Bioavailability of hop-derived iso-α-acids and reduced derivatives
Ko
Cattoor
a,
Jean-Paul
Remon
b,
Koen
Boussery
c,
Jan
Van Bocxlaer
c,
Marc
Bracke
d,
Denis
De Keukeleire
a,
Dieter
Deforce
ae and
Arne
Heyerick
*a
aUGent-Ghent University, Faculty of Pharmaceutical Sciences, Laboratory of Pharmacognosy and Phytochemistry, Harelbekestraat 72, B-9000, Ghent, Belgium. E-mail: Arne.Heyerick@UGent.be; Fax: +32 (0)9 264 8192; Tel: +32 (0)9 264 8058
bUGent-Ghent University, Faculty of Pharmaceutical Sciences, Laboratory of Pharmaceutical Technology, Harelbekestraat 72, B-9000, Ghent, Belgium
cUGent-Ghent University, Faculty of Pharmaceutical Sciences, Laboratory of Medicinal Biochemistry and Clinical Analysis, Harelbekestraat 72, B-9000, Ghent, Belgium
dGhent University Hospital, Faculty of Medicine and Health Sciences, Laboratory of Experimental Cancer Research, De Pintelaan 185, B-9000, Ghent, Belgium
eUGent-Ghent University, Faculty of Pharmaceutical Sciences, Laboratory of Pharmaceutical Biotechnology, Harelbekestraat 72, B-9000, Ghent, Belgium
First published on 17th June 2011
Abstract
Iso-α-acids (IAA) and their reduced derivatives (dihydro-iso-α-acids (DHIAA) and tetrahydro-iso-α-acids (THIAA)) have been administered to Caco-2 cell monolayers (30, 60, and 120 μM) to investigate epithelial transport, in both absorptive and secretive directions. In addition, 25 mg kg−1 IAA, DHIAA, and THIAA were applied to New Zealand white rabbits (±3–3.5 kg) in a single intravenous and oral dose. The most important pharmacokinetic parameters (Cmax, tmax, half life, clearance, and AUC0−∞) and the absolute bioavailability were determined for each class of hop acid. The results from the in vitro Caco-2 study of IAA, DHIAA, and THIAA, showed a higher membrane permeability for IAA and THIAA, both in absorptive (PappAB range 1.6–5.6 × 10−6 cm s−1) and secretive directions (PappBA range 5.7–16.3 × 10−6 cm s−1), when compared to DHIAA. Factors limiting transport of DHIAA could include phase II metabolism. After oral and i.v. dosing to New Zealand white rabbits, the absolute bioavailability for IAA was determined to be 13.0%. The reduced derivatives reached higher bioavailabilities with 28.0% for DHIAA and 23.0% for THIAA. The area under curve AUC0−∞ upon oral gavage for DHIAA and THIAA was 70.7 ± 48.4 μg h ml−1 and 57.4 ± 9.0 μg h ml−1, respectively, while that for IAA was 10.6 ± 5.3 μg h ml−1. Phase I metabolism was indicated as the main factor limiting the bioavailability of IAA. Bioavailability of DHIAA is mostly influenced by phase-II metabolism as shown by enzymatic hydrolysis of plasma samples upon administration of DHIAA.
Introduction
Scientific evidence indicates that moderate beer consumption contributes to a healthy lifestyle, reducing for example risks of cardiovascular disease and blood cholesterol levels.1 This observation is not only related to the presence of ethanol,2–4 but, more importantly, also to unique biological activities of hop-derived constituents.5–7 Especially the hop bitter acids (HBA) and derivatives are of great importance, as evidenced by the increasing number of reports on various promising bioactivities with IC50 values in the lower micromolar range (anti-inflammatory and anti-angiogenic properties, improving lipid profiles, and counteracting diabetes type 2).8–13HBA originate from female hop flowers (Humulus lupulus L.) and are used in the brewing process as the main flavouring agent and as a natural preservative. During wort boiling with hop, the α-acids, the main bitter acids present in hops, are converted into iso-α-acids thereby providing beer with its typical bitter taste and foam stability. In hop, the α-acids are present as a mixture of 3 major analogues (cohumulone, n-humulone, and adhumulone), differing in the nature of the acyl side chain (Fig. 1). For each analogue, two different beer-soluble products (cis- and trans-) can be formed, resulting in six different iso-α-acids (IAA) or isohumulones (Fig. 1).14 Due to the susceptibility of IAA to form light-induced off-flavours,15–19 light stable beers, containing reduced derivatives, such as dihydro-iso-α-acids (DHIAA) and tetrahydro-iso-α-acids (THIAA) (Fig. 1), instead of IAA, have been developed by the brewing industry. The reduced derivatives, in general, are also more stable with respect to oxidation, while THIAA, specifically, are also used to further enhance foam stability.20 DHIAA are produced by hydride reduction of IAA, introducing an additional chiral centre on the acyl side chain, leading to two epimeric reaction products for each iso-α-analogue.21 As a result, theoretically, the group of DHIAA can consist of twelve stereoisomeric products (Fig. 1). THIAA are formed by hydrogenation of the double bonds present in the side chains of IAA, thus also consisting of cis- and trans-isomeric pairs, totalling six stereoisomers.22,23
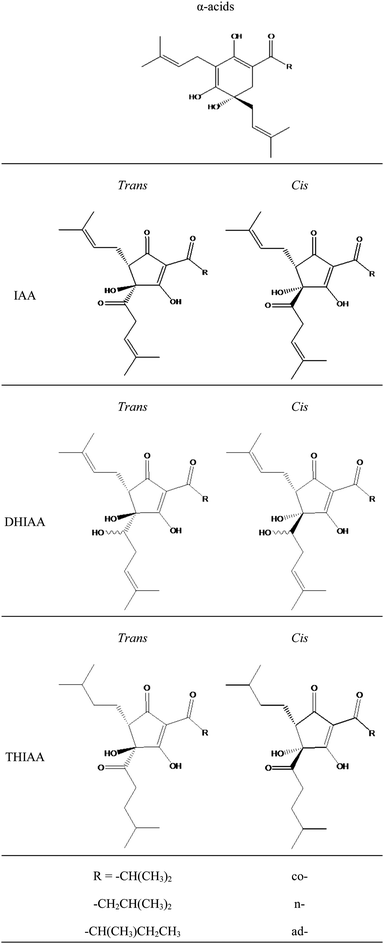 | ||
| Fig. 1 Molecular structures of hop α-acids, iso-α-acids and reduced derivatives. IAA: iso-α-acids, DHIAA: dihydro-iso-α-acids, THIAA: tetrahydro-iso-α-acids. | ||
Depending on the bitterness, the hop-derived bitter acid content in beers can vary from 10 up to 100 mg l−1,24–26 while commercially available hop-based food supplements may contain up to 400 mg of hop-derived bitter acids.27 The presence of such high concentrations of IAA and derivatives could possibly explain the positive health effects associated with ingestion of preparations containing these products.
In this area, the number of scientific publications on the possible health-benefits of HBA is increasing, while, until now, little is known on absorption, distribution, metabolism, and excretion (ADME) of these compounds. In one study on the safety, efficacy and anti-inflammatory activity of DHIAA from hops, Hall et al. reported limited data on the human absorption of DHIAA. Following oral administration of 1000 mg of DHIAA to 2 healthy human subjects, a maximum concentration in the lower micromolar range was reached at 4 h post administration.28
In the present study, different aspects of the bioavailability of the HBA present in beer were investigated. First, using a Caco-2 cell model system the intestinal absorption of IAA, DHIAA, and THIAA was studied in vitro. Differentiated human epithelial Caco-2 cells are commonly applied as a screening tool for the prediction of intestinal absorption of compounds.29 Notably, in vitro permeability coefficients measured for reference compounds obtained in the Caco-2 cell model have shown good correlation with results based on in vivo studies.
Second, pharmacokinetic parameters for IAA, DHIAA, and THIAA were investigated upon both intravenous and oral dosing in New Zealand white rabbits. Knowledge on the bioavailabilities and the pharmacokinetic parameters of hop-derived IAA, DHIAA, and THIAA is essential to understand possible health benefits associated to preparations containing hop-derived compounds such as beer and hop-based food supplements.
Materials and methods
Materials
Isohop®, Redihop®, and Tetrahop Gold® were obtained from Barth-Haas Group (Botanix, Kent, UK). LC-MS solvents (analytical grade) were from Biosolve (Valkenswaard, the Netherlands). International calibration standards for iso-α-acids (DCHA-Iso, ICS-I3), dihydro-iso-α-acids (all cis-dihydro-Iso, ICS-R2), and tetrahydro-iso-α-acids (ICS-T3) were all obtained from Labor Veritas (Zurich, Switzerland). Atenolol, propranolol, sulfatase type H1 (from Helix pomatia), 4-(2-Hydroxyethyl)-1-piperazine-ethane sulphonic acid (HEPES) sodium salt 99%, D-(+)-glucose, and all buffer constituents were purchased from Sigma-Aldrich (Bornem, Belgium). Glutamine, non-essential amino acid solution, penicillin G sodium, fungizone, amphotericin B, and fetal bovine serum (FBS) were purchased from Gibco (Invitrogen Corp., Merelbeke, Belgium).Methods
Caco-2 cell culture. Caco-2 cells (American Type Culture Collection (ATCC), Rockville, MD, USA) originating from a human colorectal carcinoma were cultured in Dulbecco's Modified Eagle Medium (DMEM) containing GLUTAMAX™, supplemented with 10% FBS, 1% non-essential amino acids, 1% L-glutamine, 100 U/ml penicillin, and 100 μg ml−1 streptomycin. Cells were grown in 25 cm2 culture flasks (Corning Costar, New York, USA) in an atmosphere of 10% CO2 and 90% relative humidity at 37 °C (Forma Scientific, Marietta, Ohio, USA). Cells were passaged every 7 days (90–95% confluence) at a split ratio of 1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. For transport studies, Caco-2 cells were seeded at a density of 1 × 105 cells cm−2 on Transwell® membrane inserts (0.4 μm pore diameter, 12 mm diameter, Corning Costar, New York, USA) and cultured until late confluence. Cell culture medium was changed every other day. Monolayers were investigated between days 18 and 24 post-seeding. Cells with passage numbers 25–50 were used. The integrity of each monolayer of differentiated cells was monitored by measuring the transepithelial electrical resistance (TEER) with a Millicel-ERS volt-ohmmeter (Millipore, Bedford, MA, USA). The TEER is a measure for the presence of tight junctions between adjacent cells. The volumes amounted to 1.5 ml at the apical side (AP) and 2.6 ml at the basolateral side (BL) of the monolayer.
:10. For transport studies, Caco-2 cells were seeded at a density of 1 × 105 cells cm−2 on Transwell® membrane inserts (0.4 μm pore diameter, 12 mm diameter, Corning Costar, New York, USA) and cultured until late confluence. Cell culture medium was changed every other day. Monolayers were investigated between days 18 and 24 post-seeding. Cells with passage numbers 25–50 were used. The integrity of each monolayer of differentiated cells was monitored by measuring the transepithelial electrical resistance (TEER) with a Millicel-ERS volt-ohmmeter (Millipore, Bedford, MA, USA). The TEER is a measure for the presence of tight junctions between adjacent cells. The volumes amounted to 1.5 ml at the apical side (AP) and 2.6 ml at the basolateral side (BL) of the monolayer.
Bidirectional transport assay. The transport medium was Hanks' buffered saline solution (HBSS) containing 10 mM HEPES + 25 mM D-(+)-glucose, adjusted to pH 7.4. The osmolarity was 350 mOsm/l, verified with an osmometer (Knauer, Berlin, Germany). Prior to the experiments, the cell culture medium was removed from both AP and BL chambers of the Transwell® plate. The cells were washed three times and pre-incubated (37 °C, 10% CO2) with transport medium for 30 min. TEER was measured before the experiments. Only cells with initial TEER values >300 Ω cm2 were used.30
In the experimental setup, different donor concentrations (30, 60, and 120 μM) were applied by adding a solution of IAA, DHIAA, or THIAA to either the AP compartment (for absorptive transport study; AP-to-BL) or to the BL compartment (for secretive transport study; BL-to-AP). Donor solutions were diluted from commercially available solutions of potassium salts of hop-derived bitter acids at pH 8–10 (Isohop®, Redihop®, and Tetrahop Gold® containing respectively 200, 300, and 90 mg ml−1 of IAA, DHIAA, and THIAA) in HBSS (dilution factor >4000). Blank transport medium was added to the other (receiving) compartment. After 1, 2, and 4 h of incubation, samples were taken out from the basolateral (for AP-to-BL transport) or apical (for BL-to-AP transport) side and the volume was replaced with blank transport medium. At the last sampling point (4 h), an aliquot of the donor compartment was included. In order to quantify the absorbed intracellular amounts, excessive transport medium was removed and monolayers were extracted with EtOH (200 μl AP; 750 μl BL) during 30 min. Each experiment was performed in triplicate (three sequential wells with Caco-2 monolayers were tested), in correspondence with several other reports on methodologies of the in vitro transport experiments across Caco-2 monolayers.31–33
Samples (apical (200 μl) and basolateral (750 μl)) taken from the Caco-2 assay at different incubation times were spiked with internal standard (IS) (1.0 μg). Having an almost identical molecular structure, THIAA were applied as internal standard in case of samples containing IAA and DHIAA. In samples following dosing of THIAA, DHIAA were used as internal standard. Next, samples were acidified (pH 2) with H3PO4 (0.1M; 1.5 volumes) followed by extraction with ethyl acetate (EtOAc; 4 volumes). Collected organic phases were evaporated (N2) and residues were reconstituted in 100μl methanol (MeOH). Samples were stored at −20 °C until LC-MS analysis.
Caco-2 control measurements. The low-permeability standard atenolol (50 μM) and the high-permeability standard propranolol (20 μM) were added to the monolayers simultaneously (in the same well) with the test compounds. TEER was measured before and after the experiments. Monolayers with low TEER values assumed to exhibit extensive leakage through imperfect occluding junctions or holes in the monolayer were discarded. Values for the atenolol flux are usually lower than 1.0 × 10−6 cm s−1 and higher than 10 × 10−6 cm s−1 for the propranolol flux.31
Enzymatic hydrolysis of Caco-2 monolayer extracts. Conjugated levels of hop-derived acids were quantified by enzymatic hydrolysis by based on a method validated by Wyns et al.34 Cell fraction extract aliquots (EtOH; 375 μl) were evaporated until dryness. Afterwards, residues were re-dissolved in 5 volumes NaOAc buffer (0.1 M, pH 5) and a preparation containing both β-glucuronidase (10
000 units/ml) and sulfatase (330 units/ml) activity from H. pomatia (30 μl) from a solution in NaOAc buffer (0.1 M, pH 5) was added. Samples were incubated for 2 h at 37 °C. Subsequently, samples were extracted as described in section ‘Bidirectional transport assay'. Replicate control samples were included with no enzyme treatment to determine the extent of glucuronidation and/or sulfation. Cellular levels of conjugated hop-derived acids were calculated by subtracting the amount of free hop acid (no enzyme treatment) from the amount of total hop-derived acids (+ β-glucuronidase/sulfatase).
Caco-2 experiment calculations. The results of the transport experiments are expressed as an apparent permeability coefficient Papp (cm s−1), which was calculated as described previously.35 Papp = δQ/δt × A−1 × C0−1 with δQ/δt (nmol s−1): the rate of transport of the compound in the receiving chamber over time, A (cm2): the surface area of the cell monolayer, C0 (nmol cm−3): the initial compound concentration in the donor compartment. Another important property was the efflux ratio (ER), which is used to represent the extent of efflux, which was calculated according the following equation: Efflux ratio = PappBA (mean)/PappAB (mean), where PappBA and PappAB are respectively the apparent permeability coefficients for transport from the basolateral to apical compartment and from the apical to basolateral compartment.
Collection of plasma samples. A first group of nine rabbits was divided in 3 groups of three animals. Each group (n = 3) received an i.v. dose in the marginal ear vein of 25 mg kg−1 (dose volume 1 ml) of IAA, DHIAA, or THIAA, respectively. Donor solutions containing 75 mg ml−1 of IAA, DHIAA, or THIAA were prepared in ammonium acetate buffer (0.1M, pH 10) by dilution of commercially available aqueous solutions (potassium salts) of hop-derived bitter acids at pH 8–10 (Isohop® 30%, Redihop® 35%, and Tetrahop Gold® 9%). The pH of the solution was increased to allow solubilizing the compounds of interest. It is not expected that this will significantly influence the pharmacokinetics upon both intravenous and oral intake as the total dosed volume was low (1.0 ml) and the recipient compartments (blood or stomach) have sufficient buffering capacity. Blood samples were collected from the ear vein at 0.5 min before and at 0.5, 2, 5, 10, 15, 30, 45, 60 and 120 min after i.v. dosing. A second group of eighteen rabbits was randomly divided in three groups of six animals. Each group (n = 6) was administered, via oral gavage using a syringe, an oral dose of 25 mg kg−1 (dose volume 1 ml) of IAA, DHIAA, or THIAA, respectively. Similar to the i.v. administration, dosing solutions were diluted in ammonium acetate buffer (0.1 M, pH 10) to a final concentration of 75 mg ml−1 from commercially available Isohop® 30%, Redihop® 35%, and Tetrahop Gold® 9%. Blood samples were collected at 0.5 min before and at 0.5, 1, 2, 4, 6, 8, 12, 16 and 24 h after oral dosing. After addition of heparin (LEO Pharma, Wilrijk, Belgium) to the blood samples, plasma was separated by centrifugation (700 × g, 5 min) and samples were stored at −20 °C until further sample processing.
Collection of faeces and urine. In a separate experiment, eighteen rabbits were divided in six groups of three animals. Each group was either administered a single oral dose or single i.v. dose in the marginal ear vein of 25 mg kg−1 solution of IAA, DHIAA, or THIAA, respectively. Donor solutions were diluted in ammonium acetate buffer (0.1M, pH 10) from commercially available solutions of potassium salts of HBA at pH 8–10 (Isohop® 30%, Redihop® 35%, and Tetrahop Gold® 9%, respectively). Urine and faeces were collected over a period of 24 h after oral and i.v. dosing. All samples were immediately frozen at −20 °C and analyzed within 2 months.
Sample extraction. Total volume of urine was determined and a sample of the total urine (500 μl) was diluted with 2.5 ml NaOAc buffer (0.1 M; pH 5.0). Faecal material was lyophilized, weighted and grinded. Samples of total faeces (1.0 g) were diluted with 3 ml H2O and homogenized. To 1.0 g of this homogenate, 5 ml NaOAc buffer (0.1 M; pH 5.0) was added. Subsequently the urine, faecal and plasma samples (300 μl) were spiked with internal standard (IS) (1.0 μg). Having an almost identical molecular structure, THIAA were applied as internal standard in case of samples containing IAA and DHIAA. In plasma samples following dosing of THIAA, DHIAA were used as internal standard. After addition of IS, samples were acidified (pH 2) with H3PO4 (0.1 M; 1.5 volumes) followed by extraction with EtOAc (4 volumes). The EtOAc phase was evaporated (N2) and residues were reconstituted in 100 μl MeOH. Samples were stored at −20 °C prior to LC-MS analysis.
Purity of donor solutions. The purity of the donor solutions from commercially available Isohop®, Redihop® and Tetrahop Gold® was evaluated as follows: Isohop®, Redihop® and Tetrahop Gold® were diluted with methanol at a concentration of 0.1 and 1.0 μg ml−1 and analyzed by LC-MS to determine quantity of IAA, DHIAA, and THIAA in each solution.
Stability in urine and plasma and extraction recovery. The stability of IAA, DHIAA, and THIAA in rabbit plasma and urine was evaluated. Blank plasma (500μl) and urine (300 μl) were spiked with IAA, DHIAA, or THIAA at a concentration of 0.1 and 1.0 μg ml−1 and left at room temperature for 1 h. Next, the samples were extracted following the procedure described in the section “Sample extraction” and analyzed using LC-MS.
Enzymatic hydrolysis of samples. The extent of phase-II conjugation of the HBA as sulphate or glucuronide in the different samples (plasma, urine, and faeces) was determinate by enzymatic hydrolysis. based on a method validated by Wyns et al.34 Plasma samples (300 μl) and a sample of total urine (500 μl) were diluted with 5 volumes NaOAc buffer (0.1 M; pH 5.0). A sample (1.0 g) of total faeces was diluted with 3 ml H2O and homogenized. To 1.0 g of this homogenate, 5 ml NaOAc buffer (0.1 M; pH 5.0) was added. To each of these mixtures, a preparation containing both β-glucuronidase (10000 units/ml) and sulfatase (330 units/ml) activity from H. pomatia (30 μl) from a solution in NaOAc buffer (0.1 M, pH 5) was added. Samples were incubated for 2h at 37 °C. Afterwards, samples were extracted as described in section ‘Sample extraction'. Replicate control samples of urine, faeces, and plasma were included with no enzyme treatment to determine the extent of glucuronidation and/or sulfation. Urine, plasma -and faecal levels of conjugated hop-derived acids were calculated by subtracting the amount of free hop acid (no enzyme treatment) from the amount of total hop-derived acids (+ β-glucuronidase/sulfatase).
| Compound | m/z (negative ionization) | retention time (min) |
|---|---|---|
| a IAA: iso-α-acids; DHIAA: dihydroiso-α-acids; THIAA: tetrahydroios-α-acids. | ||
| IAA | ||
| cis-isoco | 347 | 11.1 |
| trans-isoco | 347 | 11.7 |
| cis-isoad | 361 | 13.8 |
| cis-iso-n + trans-isoad | 361 | 14.3 |
| trans-iso-n | 361 | 14.8 |
| DHIAA | ||
| cis-DHisoco | 349 | 7.4 |
| cis-DHisoco | 349 | 9.4 |
| cis-DHisoad | 363 | 9.9 |
| cis-DHiso-n | 363 | 10.3 |
| cis-DHisoad | 363 | 11.7 |
| cis-DHiso-n | 363 | 12.3 |
| THIAA | ||
| cis-THisoco | 351 | 16.7 |
| trans-THisoco | 351 | 17.9 |
| cis-THisoad | 365 | 19.1 |
| cis-THiso-n | 365 | 19.4 |
| trans-THisoad | 365 | 20.5 |
| trans-THiso-n | 365 | 20.8 |
Pharmacokinetic calculations
The plasma concentration-time profiles were analyzed by WinNonLin® (version 5.2.1, Pharsight Corporation, Mountain View, CA, USA). The pharmacokinetic parameters were determined from the individual plasma concentration-time profiles by non-compartmental analysis. According to the plasma concentration-time curves, the area under curve (AUC0-t) was calculated by the linear trapezoidal rule from zero to the last time point showing a measurable concentration of the analyte. The terminal half-life (t1/2) was calculated as ln(2)/λz and λz was the terminal elimination rate constant and was estimated from the slope of the terminal regression line. The AUC0−∞ was calculated as AUC0-t + Ct/λz, where Ct is the last detectable plasma concentration and t is the time at which this concentration occurred.In all of the cases, the degree of extrapolation of AUC0−∞ was lower than 20%. Peak concentration (Cmax) and the time at which this occurred (tmax) were obtained from the observed data. Oral bioavailability (F) was determined by the ratio of the AUC0−∞ following oral and i.v. dosing.
Statistical analysis
SPSS release 17.0 for Windows (SPSS, Inc., Chicago, IL) was used for all statistical analyses. The Caco-2 assays had three independent observations (three sequential wells with Caco-2 monolayers were tested) for each test group. The in vivo experiments had three independent observations for each test group (an identical dose of respectively IAA, DHIAA, and THIAA was applied to three rabbits), except for the oral plasma study in which six individual rabbits received an identical oral dose. Data were expressed as means ± standard deviation. Normality of distribution was investigated using Shapiro-Wilk test and the homogeneity of variances was evaluated using Levene's test. Comparison of means between more than two groups was performed using one-way analysis of variance (ANOVA) followed by Bonferroni post-hoc comparison of statistical significance. Results were considered to be statistically significant when P < 0.05.Results
Transport of marker compounds across Caco-2 cell monolayers and monolayer integrity
The Caco-2 cell monolayers were assessed with respect to their barrier properties using reference compounds atenolol (paracellular transport) and propranolol (passive transcellular transport). Propranolol and atenolol showed Papp values of 25.3 ± 2.9 × 10−6 and 0.33 ± 0.07 × 10−6 cm s−1, respectively, which is in correspondence with reported data.30,32,37–39 The results indicate that the monolayers can be used to discriminate between compounds with low and high permeabilities.The concentrations of HBA applied in the experiments did not affect TEER or transepithelial transport, confirming the preservation of integrity of the monolayers.Bidirectional transport of IAA and reduced derivatives across Caco-2 monolayers
The present study was undertaken to investigate the in vitro transport of IAA, DHIAA, and THIAA using Caco-2 cell monolayers. The transport characteristics were determined in both the apical-to-basolateral (AP-to-BL) and basolateral-to-apical (BL-to-AP) direction. In Fig. 2A (AP-to-BL) and 2B (BL-to-AP), the cumulative concentrations (μM) transported into the receiving chamber after 4 h incubation as a function of the donor concentration of the different HBA is presented. Significantly higher concentrations (P < 0.0001) of IAA and THIAA were transported to the receiving compartment compared with DHIAA, both in absorptive (6.5 ± 0.4 μM and 7.0 ± 1.0 μM for IAA and THIAA versus 3.3 ± 0.2 μM for DHIAA) and secretive (21.5 ± 5.4 μM and 24.2 ± 2.5 μM for IAA and THIAA versus 7.1 ± 1.9 μM for DHIAA) directions, in a linear dose-dependent relationship. The cumulative concentrations (μM) transported into the receiving compartment for a dose of 120 μM as a function of time are shown in Fig. 2C (AP-to-BL) and 2D (BL-to-AP).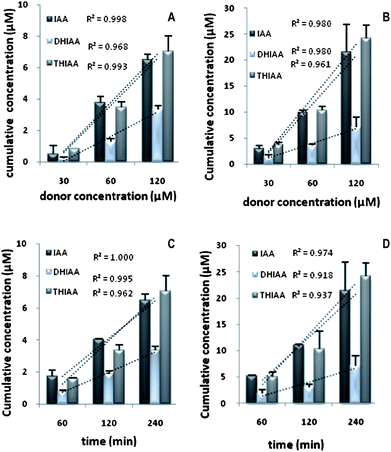 | ||
| Fig. 2 Cumulative concentrations (μM) of IAA, DHIAA, and THIAA transported across Caco-2 monolayers in absorptive (A) (AP-to-BL) and secretive (B) (BL-to-AP) directions in function of different donor concentrations for 4 h incubation and in absorptive (C) (AP-to-BL) and secretive (D) (BL-to-AP) directions in function of time for a donor concentration of 120 μM. Each point was the mean ± standard deviation of three experiments. | ||
For all the hop-derived acids, the amount transported into the receiver chambers increased linearly with time, in both absorptive and secretive directions. The rates of transport are presented in Table 2, which shows the values for the apparent permeability coefficients Papp and efflux ratios (ratio of PappBAversus PappAB) for the hop-derived acids in the AP-to-BL, as well as in the BL-to-AP direction. The PappAB values ranged from 1.58 × 10−6 to 5.57 × 10−6 cm s−1. Both IAA and THIAA showed similar absorption transport rates, since differences in their PappAB values were not statistically significant. In secretive transport, the PappBA of the hop-derived acids varied from 5.68 × 10−6 to 16.28 × 10−6 cm s−1. Efflux ratios of IAA and THIAA were similar around 3, slightly lower than the value for DHIAA which was around 3.5.
| Compound | Papp (× 10−6 cm s−1) | Efflux ratio | |
|---|---|---|---|
| AP-to-BL | BL-to-AP | ||
| a Significant difference in Papp (AP-to-BL) of DHIAA versus IAA and THIAA. P < 0.0001. b Significant difference in Papp (BL-to-AP) of DHIAA versus IAA and THIAA. P < 0.0001. | |||
| IAA | 4.62 ± 1.29 | 13.02 ± 3.13 | 2.8 ± 1.4 |
| DHIAA | 1.58 ± 0.22a | 5.68 ± 0.87b | 3.5 ± 1.0 |
| THIAA | 5.57 ± 1.22 | 16.28 ± 2.71 | 2.9 ± 1.1 |
Intracellular accumulation of IAA and reduced derivatives in Caco-2 cells
Mass balance was determined from the sum of the cumulative amount transported, the amount remaining in the donor compartment, and the amount accumulated in the cells during the transport experiment in relation to the initial amount in the donor compartment. The total amount recovered of IAA and THIAA was 85–90% in all the experiments indicating that there was no significant breakdown, metabolism, nor sorption to the surface of the Transwells®. In contrast, recovery of DHIAA was lower than 45%. The percentages of hop-derived acids associated to the cells with respect to the initial dose was found to be 1.5% with IAA, 0.9% DHIAA, and 2.0% with THIAA. This showed that hop-derived acids were not significantly accumulated in the cells during the transport experiment. In contrast, a substantial amount was transported across the monolayer.To probe possible phase-II metabolism of IAA, DHIAA, and THIAA, enzymatic hydrolysis of fractions from cell monolayers was carried out with a mixture of sulfatase and glucuronidase activities. The presence of conjugates could not be demonstrated in samples of IAA and THIAA. The amounts, quantified after deconjugation, were not significantly different from the non-hydrolyzed levels, indicating that formation of phase-II metabolites of IAA and THIAA seems unlikely. However, enzymatic hydrolysis of cellular fractions of DHIAA showed that up to 60% of the intracellular amount was conjugated as a glucuronide or a sulfate.
Purity of donor solutions
The purity of the donor solutions was evaluated by determining the quantity of IAA, DHIAA, and THIAA in the commercially available Isohop®, Redihop® and Tetrahop Gold®. In all of the solutions tested, purity was more than 98%.Stability in urine and plasma and extraction recovery
To investigate the stability of the studied compounds in rabbit plasma and urine, control experiments with IAA, DHIAA, and THIAA were carried out. Almost all (>95%) of the spiked IAA, DHIAA, and THIAA were recovered unchanged from rabbit urine and plasma, indicating no significant degradation for the IAA, DHIAA, or THIAA during the extraction conditions applied. In addition, no inter-conversion was observed between IAA, DHIAA and THIAA in the conditions of extraction used.Pharmacokinetics of IAA and reduced derivatives
Plasma level time curves following i.v. and oral administration of IAA, DHIAA, and THIAA are presented in Fig. 3 and 4, respectively. The corresponding pharmacokinetic parameters calculated from the non-compartmental analysis are shown in Table 3.| Parameter | i.v. administration | Oral administration | ||||
|---|---|---|---|---|---|---|
| IAA | DHIAA | THIAA | IAA | DHIAA | THIAA | |
| a Significant difference in CL of IAA versus DHIAA and THIAA upon i.v. dosing. P < 0.005. b Significant difference in t1/2 of IAA versus DHIAA and THIAA upon i.v. dosing. P < 0.05. c Significant difference in AUC0−∞ of IAA versus DHIAA and THIAA upon i.v. dosing. P < 0.001. d Significant difference in AUC0−∞ of IAA versus DHIAA and THIAA upon oral dosing. P < 0.05. | ||||||
| Cl (ml h−1) | 931 ± 91a | 258 ± 67 | 300 ± 7 | — | — | — |
| t1/2 (h) | 0.32 ± 0.03b | 0.88 ± 0.29 | 0.69 ± 0.07 | — | — | — |
| AUC0−∞ (h μg ml−1) | 81 ± 8c | 252 ± 7 | 250 ± 6 | 10.6 ± 5.3d | 71 ± 48 | 57 ± 9 |
| Cmax (μg ml−1) | — | — | — | 2.5 ± 1.6 | 6.6 ± 3.8 | 7.7 ± 4.3 |
| tmax (h) | — | — | — | [0.5–6.0] | [4.0–12.0] | [0.5–6.0] |
| F (%) | — | — | — | 13.0 ± 6.5 | 28.0 ± 19.4 | 23.0 ± 3.6 |
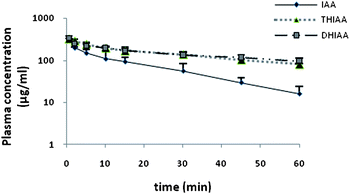 | ||
| Fig. 3 Plasma concentration curves in rabbits following intravenous (0.5–60 min; n = 3) administration of 25 mg kg−1 IAA, DHIAA, and THIAA. Values represent the mean plasma concentration and error bars represent the standard deviation. IAA: iso-α-acids; DHIAA: dihydro-iso-α-acids; THIAA: tetrahydro-iso-α-acids. | ||
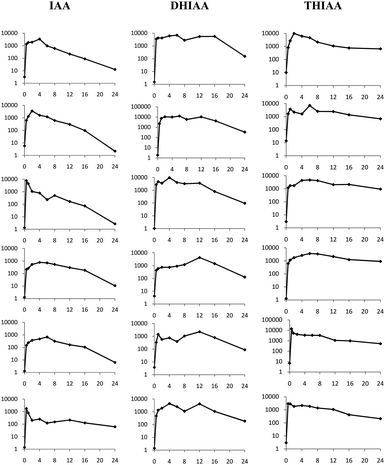 | ||
| Fig. 4 Individual plasma concentration-time profiles (log concentration (μg ml−1) versus time (h)) in rabbits following oral (0.5–24 h; n = 6) administration of 25 mg kg−1 IAA, DHIAA, and THIAA. IAA: iso-α-acids; DHIAA: dihydro-iso-α-acids; THIAA: tetrahydro-iso-α-acids. | ||
After i.v. injection of 25 mg kg−1 of IAA, DHIAA, and THIAA, the plasma level of IAA declined faster (clearance of IAA was 931 ± 91 ml h−1) than that of DHIAA and THIAA, which showed both a similar elimination slope (Fig. 3). This is also illustrated by a half-life (t1/2) of 0.32 ± 0.03 h for IAA versus 0.72 ± 0.10 h and 0.69 ± 0.07 h for DHIAA and THIAA, respectively.
When orally dosed, the plasma concentration-time profile of the individual rabbits (n = 6) was obtained as shown in Fig. 4.
Since large interindividual variation was observed for the time to reach absorption maximum, tmax was described as a range of values. For DHIAA, tmax ranged 4.0–12 h post dosing, while the tmax of IAA and THIAA varied 0.5–6 h after ingestion. For IAA, a Cmax of 2.5 ± 1.6 μg ml−1 (or equal to 6.9 ± 4.4 μM) was determined, while for DHIAA, a maximum concentration was found, 2 to 3 times higher than that of IAA (6.6 ± 3.8 μg ml−1 equalling 18.2 ± 10.5 μM). Following oral dosing of THIAA, a Cmax of 7.7 ± 4.3 μg ml−1 (equal to 21.2 ± 11.8 μM) could be distinguished. The area under curve AUC0−∞ upon oral gavage for DHIAA and THIAA was 70.7 ± 48.4 μg h ml−1 and 57.4 ± 9.0 μg h ml−1, respectively. This was almost 6 to 7 times higher than the value calculated for IAA, for which the AUC0−∞ was 10.6 ± 5.3 μg h ml−1.
The absolute bioavailability for IAA was determined to be 13.0 ± 6.5%. The reduced derivatives reached higher absolute bioavailabilities of 28.0 ± 19.4% for DHIAA, and 23.0 ± 3.6% for THIAA.
To investigate possible phase-II metabolism of IAA, DHIAA, and THIAA, enzymatic hydrolysis of plasma samples following oral treatment was carried out with a mixture of sulfatase and glucuronidase activities. The amounts of IAA and THIAA, quantified after the enzymatic reactions, were not significantly different from the non-hydrolyzed levels; hence, the presence of conjugates could not be demonstrated in samples of IAA and THIAA. However, enzymatic hydrolysis of plasma samples of DHIAA resulted in a value for the AUC0−∞ of 140 ± 43 μg h ml−1. From these results, the percentage of DHIAA being conjugated as a sulfate or a glucuronide could be estimated to be around 50%.
The pharmacokinetic data of THIAA were assessed as an example to determine a possible difference in bioavailability between the cis- and trans-stereoisomers or between the different co- versus n-homologues (Table 4 and 5). The different side chain at C1 (isopentanoyl in n- and ad-, isobutyryl in co-analogues) resulted in a higher bioavailability of the n-analogues compared to the co-analogues (47% versus 18%). On the other hand, there was no difference in bioavailability observed in favour of the cis-THIAA (17%) compared to the trans-THIAA (19%).
| cis-THIAA | trans-THIAA | |
|---|---|---|
| F (%) | 17.0 ± 3.0 | 19.0 ± 4.0 |
Apparently, a higher grade of lipophilicity (of the n- versus co-analogue) has more influence on the bioavailability than the difference in steric structure of the compounds, since the presence of the side chains on different faces of the five-membered ring of the cis- and trans-analogues did not have any effect on the bioavailability.
Fractions excreted in urine and faeces
The percentages of dose (%dose) excreted in urine and faeces following oral and i.v. administration of IAA, DHIAA, and THIAA are shown in Table 6. After oral gavage, the urinary %dose of intact IAA was 0.13 ± 0.05%. This fraction was very small when compared with the levels of THIAA and DHIAA, for which the %dose was 1.0 ± 0.1% and 12.1 ± 4.8%, respectively.| Compound | Intravenous | Oral | Fraction unabsorbed (% of dose) | ||
|---|---|---|---|---|---|
| Urine | Faeces | Urine | Faeces | ||
| a Significant difference in urinary amount of DHIAA versus IAA and THIAA upon i.v. dosing. P < 0.005. b Significant difference in faecal amount of IAA versus DHIAA and THIAA upon i.v. dosing. P < 0.05. c Significant difference in urinary amount of DHIAA versus IAA and THIAA upon oral dosing. P < 0.001. | |||||
| IAA | 0.5 ± 0.2 | 0.9 ± 0.2b | 0.13 ± 0.05 | 6.0 ± 1.8 | 5.1 ± 2.0 |
| DHIAA | 15.4 ± 1.4a | 8.4 ± 1.8 | 12.1 ± 4.8c | 13.1 ± 1.1 | 4.7 ± 2.9 |
| THIAA | 0.8 ± 0.2 | 10.1 ± 2.1 | 1.0 ± 0.1 | 25.6 ± 3.6 | 15.5 ± 4.9 |
In the faeces, the %dose of intact THIAA following oral application was 25.6 ± 7.6%, while the %dose of DHIAA and IAA were respectively 13.1 ± 1.1% and 6.0 ± 1.8%. Following i.v. administration, urinary %dose of intact DHIAA were determined to be 15.4 ± 1.4%. The levels of IAA and THIAA were comparable, 0.50 ± 0.2% and 0.8 ± 0.2%, respectively.
Faecal %dose of intact IAA after i.v. dosing, were yet again very low compared to these of the reduced derivatives, 0.9 ± 0.2%. For DHIAA and THIAA, comparable percentages could be determined; 8.4 ± 1.8% and 10.1 ± 2.1% for DHIAA and THIAA, respectively. From these data, it was possible to calculate the unabsorbed fraction as the difference in the faecal %dose following oral and i.v. administration. This was around 5% of the ingested dose for IAA and DHIAA, in contrast with THIAA, for which an unabsorbed fraction of 15.5 ± 4.9% was calculated. Large differences between the different classes of hop-derived acids could be observed: urinary excretion of DHIAA upon both oral and i.v. dosing exceeded the values of IAA and THIAA, which were significantly lower. The differences in urinary excretion between IAA and THIAA were not statistically significant. Samples of faeces and urine were also subjected to enzymatic hydrolysis with a mixture of sulfatase and glucuronidase activities to screen for the presence of possible phase-II conjugates of IAA, DHIAA, and THIAA. Subtracting the amounts upon enzymatic hydrolysis from the control samples (with no enzyme treatment), showed no significant conjugation of IAA or THIAA in urine or faeces. However, enzymatic hydrolysis of urine samples of DHIAA following oral ingestion, showed that up to 22% of the excreted amount of DHIAA was conjugated as a sulfate or a glucuronide.
Discussion
Hop-derived bitter acids, the main flavoring agents in beer, have been reported to show important bioactive properties, including anti-inflammatory, anti-angiogenic properties, lipid metabolism enhancement, and counteracting diabetes type 2. However, a review of the literature shows that data on the pharmacokinetics of these compounds are lacking. In this study, the in vitro transport characteristics, pharmacokinetic parameters, and oral bioavailability, of IAA, DHIAA, and THIAA were assessed. The doses of hop acid applied in the present Caco-2 assay (30–120 μM) are relevant as beer may contain up to 100 mg l−1 hop-derived bitter acids (depending on beer type and brand) equaling 250 μM HBA.40The dose solutions used in the rabbit trials are diluted from commercially available Isohop®, Redihop® and Tetrahop Gold®, which are stable solutions of potassium salts of respectively IAA, DHIAA, and THIAA at pH 8–10. For stability and solubility reasons, dose formulations were obtained by dilution with ammonium acetate buffer with pH 10.Nevertheless, a potential influence on bioavailability by the use of non-physiological pH, should be considered for further investigation.
An animal dose of 25 mg kg−1 in the pharmacokinetic study can be translated to a human equivalent dose (HED) using the following formula: HED = 25 mg kg−1 × Km factor (rabbit)/Km factor (human), in which the Km factor (kg m−2) is calculated from the ratio of the body weight (kg) and body surface area (m2) of a species. The FDA draft guidelines report values for the Km of 12 for rabbits and 37 for humans, based on the ratio of the body weight and the body surface area (BSA).41,42 In this way, a HED of 8 mg kg−1 could be calculated, which would correspond with a dose around 500 mg for a human weight of 60–70 kg. This is in the line with the amounts of hop-derived acids present in commercially available dietary supplements (usually 400 mg or more) and the doses (single or frequent dosing) used in clinical trials with animals8,43,44 or humans.9,10,28 However, this dose is not practical for moderate beer consumption, since a provided amount of 400–500 mg hop-derived acids would require at least 5 litres of beer intake.
In all of the publications cited, no adverse effects were reported. In one report, Chappel et al. conducted a study to determine the effect associated with subchronic oral administration of THIAA (as well as hexahydroiso-α-acids) in the dog. Most of the materials were excreted in the faeces and the no-observed-adverse-effect level (NOAEL) of the compounds was 100 and 50 mg kg−1 body weight, for THIAA and hexahydroiso-α-acids, respectively. As for dogs, the observations showed that these compounds were generally well tolerated.45
Also, in an observational human trial to investigate the efficacy of a formula containing DHIAA (Meta050) (440 mg daily for eight weeks) on pain in patients with rheumatic disease, did not result in clinically relevant changes in blood pressure, complete blood counts, or liver and kidney function. Furthermore, there was no negative impact on gastrointestinal markers normally affected by selective COX-2 enzyme inhibitors, as concluded from normal fecal calprotectin excretion. Similar data were obtained after administration of pure DHIAA (450 mg daily for 2 weeks).10,46
In this study, the oral bioavailability of IAA was found to be less than 15%, while bioavailability of the reduced derivatives was higher (23% for THIAA and 28% for DHIAA). However, differences in bioavailability were determined to be not statistically significant, because of large inter-individual variation in the animals. Factors limiting a high bioavailability can be diverse, but typically include inefficient absorption and rapid metabolism (i.e., the first-pass effect).
Results from the Caco-2 experiments showed that permeability (AP-to-BL) mechanisms other than passive diffusion seem unlikely, as indicated by the linear dose-transport and time-transport relationships, and the lack of saturation effects. A passive diffusion transport mechanism occurs most probably transcellularly, since the paracellular pathway is restricted by the tight junctions of intestinal epithelium.47 Also, the surface area of the luminal cell membrane of the intestinal epithelium is 1000-fold larger than that of the paracellular space.48 Based on the PappAB, a ranking in absorption is suggested as follows, THIAA ≈ IAA > DHIAA. Because of the linear dose-transport relationship, Papp values were independent of the dose applied for all the HBA tested. In studies attempting to correlate passive drug permeability in Caco-2 experiments with drug absorption in humans after oral administration, moderately to well absorbed compounds (20–80% fraction absorbed) were found to have permeability coefficients 10−6 < Papp < 10 × 10−6 cm s−1 whereas poorly absorbed drugs had Papp < 0.1 × 10−6 cm s−1 (< 20% fraction absorbed).35,49 This is supported by the calculated unabsorbed fractions (4% for IAA and DHIAA and 15% for THIAA), obtained from the amounts determined in the faeces upon oral and i.v. application, suggesting efficient absorption of IAA, DHIAA, and THIAA. However, this calculation of unabsorbed fraction is likely an underestimated value, since a significant fraction of the administered compounds can be unabsorbed but potentially metabolized/degraded by the microbiota into diverse metabolites which were not detected in the analysis.
For all different hop-derived acids tested, secretion (from BL-to-AP) showed a linear relationship between dose and amount transported suggesting that secretion is also expected to occur by passive diffusion. Efflux would only become important when concentrations at the luminal side attain 20–50% of that at the blood side, independent of the involvement of active transporters (since diffusion is forced by a concentration gradient). Most likely, this is only reached just before complete absorption. However, efflux permeability coefficients (BL-to-AP) (PappBA ranged 6 – 16 × 10−6 cm s−1) and efflux ratios (around 3–3.5) were substantial, so additional experiments using specific inhibitors for efflux pumps (Pgp, MRP-2, and BCRP) providing proof of possible active efflux mechanism involvement would be important. In earlier presented work, the in vitro transport of hop α-acids and β-acids across Caco-2 monolayers has been studied, showing efficient epithelial transport of hop α-acids (Papp > 10 × 10−6 cm s−1), whereas the permeability of β-acids was limited by the involvement of Pgp and MRP-2 type efflux transporters and phase-II metabolism.50
The lower Papp value of DHIAA compared to that of IAA and THIAA could be explained by a substantial conjugation of DHIAA, following absorption in the Caco-2 cells, from where the major fraction being conjugated can transfer to the BL compartment, or back into the AP compartment, whether or not with the involvement of active transport mechanisms, most often of the MRP-type family.51 Although the Caco-2 cell model is recognized rather as a model of human intestinal absorption than of phase II intestinal metabolism, there are many examples of conjugation reactions of xenobiotics by Caco-2 cells in the literature,52–54 including flavonoids originating from hops, which are known to be extensively conjugated by Caco-2 cells.55 Nevertheless, absorption of hop-derived acids is suggested to be efficient in rabbits, indicated by substantial plasma concentrations in the μg ml−1 (or lower μM) scale. From the pharmacokinetic data of DHIAA obtained following oral application, an AUC0−8h of 30.6 h μg ml−1 could be determined, which is in accordance with the results published by Hall et al., taken into account an HED of 560 mg. Following oral application of a dose of 700 mg DHIAA administered to 2 healthy human subjects, an AUC0−8h of 26 μg h ml−1 was calculated, which is in line with the results of our study.28
Prior to absorption and introduction of a compound in the systemic circulation and exposure to liver enzymes, intestine epithelial cells (enterocytes) provide the first site for CYP-catalyzed and phase-II metabolism,56 since the highest catalytic activity resides in the proximal region of the small intestine.57–59 Following oral dosing, only minor amounts of intact IAA were determined in urine and faeces. The mass balance (a summed total of %dose in urine and faeces of intact and conjugated forms over 24h) of IAA was ≤ 6% in our study, indicating metabolism/degradation of IAA to be the most important path of elimination and factor influencing its bioavailability. This seems consistent with the low levels recovered in urine and faeces after i.v. administration of IAA. Compared to IAA, the mass balance of the reduced derivatives following oral dosing was substantially higher, totalling 47% for DHIAA and 25% for THIAA of the dose administered, showing a substantial part of orally administered DHIAA and THIAA escapes metabolism/degradation in vivo in rabbits.
Previous experiments reported by Aniol et al. showed that hop α-acids and β-acids are totally degraded when incubated with peroxidase enzymes from plant extracts.60 This could suggest the involvement of P450 enzymes in the metabolism of HBA. Furthermore, two reports describe the induction of quinine reductase activity by humulones and isohumulones and the activation of CYP3A4, CYP2B6 and some multidrug resistance (MDR1) levels in human hepatocytes indicating that these hop-derived acids can stimulate both phase-I and phase-II detoxification processes.5,61 The recent reports of Intelmann and Hofmann on the identification of degradation products of IAA formed upon beer ageing62 indicates the proneness of IAA to oxidative degradation, partly based on findings already established much earlier by several authors.63–70
Although phase-II conjugation of IAA and THIAA cannot be completely ruled out, in none of the plasma, urine, and faecal samples proof for sulfation or glucuronidation could be demonstrated. This corresponds also with the results of the enzymatic hydrolysis of Caco-2 monolayer samples. Other possible phase-II metabolism reactions (conjugation of glutathione or amino acids…) should, however, be further investigated. On the contrary, in the plasma and urine samples following DHIAA ingestion, substantial sulfation or glucuronidation was evident. The presence of an accessible alcoholic group in the acyl side chain of the molecular structure of DHIAA explains the absence of conjugation of IAA and THIAA. The enolic group, present in the molecular structures of IAA and THIAA, may be inactive for conjugation in view of its acidity or due to intramolecular hydrogen bonding with the adjacent carbonyl group in the acyl side chain.
Conclusions
The bioavailabilities in rabbits of hop-derived bitter acids, which have been reported to show important bioactive properties, have been investigated in the present work. At least in rabbits, the maximum exposure levels reached for IAA, DHIAA and THIAA are in the range of 2–8 μg ml−1 (or equal to 7–20 μM), which is in line with bioactive concentrations previously suggested elsewhere.28,43,44,71 The bioavailability of IAA was lower compared to the reduced derivatives and largely affected by phase-I metabolic stability. The bioavailability of DHIAA was influenced by substantial phase-II metabolism. Further research on the metabolism of hop-derived bitter acids is in progress.Acknowledgements
Ko Cattoor is a recipient of an academic scholarship ‘malting and brewing’ provided by the InBev-Baillet Latour Fund, Leuven, Belgium. The authors gratefully acknowledge the technical assistance of Daniel Tensy.Notes and references
- The Brewers of Europe, Brussels, Belgium, 2008, pp. 8–22 Search PubMed.
- R. Doll, R. Peto, E. Hall, K. Wheatley and R. Gray, Br. Med. J., 1994, 309, 911–918 Search PubMed.
- M. J. Thun, R. Peto, A. D. Lopez, J. H. Monaco, S. J. Henley, C. W. Heath and R. Doll, N. Engl. J. Med., 1997, 337, 1705–1714 Search PubMed.
- S. R. Hamilton, J. Hyland, D. McAvinchey, Y. Chaudhry, L. Hartka, H. T. Kim, P. Cichon, J. Floyd, N. Turjman, G. Kessie, P. P. Nair and J. Dick, Cancer Res., 1987, 47, 1551–1559 Search PubMed.
- G. Bohr, K. Klimo, J. Zapp, H. Becker and C. Gerhauser, Nat. Prod. Commun., 2008, 3, 1971–1976 Search PubMed.
- C. Gerhauser, Eur. J. Cancer, 2005, 41, 1941–1954 Search PubMed.
- M. Shimamura, T. Hazato, H. Ashino, Y. Yamamoto, E. Iwasaki, H. Tobe, K. Yamamoto and S. Yamamoto, Biochem. Biophys. Res. Commun., 2001, 289, 220–224 Search PubMed.
- V. R. Konda, A. Desai, G. Darland, J. S. Bland and M. L. Tripp, J. Inflamm. (Lond), 2009, 6 Search PubMed.
- D. M. Minich, J. S. Bland, J. Katke, G. Darland, A. Hall, R. H. Lerman, J. Lamb, B. Carroll and M. Tripp, in 1st Annual Meeting of the Natural Health Products-Research Society of Canada, Natl Research Council Canada-N R C Research Press, Montreal, Canada, 2004, pp. 872–883 Search PubMed.
- D. Lukaczer, G. Darland, M. Tripp, D. Liska, R. H. Lerman, B. Schiltz and J. S. Bland, Phytother. Res., 2005, 19, 864–869 CrossRef CAS.
- P. Zanoli and M. Zavatti, J. Ethnopharmacol., 2008, 116, 383–396 Search PubMed.
- M. Van Cleemput, K. Cattoor, K. De Bosscher, G. Haegeman, D. De Keukeleire and A. Heyerick, J. Nat. Prod., 2009, 72, 1220–1230 Search PubMed.
- K. Obara, M. Mizutani, Y. Hitomi, H. Yajima and K. Kondo, Clin. Nutr., 2009, 28, 278–284 Search PubMed.
- R. Stevens, Chem. Rev., 1967, 67, 19–71 Search PubMed.
- K. Huvaere, M. L. Andersen, K. Olsen, L. H. Skibsted, A. Heyerick and D. De Keukeleire, Chem.–Eur. J., 2003, 9, 4693–4699 CrossRef CAS.
- A. Heyerick, K. Huvaere, D. De Keukeleire and M. D. E. Forbes, Photochem. Photobiol. Sci., 2005, 4, 412–419 RSC.
- K. Huvaere, M. L. Andersen, L. H. Skibsted, A. Heyerick and D. De Keukeleire, J. Agric. Food Chem., 2005, 53, 1489–1494 CrossRef CAS.
- K. Huvaere, M. L. Andersen, M. Storme, J. Van Bocxlaer, L. H. Skibsted and D. De Keukeleire, Photochem. Photobiol. Sci., 2006, 5, 961–969 RSC.
- C. S. Burns, A. Heyerick, D. De Keukeleire and M. D. E. Forbes, Chem.–Eur. J., 2001, 7, 4553–4561 CrossRef CAS.
- T. Kunimune and T. H. Shellhammer, J. Agric. Food Chem., 2008, 56, 8629–8634 Search PubMed.
- M. Moir, J. Am. Soc. Brew. Chem., 2000, 58, 131–146 Search PubMed.
- P. L. P. Ting and H. Goldstein, J. Am. Soc. Brew. Chem., 1996, 54, 103–109 Search PubMed.
- P. M. Brown, G. A. Howard and A. R. Tatchell, J. Chem. Soc., 1959, 545–551 RSC.
- S. Malfliet, F. Van Opstaele, J. De Clippeleer, E. Syryn, K. Goiris, L. De Coornan and G. Aerts, J. Inst. Brew., 2008, 114, 180–192 Search PubMed.
- S. Araki, M. Takashio and K. Shinotsuka, J. Am. Soc. Brew. Chem., 2002, 60, 26–30 Search PubMed.
- A. Forster, B. Beck, R. Schmidt, C. Jansen and A. Mellenthin, Monatsschrift Fur Brauwissenschaft, 2002, 55, 98–104 Search PubMed.
- Metagenics, www.metagenics.com/products, 2010.
- A. J. Hall, J. G. Babish, G. K. Darland, B. J. Carroll, V. R. Konda, R. H. Lerman, J. S. Bland and M. L. Tripp, Phytochem., 2008, 69, 1534–1547 Search PubMed.
- P. Stenberg, U. Norinder, K. Luthman and P. Artursson, J. Med. Chem., 2001, 44, 1927–1937 CrossRef CAS.
- M. C. Gres, B. Julian, M. Bourrie, V. Meunier, C. Roques, M. Berger, X. Boulenc, Y. Berger and G. Fabre, Pharm. Res., 1998, 15, 726–733 Search PubMed.
- B. Press and D. Di Grandi, Curr. Drug Metab., 2008, 9, 893–900 Search PubMed.
- S. Yamashita, T. Furubayashi, M. Kataoka, T. Sakane, H. Sezaki and H. Tokuda, Eur. J. Pharm. Sci., 2000, 10, 195–204 Search PubMed.
- C. A. Bailey, P. Bryla and A. W. Malick, Adv. Drug Delivery Rev., 1996, 22, 85–103 Search PubMed.
- C. Wyns, S. Bolca, D. De Keukeleire and A. Heyerick, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2010, 878, 949–956 CrossRef CAS.
- P. Artursson and J. Karlsson, Biochem. Biophys. Res. Commun., 1991, 175, 880–885 CrossRef CAS.
- Institutional Animal Care and Use Committee Guidebook, Office of Laboratory Animal Welfare (NIH), USA, 2002.
- P. Augustijns and R. Mols, J. Pharm. Biomed. Anal., 2004, 34, 971–978 CrossRef CAS.
- J. Karlsson and P. Artursson, Int. J. Pharm., 1991, 71, 55–64 Search PubMed.
- S. H. Chong, S. A. Dando, K. M. Soucek and R. A. Morrison, Pharm. Res., 1996, 13, 120–123 Search PubMed.
- B. Jaskula, K. Goiris, G. De Rouck, G. Aerts and L. De Cooman, J. Inst. Brew., 2007, 113, 381–390 Search PubMed.
- S. Reagan-Shaw, M. Nihal and N. Ahmad, FASEB J., 2008, 22, 659–661 Search PubMed.
- U.S. Food and Drug Administration, Rockville, Maryland, USA, 2002 Search PubMed.
- H. Yajima, E. Ikeshima, M. Shiraki, T. Kanaya, D. Fujiwara, H. Odai, N. Tsuboyama-Kasaoka, O. Ezaki, S. Oikawa and K. Kondo, J. Biol. Chem., 2004, 279, 33456–33462 Search PubMed.
- T. Namikoshi, N. Tomita, S. Fujimoto, Y. Haruna, M. Ohzeki, N. Komai, T. Sasaki, A. Yoshida and N. Kashihara, Hyperten. Res., 2007, 30, 175–184 Search PubMed.
- C. I. Chappel, S. Y. Smith and M. Chagnon, Food Chem. Toxicol., 1998, 36, 915–922 Search PubMed.
- D. M. Minich, J. S. Bland, J. Katke, G. Darland, A. Hall, R. H. Lerman, J. Lamb, B. Carroll and M. Tripp, Can. J. Physiol. Pharmacol., 2007, 85, 872–883 Search PubMed.
- J. Hochman and P. Artursson, J. Controlled Release, 1994, 29, 253–267 Search PubMed.
- J. R. Pappenheimer and K. Z. Reiss, J. Membr. Biol., 1987, 100, 123–136 Search PubMed.
- W. L. Chiou and A. Barve, Pharm. Res., 1998, 15, 1792–1795 Search PubMed.
- K. Cattoor, M. Bracke, D. Deforce, D. De Keukeleire and A. Heyerick, J. Agric. Food Chem., 2010, 58, 4132–4140 Search PubMed.
- T. Murakami and M. Takano, Expert Opin. Drug Metab. Toxicol., 2008, 4, 923–939 Search PubMed.
- U. K. Walle, A. Galijatovic and T. Walle, Biochem. Pharmacol., 1999, 58, 431–438 Search PubMed.
- S. Kobayashi, S. Tanabe, M. Sugiyama and Y. Konishi, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 33–41 Search PubMed.
- J. B. Vaidyanathan and T. Walle, Pharm. Res., 2001, 18, 1420–1425 CrossRef CAS.
- D. Nikolic, Y. M. Li, L. R. Chadwick and R. B. van Breemen, Pharm. Res., 2006, 23, 864–872 Search PubMed.
- P. B. Watkins, Adv. Drug Delivery Rev., 1997, 27, 161–170 Search PubMed.
- M. F. Paine, M. Khalighi, J. M. Fisher, D. D. Shen, K. L. Kunze, C. L. Marsh, J. D. Perkins and K. E. Thummel, J. Pharmacol. Exp. Ther., 1997, 283, 1552–1562 Search PubMed.
- Q. Y. Zhang, D. Dunbar, A. Ostrowska, S. Zeisloft, J. Yang and L. S. Kaminsky, Drug Metab. Dispos., 1999, 27, 804–809 Search PubMed.
- T. Nakamura, K. Okada, K. Nagata and Y. Yamazoe, Jpn. J. Pharmacol., 2000, 82, 232–239 Search PubMed.
- M. Aniol, A. Bartmanska, E. Huszcza, W. Maczka and A. K. Zolnierczyk, Przem. Chem., 2009, 88, 392–395 Search PubMed.
- D. G. Teotico, J. J. Bischof, L. Peng, S. A. Kliewer and M. R. Redinbo, Mol. Pharmacol., 2008, 74, 1512–1520 Search PubMed.
- D. Intelmann and T. Hofmann, J. Agric. Food Chem., 58, pp. 5059–5067 Search PubMed.
- D. W. Diffor, E. S. A. Ramos and G. L. Hansen, Proc. Am. Soc. Brew. Chem., 1972, 109–114 Search PubMed.
- B. J. Clarke and R. P. Hilldebrand, J. Inst. Brew., 1967, 73, 282–293 Search PubMed.
- W. G. Schulze, P. L. Ting, L. A. Henckel and H. Goldstein, J. Am. Soc. Brew. Chem., 1981, 39, 12–15 Search PubMed.
- D. De Keukeleire, Cerevisia, 1981, 73–80 Search PubMed.
- M. Vanhoey, M. Vandewal and M. Verzele, Bull. Soc. Chim. Belg., 1970, 79, 499–509 Search PubMed.
- N. Hashimoto and T. Eshima, J. Inst. Brew., 1979, 85, 136–140 Search PubMed.
- O. Corzo and N. Bracho, J. Food Sci., 2004, 69, S285–S289 Search PubMed.
- M. R. Cann, A. M. Davis and P. V. R. Shannon, J. Chem. Soc., Perkin Trans. 1, 1982, 375–383 RSC.
- H. Nozawa, W. Nakao, F. Zhao and K. Kondo, Mol. Nutr. Food Res., 2005, 49, 772–778 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |