Dietary vitamin K alleviates the reduction in testosterone production induced by lipopolysaccharide administration in rat testis
Naofumi
Takumi
a,
Hitoshi
Shirakawa
*a,
Yusuke
Ohsaki
a,
Asagi
Ito
a,
Takaya
Watanabe
a,
Puspo E.
Giriwono
a,
Toshiro
Sato
b and
Michio
Komai
a
aLaboratory of Nutrition, Graduate School of Agricultural Science, Tohoku University, Sendai, 981-8555, Japan. E-mail: shirakah@biochem.tohoku.ac.jp; Fax: +81 22 717 8813; Tel: +81 22 717 8812
bFine Chemical & Food Laboratories, J-Oil Mills, Inc., Fukuroi, Shizuoka 437-1111, Japan
First published on 13th June 2011
Abstract
Vitamin K is essential for the posttranslational modifications of blood coagulation factors and proteins present in the bone matrix. Vitamin K is distributed not only in the liver and bones but is also abundant in the brain, kidney, and gonadal tissues. However, the function of extra-hepatic/bone vitamin K has not been fully elucidated. Previously, we observed that dietary supplementation with vitamin K suppresses inflammation, and vitamin K deficiency decreases testicular testosterone production in rats. Here, we examined whether the dietary vitamin K state affects testicular steroidogenesis in lipopolysaccharide (LPS)-treated rats because vitamin K has anti-inflammatory activity. Male Wistar rats were fed either vitamin K-free or control diets for 35 d, and then intraperitoneally administered LPS (0.5 mg kg−1 body weight) to induce inflammation for 6 h. Vitamin K deficiency symptoms were not observed in the vitamin K-free diet group; however, the vitamin K levels in the testis were significantly lower in the vitamin K-free diet group than in the control diet group. After LPS treatment, plasma testosterone levels were significantly reduced in the vitamin K-free diet group compared with the control diet group. Testicular mRNA and protein levels of Cyp11a, a rate-limiting enzyme in steroidogenesis, corresponded to plasma testosterone levels. However, plasma luteinizing hormone levels were unaffected by diet and LPS. Phosphorylated nuclear factor κB p65 in the testis was significantly increased in the LPS-treated, vitamin K-free diet group compared with control. These results indicate that dietary vitamin K affects testicular vitamin K levels and ameliorates the LPS-induced reduction in testicular testosterone synthesis. Testicular vitamin K might facilitate the inhibition of inflammation signal transduction and maintain steady levels of testosterone.
Introduction
Vitamin K is known to be essential for blood coagulation and bone metabolism in mammals. Vitamin K functions as a cofactor for microsomal γ-glutamylcarboxylase (GGCX) during the posttranslational synthesis of γ-carboxyglutamate from glutamate residues present in vitamin K-dependent precursor proteins.1,2 There are two types of naturally occurring vitamin K forms, phylloquinone (vitamin K1), synthesized in plants, and menaquinone (vitamin K2 or MK-n), produced mainly by microorganisms. Significant amounts of menaquinones are synthesized by intestinal flora in the lower intestine and subsequently absorbed. Intestinally synthesized menaquinones can contribute to maintenance of vitamin K levels in the host, but their relative contributions to vitamin K as a nutrient are debatable.3,4 After absorption from the intestine, vitamin K is transported to the liver by triglyceride-rich lipoprotein chylomicrons to activate vitamin K-dependent proteins. Although a significant portion of vitamin K is redistributed to extrahepatic tissues, such as the heart, kidney, brain, and gonadal tissues,5–7 it remains unclear whether redistributed vitamin K primarily serves as a cofactor for GGCX in these tissues.Several studies have indicated that vitamin K has novel functions other than protein γ-carboxylation. Menaquinone-4, a form of vitamin K2 that contains a geranylgeranyl group as an isoprenyl side chain at position 3 of 2-methylnaphthoquinone, possesses unique functions: induction of programmed cell death of osteoclasts and several tumor cellsin vitro,8inhibition of osteoclast differentiation9–12 and osteoblast apoptosis,13 and regulation of expression of specific genes by activating the nuclear receptor PXR (SXR).14–17 Furthermore, vitamin K1 and menaquinone-4 suppress LPS-induced inflammation in cultured cells and rats,18–20 and prevent cell death induced by oxidative stress in oligodendrocytes and neurons.21 They also activate protein kinase A (PKA) in neuron-like cells, hepatoma cells, and osteosarcoma cells.22–24 These functions are not mediated viaprotein γ-carboxylation.
Previously, we discovered that testosterone production in the testis was down-regulated in the vitamin K-deficient state accompanying decreased Cyp11a gene expression.25 Cyp11a is the first enzyme in the steroidogenesis pathway in the testis and is rate limiting; its gene expression is controlled by luteinizing hormone (LH) secreted from the pituitary gland. Steroidogenic factor 1 (SF-1), a member of the nuclear receptor superfamily, and cyclic AMP response element-binding protein (CREB) are responsible for the regulation of steroidogenic genes, including Cyp11a. Their transcriptional activities are stimulated by PKAvia the LH receptor and inhibited by nuclear factor κB (NFκB) vialipopolysaccharide (LPS) and/or inflammatory cytokines.26,27 Therefore, Cyp11a expression is reduced by inflammatory mediators, and consequently, steroidogenesis in the testis is inhibited. On the other hand, vitamin K can repress the activation of NFκB in human hepatocarcinoma and macrophage-like cells28,20 as well as in mouse osteoblasts and osteoclasts.29 Thus, we hypothesize that vitamin K functions in the testes to maintain adequate levels of Cyp11a expression via the repression of NFκB activation. In this study, we attempted to clarify the role of testicular vitamin K on steroidogenesis under induction of inflammation in rats.
Materials and methods
Animals and diets
Male Wistar rats (8 weeks of age) were purchased from Japan SLC, (Shizuoka, Japan) and housed at 23 °C ± 2 °C in a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :12 light : dark cycle (lights switched on at 8:00 AM) in a specific pathogen-free animal house.
:12 light : dark cycle (lights switched on at 8:00 AM) in a specific pathogen-free animal house.
Control and low vitamin K (VK-free) diets were prepared according to the AIN-93G formula30 with or without vitamin K1 (final concentration, 0.75 mg kg−1 diet; Wako Pure Chemicals, Osaka, Japan). After rats freely fed on either experimental diet for 35 d, they were intraperitoneally administered LPS (Escherichia coli serotype O111:B4, L2630, Sigma Chemical, St. Louis, MO) dissolved in saline (0.5 mg kg−1 body weight). Six h after administration, the rats were sacrificed, and their blood, livers, and testes were obtained for further analyses.
Ethical guidelines
The experimental plan of the present study was approved by the Animal Research-Animal Care Committee of the Graduate School of Agricultural Science, Tohoku University. All experiments were performed by following the guidelines framed by this committee in accordance with Japanese governmental legislation (1980). The same committee supervised the care and use of the rats in this study.Plasma total and undercarboxylated osteocalcin
Plasma total osteocalcin (OC) levels were determined using a commercially available enzyme-linked immunosorbent assay (ELISA) kit (Biomedical Technologies Inc., Stoughton, MA). The levels of undercarboxylated OC (ucOC) were measured as described previously31 with slight modifications. A 100-μL fraction of the plasma sample was incubated at 4 °C for 4 h with 6 mg hydroxyapatite suspended in a 60 μL of a 0.85% NaCl solution containing 2 mM CaCl2, and the mixture was subsequently centrifuged. The ucOC levels in the supernatant were determined by ELISA.High-performance liquid chromatography (HPLC) for measurements of vitamin K in tissues
Tissue samples (1 g) were homogenized in 5 volumes of 66% 2-propanol. Vitamin K was extracted from the homogenate using six volumes of n-hexane as described previously19 and measured using a fluorescent HPLC system (Waters 600E system; Puresil 5C18 column; Waters, Milford, MA; RC 10-3 PtO2 column; Shiseido-IRICA, Kyoto, Japan; Hitachi F-1000 fluorescence detector, excitation at 240 nm, emission at 430 nm; Hitachi D-2000 data processor, Tokyo, Japan). The concentrations of vitamin K1 and menaquinone-4 were calculated using their relative fluorescent intensities of menaquinone-3 (Eisai Co., Ltd., Tokyo, Japan) as an internal standard.19RNA preparation and quantitative reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from the whole testes using the guanidine/isothiocyanate-based reagent Isogen (Nippon Gene, Tokyo, Japan), according to the instruction manual. For cDNA synthesis, 2 μg total RNA was used as a template. The RNA was incubated in RT buffer (50 mM Tris-HCl, pH 8.3, 75 mM KCl, 3 mM MgCl2, and 5 mM DTT) containing 50 units Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA), 40 units RNaseOUT RNase inhibitor (Invitrogen), 1 mM dNTP, and 0.5 μg oligo-dT (GE Healthcare Bio-Science, Tokyo, Japan) at 50 °C for 60 min. An aliquot of cDNA was used as a template for quantitative PCR using the ABI 7300 Real Time PCR System (Applied Biosystems, Foster City, CA). Cyp11a cDNA was amplified by cDNA-specific primers (forward, GAGAAGCCTATCTTCTTCAACTTCCA; reverse, TGCAGCCTGCAATTCATACAGT) using the SYBR Premix Ex Taq solution (Takara Bio, Otsu, Japan). Relative mRNA expression levels were normalized to the amount of eukaryotic translation elongation factor 1α1 mRNA (forward, GATGGCCCCAAATTCTTGAAG; reverse, GGACCATGTCAACAATGGCAG).Western blot analysis
The testis was homogenized in 5 volumes of phosphate-buffered saline and centrifuged at 1700 × g at 4 °C for 5 min. The supernatant collected was denatured in sodium dodecyl sulfate (SDS) gel loading buffer (62.5 mM Tris-HCl, pH 6.8, 2% SDS, 50 mM DTT, and 6% glycerol, final) and electrophoresed in a 12.5% SDS-polyacrylamide gel. Protein concentrations of the lysates were determined by the Bradford method using the BioRad protein assay kit (Hercules, CA) and bovine serum albumin (BSA, Sigma Chemical) as a standard. The proteins were transferred onto Immobilon-P membranes (Millipore, Billerica, MA). After blocking in Tris-buffered saline containing Tween 20 (TBS-T; 10 mM Tris-HCl pH 7.4, 150 mM NaCl, and 0.1% Tween 20) and either 5% dried milk or BSA, the membranes were reacted with anti-rat Cyp11a antibody (1/5000 dilution in TBS-T containing 5% dried milk; Chemicon, Temecula, CA), anti-phosphorylated NFκB p65 antibody (1/1000 dilution in TBS-T containing 5% BSA; Cell Signaling Technology, Danvers, MA) or anti-NFκB p65 antibody (1/1,000 dilution in TBS-T containing 5% BSA; Cell Signaling Technology) for 1 h at room temperature, washed, and incubated with anti-rabbit IgG peroxidase conjugate (0.16 μg mL−1 in TBS-T containing 5% dried milk; Pierce, Rockford, IL) for another hour. The bound antibodies were detected using the ECL Plus western blotting detection reagent (GE Healthcare Bio-Science) and a LAS-4000 mini luminescent image analyzer (Fujifilm, Tokyo, Japan). The relative expression levels of each protein were normalized according to the amount of β-actin detected using an anti-β-actin antibody (Abcam, Tokyo, Japan).Measurement of testosterone and luteinizing hormone in plasma
Testosterone concentrations in the plasma were determined using an ELISA kit purchased from Cayman Chemical (Ann Arbor, MI). Samples were prepared according to the manufacturer's instructions. In brief, testosterone from the plasma was extracted twice in 5 volumes of diethyl ether. After extraction, the organic phase was collected by centrifugation at 1500 × g for 5 min and evaporated using a vacuum centrifugal evaporator. The extract was resuspended in an adequate volume of EIA buffer that was present in the kit. Luteinizing hormone concentrations in plasma were determined using the rodent LH ELISA test purchased from Endocrine Technologies (Newark, CA).Statistical analysis
The results are expressed as the mean ± standard error (SEM). The data in Fig. 1 and 2 were analyzed by one-way analysis of variance, and multiple comparisons between the groups were performed using the Tukey-Kramer test. Data in Tables 1–3 and Fig. 3 were analyzed using the Student's t test. Statistical analyses were performed using the StatcelQC program (OMS Publishing, Saitama, Japan).Results
Effects of dietary vitamin K on plasma osteocalcin, inflammatory markers, and organ vitamin K levels
After rats were fed the experimental diets for 35 d, their body and organ weights did not differ between the VK-free and control groups (data not shown). Since the VK-free diet was prepared without the addition of any vitamin K, we measured hepatic vitamin K levels and plasma levels of total osteocalcin (OC) and undercarboxylated osteocalcin (ucOC). In the liver, vitamin K1 levels were strongly affected by diet and were significantly decreased in the VK-free diet group compared with the control group, while menaquinone-4 and other vitamin K analogues were undetectable in either group (Table 1). The plasma ucOC/total OC ratio is used to assess the state of vitamin K deficiency in a more effective manner than blood coagulation time.32,33 No significant differences in plasma total OC levels, ucOC levels, and ucOC/total OC ratios were observed between the groups (Table 2). We observed that plasma aspartate aminotransferase (AST) activity was significantly elevated in vitamin K-deficient animals with internal bleeding noted in several organs (Shirakawa et al., unpublished results). In the present experiment, no differences were observed in plasma AST activities or levels of tumor necrosis factor-α (TNF-α), an inflammatory cytokine, between either group; these values were within the normal range. In the testis, we detected only vitamin K1 and menaquinone-4, not other vitamin K analogues. Testis vitamin K, which is composed of more than 97% menaquinone-4, was significantly lower in the VK-free diet group compared with the control group (Table 1). These results clearly indicate that the VK-free diet used in this experiment did not induce vitamin K-deficient symptoms within 35 d of feeding even though the levels of vitamin K in organs were significantly reduced.| Group | Control | VK-free |
|---|---|---|
| a VK-free, vitamin K-free group; nd, not detected. Mean ± SEM (pmol g−1 tissue), n = 8. *P < 0.01 vs. control group. | ||
| Liver | ||
| Vitamin K1 | 313.33 ± 28.55 | 46.55 ± 3.02* |
| Menaquinone-4 | nd | nd |
| Testis | ||
| Vitamin K1 | 4.61 ± 0.94 | 0.53 ± 0.37* |
| Menaquinone-4 | 186.58 ± 6.65 | 105.48 ± 4.07* |
| Total | 191.20 ± 6.19 | 106.00 ± 3.86* |
| Group | Control | VK-free |
|---|---|---|
| a VK-free, vitamin K-free group; ucOC, undercarboxylated osteocalcin; OC, osteocalcin; AST, aspartate aminotransferase; TNF-α, tumor necrosis factor-α. Mean ± SEM, n = 7–8. | ||
| ucOC (ng mL−1) | 6.03 ± 0.63 | 6.43 ± 0.50 |
| Total OC (ng mL−1) | 97.32 ± 5.14 | 85.22 ± 5.42 |
| ucOC/total OC | 6.30 ± 0.70 | 7.67 ± 0.59 |
| AST activity (Karmen Unit) | 23.42 ± 2.95 | 21.63 ± 3.60 |
| TNF-α (pg mL−1) | 1.71 ± 0.61 | 0.79 ± 0.25 |
Effects of dietary vitamin K on plasma testosterone and luteinizing hormone levels after LPS administration
Plasma testosterone levels display circadian variations,34 and maximum levels were observed at approximately 6:00 PM (data not shown). Hence, we administered LPS at 12:00 AM and evaluated plasma testosterone levels 6 h after administration. With regard to plasma testosterone levels, no differences were observed between the VK-free and control diet groups without LPS treatment; however, levels were significantly reduced in the LPS-treated groups compared to the LPS-untreated groups (Fig. 1A). Furthermore, testosterone levels in the LPS-treated VK-free group were significantly reduced compared to the LPS-treated control group. Therefore, dietary vitamin K status influenced plasma testosterone levels only under LPS-treated conditions. On the other hand, plasma levels of luteinizing hormone (LH), a positive regulator of testosterone synthesis released from the pituitary, did not differ among the groups (Fig. 1B); hence, dietary vitamin K levels did not affect the release of the pituitary gonadotropin and did not likely influence the pituitary-testis gonadotropin axis.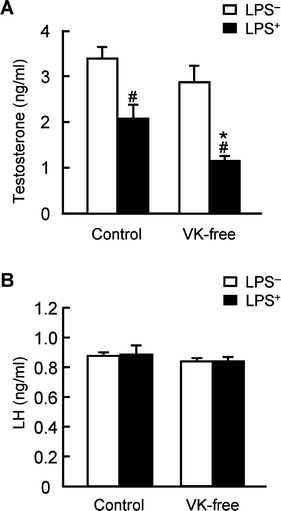 | ||
| Fig. 1 Effects of dietary vitamin K on testosterone and luteinizing hormone concentrations in the plasma. (A) Testosterone present in the plasma was extracted with diethyl ether and measured using ELISA, as described in the Materials and methods section. (B) Plasma luteinizing hormone (LH) was measured using ELISA. The data are expressed as the mean ± SEM, n = 8. * P < 0.01 vs. control group. # P < 0.01 vs. LPS-untreated group (LPS−). | ||
Effects of dietary vitamin K on Cyp11a expression in the testis
The amount of mRNA of the mitochondrial enzyme Cyp11a, a rate-limiting enzyme in testosterone synthesis in the testis, was measured by quantitative RT-PCR. After the Cyp11a mRNA level was normalized to levels of the eukaryotic translation elongation factor 1α1 mRNA as an internal standard, we observed that LPS administration significantly reduced relative Cyp11a mRNA levels in both dietary groups (Fig. 2A). LPS administration significantly reduced the levels of Cyp11a protein as determined by western blot analysis (Fig. 2B). Furthermore both mRNA and protein levels of Cyp11a in the LPS-treated VK-free group were significantly decreased compared with LPS-treated control group; these levels corresponded well to plasma testosterone levels. LPS treatment is known to increase plasma AST activity and TNF-α levels; neither differed between the groups (Table 3). Phosphorylation of NFκB p65 (activated form) is markedly increased after inflammation stimulation (e.g., by LPS).35 The levels of total and phosphorylated NFκB p65 in the testis before LPS treatment were not different between the groups (data not shown). The levels of phosphorylated NFκB p65 in the testes in the LPS-treated VK-free group were significantly increased compared with the control group although total p65 levels were not different between the groups (Fig. 3). These results indicate that dietary vitamin K suppresses the reduction in Cyp11a expression after LPS treatment and alleviates the inhibitory effects of inflammatory stimuli on testosterone synthesis.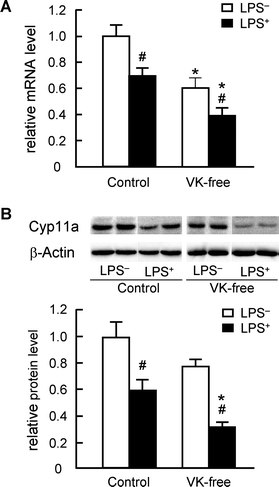 | ||
| Fig. 2 Effects of dietary vitamin K on Cyp11a mRNA and protein levels in the testis. (A) The amount of Cyp11a mRNA in the testis was determined by quantitative RT-PCR as described in the Materials and methods and is expressed as the fold-change of control LPS-untreated group (LPS−) values. (B) The amount of testicular Cyp11a protein was measured by western blot analysis as described in the Materials and methods section. Whole testis lysate (10 μg) was separated in a 12.5% SDS-polyacrylamide gel and blotted onto a PVDF membrane; Cyp11a was then detected using anti-rat Cyp11a antibody. β-Actin was used as the loading control. The data are expressed as the mean ± SEM, n = 8. * P < 0.01 vs. control groups. # P < 0.01 vs. LPS−. | ||
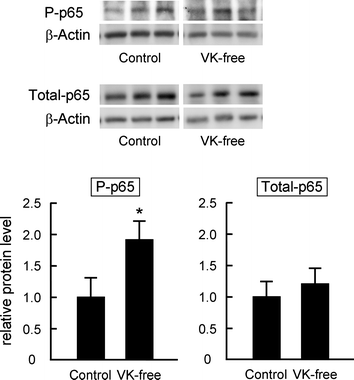 | ||
| Fig. 3 Effects of dietary vitamin K on the phosphorylation levels of NFκB p65 in the testis after LPS administration. The amount of phosphorylated (P-p65) and total NFκB p65 (Total-p65) proteins in the testis was measured by western blot analysis as described in the Materials and methods section. Whole testis lysate (10 μg) was separated in a 12.5% SDS-polyacrylamide gel and blotted onto a PVDF membrane; phosphorylated and total NFκB p65 were then detected using anti-phospho-p65 and anti-p65 antibodies. β-Actin was used as the loading control. The data are expressed as the mean ± SEM, n = 6–7. * P < 0.05 vs. control group. | ||
Discussion
Previously, we found that plasma testosterone levels in vitamin K-deficient rats were decreased, accompanying the decrease in Cyp11a mRNA and protein levels in the testis.25 Interestingly, vitamin K concentrations in the testis correlated positively and significantly with Cyp11a mRNA levels (r = 0.861, P < 0.01); however, plasma LH levels did not change under vitamin K-deficient conditions. Therefore, testicular vitamin K seems to be involved in testosterone biosynthesisvia the regulation of Cyp11a expression. Cyp11a catalyzes the synthesis of pregnenolone from cholesterol in the mitochondria, and its gene expression is regulated by the nuclear receptor SF-1 and CREB in the Leydig cells of the testis.26 The transcriptional ability of both transcription factors is stimulated by cAMP signaling from the LH receptor followed by induction of their phosphorylation and/or enhancement of their interactions with other transcription factors and coactivators.36–39 On the other hand, activated nuclear NFκB interferes with the transcriptional activity of SF-1 as a corepressor40,41 and competes with CREB for CBP, a typical coactivator of both NFκB and CREB.42 Characteristically, the vitamin K-deficient state enhances systemic inflammation19 and may stimulate NFκB activation. Additionally, vitamin K inhibits NFκB activationvia inflammatory stimuli.20,28,29 Therefore, reduced testosterone synthesis in the vitamin K-deficient state may be caused by the inactivation of SF-1 and CREBvia activated NFκB. In order to clarify this possibility, we developed an experimental model that has relatively low levels of vitamin K in the testis but does not demonstrate the symptoms associated with severe vitamin K deficiency that accompanies the inflammation. After the rats were fed with a VK-free diet for 35 d, it was observed that the ucOC/total OC ratio, a sensitive marker of the vitamin K-deficient state, TNF-α levels and AST activity in plasma did not differ between the VK-free and control diet groups (Table 2). On the other hand, vitamin K levels in the testis were significantly reduced in the VK-free diet group (55% of the control group, Table 1). Since our developed model had low vitamin K levels in the testis and did not exhibit vitamin K-deficient symptoms with systemic inflammation, we used this model for further analyses.Plasma testosterone levels were significantly reduced in both groups after LPS treatment, supporting former observations.27,43,44 Dietary vitamin K levels did not affect plasma testosterone in the LPS-untreated groups; however, in the LPS-treated groups, testosterone levels in the VK-free group were significantly lower than the control group (Fig. 1A). Previously, we reported that dietary supplementation with vitamin K suppressed the inflammation induced by LPS administration in rats, and vitamin K treatment reduced LPS-induced IL-6 mRNA expression in human macrophagic THP-1 cells.19,20IκB interacts with NFκB and retains NFκB in the cytoplasm under conditions lacking inflammatory stimuli. LPS and its induced inflammatory cytokines cause IκB phosphorylation and degradation following activation of the upstream kinase, IKK (IKKα/β phosphorylation). After degradation of IκB, free NFκB is subsequently translocated to the nucleus and stimulates transcription of its target genes. Vitamin K inhibits LPS- and cytokine-induced NFκB activationvia suppression of IKK activity in hepatocarcinoma,28 suppression of IKKα/β phosphorylation in macrophagic cells,20 and increase of IκB mRNA in osteoblasts and osteoclasts.29Phosphorylation of NFκB p65 (i.e., the activated form of NFκB) in the testis of the VK-free group was significantly induced by LPS treatment compared with that in the control group (Fig. 3) even though there were no obvious differences in plasma AST activity and TNF-α levels between either group (Table 3). Therefore, testicular vitamin K probably contributes to blockade of NFκB activation by LPS and/or LPS-induced inflammatory cytokines, thereby alleviating the reduction in testosterone synthesis. PKA signaling can influence NFκB activation.45,46Vitamin K stimulates PKA activity in neuron-like cells, hepatoma cells, and osteosarcoma cells by an unknown mechanism.22–24 The level of phosphorylated PKA catalytic subunit (activated form) in testis before LPS treatment showed decreasing tendency in VK-free group (data not shown). Thus it is possible that activated PKA might inhibit NFκB activation in testis. A detailed mechanism should be revealed after further analysis.
Local and chronic inflammation contributes to the age-related reduction in testosterone synthesis. Lowering of testosterone levels in the blood is considered a pathogenic factor of age-related diseases, such as cancer, osteoporosis, atherosclerosis, and mental disease. Dietary vitamin K predominantly accumulates in the testis and may contribute to the blockade of inflammatory signaling and maintenance of adequate levels of testosterone.
Abbreviations
| CREB | Cyclic AMP response element-binding protein |
| GGCX | γ Glutamyl carboxylase |
| LH | Luteinizing hormone |
| LPS | Lipopolysaccharide |
| NFκB | Nuclear factor κB |
| OC | Osteocalcin |
| SF-1 | Steroidogenic factor 1 |
| ucOC | Undercarboxylated osteocalcin |
Acknowledgements
This work was partially supported by a grant-in-aid for scientific research from the Japan Society for the Promotion of Science, 16580095 for H.S.References
- S. Shuman and S. L. Booth, Annu. Rev. Nutr., 2009, 29, 89–110 Search PubMed.
- K. L. Berkner, Vitam. Horm., 2008, 78, 131–156 Search PubMed.
- J. W. Suttie, Annu. Rev. Nutr., 1995, 15, 399–417 Search PubMed.
- B. L. Sarah and A. A. Rajabi, Vitam. Horm., 2008, 78, 1–22 Search PubMed.
- R. Yamamoto, M. Komai, K. Kojima, Y. Furukawa and S. Kimura, J. Nutr. Sci. Vitaminol. (Tokyo), 1997, 43, 133–143 Search PubMed.
- J. E. Ronden, H. H. Thijssen and C. Vermeer, Biochim. Biophys. Acta, Gen. Subj., 1998, 1379, 16–22 Search PubMed.
- A. M. Huber, K. W. Davidson, M. E. O'Brien-Morse and J. A. Sadowski, J. Nutr., 1999, 129, 1039–1044 Search PubMed.
- T. Shibayama-Imazu, T. Aiuchi and K. Nakaya, Vitam. Horm., 2008, 78, 211–226 Search PubMed.
- Y. Akiyama, K. Hara, T. Tajima, S. Murota and I. Morita, Eur. J. Pharmacol., 1994, 263, 181–185 Search PubMed.
- K. Hara, Y. Akiyama, T. Nakamura, S. Murota and I. Morita, Bone, 1995, 16, 179–184 Search PubMed.
- Y. Takeuchi, M. Suzawa, S. Fukumoto and T. Fujita, Bone, 2000, 27, 769–776 Search PubMed.
- H. Taira, Y. Fujikawa, O. Kudo, I. Itonaga and T. Torisu, Calcif. Tissue Int., 2003, 73, 78–85 Search PubMed.
- S. Urayama, A. Kawakami, T. Nakashima, M. Tsuboi, S. Yamasaki, A. Hida, Y. Ichinose, H. Nakamura, E. Ejima, T. Aoyagi, T. Nakamura, K. Migita, Y. Kawabe and K. Eguchi, J. Lab. Clin. Med., 2000, 136, 181–193 Search PubMed.
- M. M. Tabb, A. Sun, C. Zhou, F. Grun, J. Errandi, K. Romero, H. Pham, S. Inoue, S. Mallick, M. Lin, B. M. Forman and B. Blumberg, J. Biol. Chem., 2003, 278, 43919–43927 Search PubMed.
- T. Ichikawa, K. Horie-Inoue, K. Ikeda, B. Blumberg and S. Inoue, J. Biol. Chem., 2006, 281, 16927–16934 Search PubMed.
- M. Igarashi, Y. Yogiashi, M. Mihara, I. Takada, H. Kitagawa and S. Kato, Mol. Cell. Biol., 2007, 27, 7947–7954 Search PubMed.
- K. Azuma, T. Urano, Y. Ouchi and S. Inoue, Endocr. J., 2009, 56, 843–849 Search PubMed.
- K. Reddi, B. Henderson, S. Meghji, M. Wilson, S. Poole, C. Hopper, M. Harris and S. J. Hodges, Cytokine, 1995, 7, 287–290 Search PubMed.
- Y. Ohsaki, H. Shirakawa, K. Hiwatashi, Y. Furukawa, T. Mizutani and M. Komai, Biosci., Biotechnol., Biochem., 2006, 70, 926–932 Search PubMed.
- Y. Ohsaki, H. Shirakawa, A. Miura, P. E. Giriwono, S. Sato, A. Ohashi, M. Iribe, T. Goto and M. Komai, J. Nutr. Biochem., 2010, 21, 1120–1126 Search PubMed.
- J. Li, J. C. Lin, H. Wang, J. W. Peterson, B. C. Furie, B. Furie, S. L. Booth, J. J. Volpe and P. A. Rosenberg, J. Neurosci., 2003, 23, 5816–5826 Search PubMed.
- C. K. Tsang and Y. Kamei, Neurosci. Lett., 2002, 323, 9–12 Search PubMed.
- M. Otsuka, N. Kato, R. X. Shao, Y. Hoshida, H. Ijichi, Y. Koike, H. Taniguchi, M. Moriyama, Y. Shiratori, T. Kawabe and M. Omata, Hepatology, 2004, 40, 243–251 Search PubMed.
- T. Ichikawa, K. Horie-Inoue, K. Ikeda, B. Blumberg and S. Inoue, J. Mol. Endocrinol., 2007, 39, 239–247 Search PubMed.
- H. Shirakawa, Y. Ohsaki, Y. Minegishi, N. Takumi, K. Ohinata, Y. Furukawa, T. Mizutani and M. Komai, Biochim. Biophys. Acta, Gen. Subj., 2006, 1760, 1482–1488 Search PubMed.
- I. C. Guo, M. C. Hu and B. C. Chung, J. Biomed. Sci., 2003, 10, 593–598 Search PubMed.
- D. B. Hales, J. Reprod. Immunol., 2002, 57, 3–18 Search PubMed.
- I. Ozaki, H. Zhang, T. Mizuta, Y. Ide, Y. Eguchi, T. Yasutake, T. Sakamaki, R. G. Pestell and K. Yamamoto, Clin. Cancer Res., 2007, 13, 2236–2245 Search PubMed.
- M. Yamaguchi and M. N. Weitzmann, Int. J. Mol. Med., 2011, 27, 3–14 Search PubMed.
- P. G. Reeves, F. H. Nielsen and G. C. Fahey Jr, J. Nutr., 1993, 123, 1939–1951 CAS.
- T. Sato, Y. Ohtani, Y. Yamada, S. Saitoh and H. Harada, Br. J. Nutr., 2007, 87, 307–314 Search PubMed.
- P. A. Price and Y. Kaneda, Thromb. Res., 1987, 46, 121–131 Search PubMed.
- S. L. Booth, L. Martini, J. W. Peterson, E. Saltzman, G. E. Dallal and R. J. Wood, J. Nutr., 2003, 133, 2565–2569 Search PubMed.
- A. I. Esquifino, F. Chacon, V. Jimenez, C. F. Reyes Toso and D. P. Cardinali, J. Circadian Rhythms, 2004, 2, 1 Search PubMed.
- P. Viatour, M. P. Merville, V. Bours and A. Chariot, Trends Biochem. Sci., 2005, 30, 43–52 Search PubMed.
- G. D. Hammer, I. Krylova, Y. Zhang, B. D. Darimont, K. Simpson, N. L. Weigel and H. A. Ingraham, Mol. Cell, 1999, 3, 521–526 Search PubMed.
- D. L. Carlone and J. S. Richards, J. Steroid Biochem. Mol. Biol., 1997, 61, 223–231 Search PubMed.
- Z. Liu and E. R. Simpson, Mol. Cell. Endocrinol., 1999, 153, 183–196 Search PubMed.
- D. Monté, F. DeWitte and D. W. Hum, J. Biol. Chem., 1998, 273, 4585–4591 Search PubMed.
- C. Y. Hong, J. H. Park, K. H. Seo, J. M. Kim, S. Y. Im, J. W. Lee, H. S. Choi and K. Lee, Mol. Cell. Biol., 2003, 23, 6000–6012 Search PubMed.
- C. Y. Hong, J. H. Park, R. S. Ahn, S. Y. Im, H. S. Choi, J. Soh, S. H. Mellon and K. Lee, Mol. Cell. Biol., 2004, 24, 2593–2604 Search PubMed.
- R. Shenkar, H.-K. Yum, J. Arcaroli, J. Kupfner and E. Abraham, Am. J. Physiol. Lung Cell. Mol. Physiol., 2001, 281, L418–L426 Search PubMed.
- M. K. O'Bryan, S. Schlatt, D. J. Phillips, D. M. de Kretser and M. P. Hedger, Endocrinology, 2000, 141, 238–246 Search PubMed.
- K. Held Hales, T. Diemer, S. Ginde, B. K. Shankar, M. Roberts, H. B. Bosmann and D. B. Hales, Endocrinology, 2000, 141, 4000–4012 Search PubMed.
- N. Takahashi, T. Tetsuka, H. Uranishi and T. Okamoto, Eur. J. Biochem., 2002, 269, 4559–4565 Search PubMed.
- A. Oeckinghaus and S. Ghosh, Cold Spring Harbor Perspect. Biol., 2009, 1, a000034 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |