J. O'Brien and C. A. Rice-Evans, Biochem. Biophys. Res. Commun., 2001, 282, 1161–1168 CrossRef.
- M. Leopoldini, I. P. Pitarch, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 92–96 CrossRef CAS.
- D. Amic and B. Lucic, Bioorg. Med. Chem., 2010, 18, 28–35 CrossRef CAS.
- T. Denisova and E. Denisov, Russ. Chem. Bull., 2008, 57, 1858–1866 CrossRef CAS.
- J. Zhang, F. Du, B. Peng, R. Lu, H. Gao and Z. Zhou, THEOCHEM, 2010, 955, 1–6 CrossRef CAS.
- M. Leopoldini, T. Marino, N. Russo and M. Toscano, J. Phys. Chem. A, 2004, 108, 4916–4922 CrossRef CAS.
- M. Leopoldini, N. Russo and M. Toscano, Food Chem., 2011, 125, 288–306 CrossRef CAS.
- Q. Jia and T. M. Farrow, US Pat., 2010, 2010/0168223 Search PubMed.
- R. H. Erickson, K. J. Natalie, W. Bock, Z. Lu, F. Farzin, R. G. Sherrill, D. J. Meloni, R. J. Patch and W. J. Rzesotarski, J. Med. Chem., 1992, 35, 1526–1535 CrossRef CAS.
- R. J. Williams, J. P. E. Spencer and C. Rice-Evans, Free Radical Biol. Med., 2004, 36, 838–849 CrossRef CAS.
- W. Brand-Williams, M. E. Cuvelier and C. Berset, LWT–Food Sci. Technol., 1995, 28, 25–30 CrossRef CAS.
- O. Dangles, G. Fargeix and C. Dufour, J. Chem. Soc., Perkin Trans. 2, 1999, 1387–1395 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, GAUSSIAN 03 (Revision D.01), Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Chem. Phys. Lett., 1989, 157, 200–206 CrossRef CAS.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- C. Peng, P. Ayala, H. Schlegel and M. Frisch, J. Comput. Chem., 1996, 17, 49–56 CrossRef CAS.
- L. F. Wang and H. Y. Zhang, Bioorg. Med. Chem. Lett., 2004, 14, 2609–2611 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- V. Barone, Recent Advances in Density Functional Methods, Part I, ed. D. P. Chong, World Scientific Publishing Co., Singapore, 1996 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |