DOI:
10.1039/C1FO10033E
(Paper)
Food Funct., 2011,
2, 259-264
Phenolic tyrosinase inhibitors from the stems of Cudrania cochinchinensis
Received
2nd March 2011
, Accepted 29th March 2011
First published on 12th April 2011
Abstract
The phytochemcal profiles of Cudrania cochinchinensis leaf, twig, stem and root were compared by HPLC analysis. It was found that C. cochinchinensis stem extract contained some unknown natural products with potential tyrosinase inhibitory activities. Therefore, the chemical constitutes in extract (95% ethanol) of C. cochinchinensis stem were further investigated in this study. A new racemic mixture, (±)2,3-cis-dihydromorin, and fifteen known phenolic compounds, dihydrokaempferol 7-O-β-D-qlucopyranoside, skimmin, quercetin-7-O-β-D-glucoside, 2,3-dihydroquercetin 7-O-β-D-glucoside, kaempferol-7-O-β-glucopyranoside, quercetin-3,7-di-O-β-D-glucoside, morin-7-O-β-D-glucoside, 1,3,5,8-tetrahydroxyxanthen-9-one, 2,3-trans-dihydromorin, aromadendrin, oxyresveratrol, genistin, protocatechuic acid, kaempferol 3,7-di-O-β-glucopyranoside, and naringenin were isolated. Spectral techniques (MS, 1H NMR and 13C NMR) were utilized for their structural identification and their inhibitory activities on mushroom tyrosinase were also evaluated. The results showed that tyrosinase inhibitory activities of (±)2,3-cis-dihydromorin (IC50 = 31.1 μM), 2,3-trans-dihydromorin (IC50 = 21.1 μM), and oxyresveratrol (IC50 = 2.33 μM), were more potent than that of kojic acid (IC50 = 50.8 μM), a well-known tyrosinase inhibitor, indicating that Cudrania cochinchinensis stem will be a great potential agent for the development of effective natural tyrosinase inhibitors.
Introduction
Tyrosinase (EC 1.14.18.1), a copper-containing polyphenol oxidase (PPO), is widely distributed in microorganisms, plants, and mammals.1 It is responsible for enzymatic browning of the plant-derived foods and the formation of melanin pigments in mammals.1 In the food industry, tyrosinase catalyzes the postharvest oxidation of polyphenolic compounds in fresh fruits and vegetables during storage and processing and consequently leads to the loss in appearance, flavor, and nutritional quality.2 Therefore, various approaches have been adopted to inhibit the activity of tyrosinase in foods including the addition of natural tyrosinase inhibitors as chemical additives.3 The suppression of melanin formationvia the inhibition of tyrosinase activity is also applicable to skin-lightening and depigmenting in the cosmetic field.4 Furthermore, some evidences showed tyrosinase might play a role in neuromelanin production and damaged neurons associated with Parkinson's disease,5 rendering tyrosinase inhibitors of even greater importance. A broad spectrum of potent tyrosinase inhibitors have been obtained from the natural products, including ascorbic acid and its derivatives, thiol-containing amino acids (such as cysteine), kojic acid, citric acid, oxalic acid,6 and different classes of phenolic compounds (such as oxyresveratrol and its derivatives, chlorophorin and 4-prenyloxyresveratrol,7isoflavan derivatives, glabridin and glyasperin C).8 In our previous studies, several flavonoids, trans-dihydromorin, steppogenin, norartocarpetin, artocarpanone, artocarpesin, and isoartocarpesin, were isolated from the wood of Artocarpus heterophyllus with potent tyrosinase inhibitory activities.9 Subsequent content analysis showed that the wood contained these active compounds about 0.15%,10 suggesting it is a promising candidate as tyrosinase inhibitor with real commercial application. Although great progress has already been made in the discovery of natural tyrosinase inhibitors, the constant searching for novel and strong ones is still a continuing focus of phytochemical researches.
Cudrania cochinchinensis is distributed in the southern part of China, Japan and Korea. Its roots are used to treat rheumatism, hepatitis, gonorrhea, bruising, and contused wounds in China.11 Many phenolics, including xanthones, flavonols, flavonol salt, isoflavones and flavanones, were isolated from its bark and roots, with cytotoxic, anti-lipid peroxidation, and anti-microbial activities, etc.11,12 Previous studies mostly focused on the roots and bark, while the components and medicinal properties of the stems have not been reported. In the present study, the HPLC profiles of different parts of C. cochinchinensis were compared and the extract of C. cochinchinensis stems was subjected to detailed phytochemical analysis because of its potent inhibitory activity against mushroom tyrosinase (IC50 = 36.3 μg mL−1). Phytochemical analysis of the 95% ethanol extract of C. cochinchinensis stems led to the isolation and purification of one new and fifteen known phenolic compounds. Systematic chromatographic and spectral approaches in characterization of these active components are described in detail in the following sections.
Materials and methods
Chemicals and instruments
Mushroom tyrosinase (5370 units mg−1), L-tyrosine, formic acid, kojic acid were purchased from Sigma Chemical Co (St. Louis, USA). HPLC grade solvents were all purchased from BDH (Poole, UK). Sephadex LH-20 was purchased from GE Healthcare Bio-Sciences AB (Uppsala, Sweden). Silica gel (200–300 mesh) for column chromatography was purchased from Qingdao Marine Chemical Company (Qingdao, P R China). TLC plates 25 DC-platten Kieselgel 60 F254) were obtained from Merck (Darmstadt, Germany). Ultimate XB-C18 column (250 × 4.6 mm, 5μm) was purchased from Welch Materials, Inc. (Shanghai, P R China). Semi-preparative HPLC system was carried out on a Waters 600 system equipped with a 2487 dual-wavelength detector, a Masslynx V4.0 software and a Phenomenex Luna C18 column (250 × 21.2 mm, 5 μm). 1H NMR and 13C NMR were obtained on a Bruker 400 or 500 DRX NMR spectrometer. Mass spectrum was run on an Applied Biosystems Q-trap 2000 mass spectrometer. HR Mass spectrum was run on Agilent 6210 LC/MSD TOF. The tyrosinase inhibitory activity was monitored on a UV-1206 Spectro-photometer (Shimadzu Corporation, Japan).
Plant materials
The fresh stems, leaves, twigs and roots of C. cochinchinesis were collected at Nanning, Guangxi Province, P R China, in June 2008. The voucher specimen (accession number 20080601, 20080602, 20080603, 20080604) was deposited at School of Biological Sciences, The University of Hong Kong.
HPLC analysis of leaves, twigs, stems, and roots of C. cochinchinensis
Ground plant materials were extracted with methanol by sonication for 60 min. After cooling, the extract solutions were filtrated and then analyzed by HPLC. The analyses were carried out on an Ultimate C18 column eluted at room temperature. The mobile phases were solvent A, water containing 0.2% formic acid, and solvent B, acetonitrile. The elution program was as follows: 0–10 min, 5%–15% B; 10–20 min, 15%–25% B; 20–30 min, 25%–40% B; 30–40 min, 40%–55% B; 40–50 min, 55%–70% B; 50–60 min, 70%–100% B; 60–75 min, 5% B. The flow rate was set at 1.0 mL min−1 and the detection system monitored absorbance at 254 nm. The sample injection volume was 10 μL.
Extraction and isolation of C. cochinchinensis stem phytochemicals
The stems of C. cochinchinensis (850 g) were ground into powder form using a mini-grinder and packed into an extraction bag, then soaked with 95% ethanol (3 × 6 L) for 24 h at room temperature in a traditional Chinese medicine extractor. The extract was concentrated under vacuum at 40 °C with a rotary evaporator to dryness and 21.8 g residue was obtained. The dried extract was dissolved in methanol and then subjected to silica gel (200–300 mesh) column chromatography by successive elution with different proportions of n-hexane–ethyl acetate (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 3:1), ethyl acetate, and ethyl acetate–methanol (10:1, 5:1) to offer 7 fractions (Frs.1–7). Fr.1 (0.96 g) was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.A1 and A2). Fr.A1 was purified by semi-preparative HPLC [solvent, ACN-H2O (12:88), detection wavelength at 254 nm, flow rate at 8.0 mL min−1] to give compound 2 (33.8 mg). Fr.A2 was isolated by semi-preparative HPLC [solvent, ACN-H2O (12:88) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 3 (2.7 mg). Fr.2 was chromatographed over Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 4 (3.6 mg) and compound 5 (27.3 mg). Fr.3 was subjected to Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.B1 and B2). Fr.B1 was purified by semi-preparative HPLC [solvent, MeOH–H2O (45:55) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 6 (2.3 mg). Fr.4 was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 13 (0.5 mg). Fr.5 was chromatographed over Sephadex LH-20 [MeOH–H2O (1:1)] to offer 6 sub-fractions (Fr.C1–C6). Fr.C1 was subjected to silica gel [n-hexane–ethyl acetate (2:3)], then purified by semi-preparative HPLC [solvent, MeOH–H2O (43:57) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 10 (2.1 mg) and compound 1 (7.5 mg). Fr.C2 was subjected to Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 9 (1.5 mg). Fr.C3 was isolated by silica gel [n-hexane–ethyl acetate (1:1)] and then purified by semi-preparative HPLC [solvent, MeOH–H2O (43:57) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 12 (102.6 mg). Fr.C4 was subjected to silica gel [n-hexane–ethyl acetate (3:5)] to give compound 11 (13.1 mg). Fr.C6 was chromatographed on silica gel [n-hexane–ethyl acetate (1:2)] to offer compound 14 (4.4 mg). Fr.6 was filtrated and the filtrate was subjected to Sephadex LH-20 [MeOH–H2O (1:1)], then purified by silica gel [ethyl acetate–methanol–water (4: 1: 0.2)] to give compound 8 (2.3 mg). The insoluble part of Fr. 6 was dissolved in DMSO and purified by semi-preparative HPLC [solvent, MeOH–H2O (32:68), with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 7 (3.9 mg) and compound 15 (4.4 mg). Fr.7 was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.D1 and D2). Fr.D2 was purified by semi-preparative HPLC [solvent, ACN-H2O (40:60) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 16 (8.5 mg).
:1, 3:1), ethyl acetate, and ethyl acetate–methanol (10:1, 5:1) to offer 7 fractions (Frs.1–7). Fr.1 (0.96 g) was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.A1 and A2). Fr.A1 was purified by semi-preparative HPLC [solvent, ACN-H2O (12:88), detection wavelength at 254 nm, flow rate at 8.0 mL min−1] to give compound 2 (33.8 mg). Fr.A2 was isolated by semi-preparative HPLC [solvent, ACN-H2O (12:88) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 3 (2.7 mg). Fr.2 was chromatographed over Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 4 (3.6 mg) and compound 5 (27.3 mg). Fr.3 was subjected to Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.B1 and B2). Fr.B1 was purified by semi-preparative HPLC [solvent, MeOH–H2O (45:55) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 6 (2.3 mg). Fr.4 was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 13 (0.5 mg). Fr.5 was chromatographed over Sephadex LH-20 [MeOH–H2O (1:1)] to offer 6 sub-fractions (Fr.C1–C6). Fr.C1 was subjected to silica gel [n-hexane–ethyl acetate (2:3)], then purified by semi-preparative HPLC [solvent, MeOH–H2O (43:57) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to give compound 10 (2.1 mg) and compound 1 (7.5 mg). Fr.C2 was subjected to Sephadex LH-20 [MeOH–H2O (1:1)] to give compound 9 (1.5 mg). Fr.C3 was isolated by silica gel [n-hexane–ethyl acetate (1:1)] and then purified by semi-preparative HPLC [solvent, MeOH–H2O (43:57) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 12 (102.6 mg). Fr.C4 was subjected to silica gel [n-hexane–ethyl acetate (3:5)] to give compound 11 (13.1 mg). Fr.C6 was chromatographed on silica gel [n-hexane–ethyl acetate (1:2)] to offer compound 14 (4.4 mg). Fr.6 was filtrated and the filtrate was subjected to Sephadex LH-20 [MeOH–H2O (1:1)], then purified by silica gel [ethyl acetate–methanol–water (4: 1: 0.2)] to give compound 8 (2.3 mg). The insoluble part of Fr. 6 was dissolved in DMSO and purified by semi-preparative HPLC [solvent, MeOH–H2O (32:68), with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 7 (3.9 mg) and compound 15 (4.4 mg). Fr.7 was chromatographed on Sephadex LH-20 [MeOH–H2O (1:1)] to offer 2 sub-fractions (Fr.D1 and D2). Fr.D2 was purified by semi-preparative HPLC [solvent, ACN-H2O (40:60) with flow rate at 8.0 mL min−1, detection wavelength at 254 nm] to offer compound 16 (8.5 mg).
HPLC analysis of compounds 1 and 10
The analyses were carried out on an Ultimate C18 column eluted at room temperature. The mobile phases were solvent A, water consisting of 0.2% formic acid, and solvent B, acetonitrile. For the elution program, the gradient elution was as follows: initial 10% B; 0–10 min, 10%–30% B; 10–20 min, 30%–50% B; 20–30 min, 50%–70% B; 30–40 min, 70%–100% B. The flow rate was set at 1.0 mL min−1 and the detection system monitored absorbance at 280 nm. The sample injection volume was 10 μL.
Mushroom tyrosinase inhibitory assay
Tyrosinase inhibitory activities of plant extract and isolated compounds were determined by spectrophotometric approach using L-tyrosine as the substrate. The procedure was adopted according to our previous studies.10 Firstly, all samples and compounds were dissolved in DMSO at 1.0 mg mL−1 and then diluted to different concentrations. Secondly, each sample (30 μL) was diluted with 970 μL 0.05 mM sodium phosphate buffer (pH 6.8) in the test tubes, then 1 mL L-tyrosine solution was added and finally 1.0 mL mushroom tyrosinase (200 units mL−1) was added. Thirdly, the test mixture (3.0 mL) was mixed by Vortex and absorbance at 490 nm was measured, then it incubated for 20 min at 37 °C and absorbance at 490 nm was measured the second time. The absorbance of the same mixture without the test sample was used as the blank. Fourthly, the IC50, the concentration of plant extracts or compounds at which half the original tyrosinase activity is inhibited, was determined for plant extracts and each compound. The percent inhibition of tyrosinase activity was calculated as follows:
| % inhibition = [(A2 − A1) − (B2 − B1)]/(A2 − A1) × 100 |
A1 is the absorbance at 490 nm of the blank at 0 min, A2 is the absorbance at 490 nm of the blank at 20 min; B1 is the absorbance at 490 nm of test sample at 0 min, B2 is the absorbance at 490 nm of test sample at 20 min.
Statistical analysis
The analysis of variance (ANOVA) and Tukey's multiple range test for comparison of means and least significant differences were performed with the obtained data using the SAS system (SAS Institute, Inc, Cary, NC). P < 0.05 was selected as the level decision for significant differences.
Results and discussion
HPLC analysis of leaves, twigs, stems, and roots of C. cochinchinensis
The tyrosinase inhibition assay showed that the stem extract had the highest inhibitory activity, followed by the root and the twig, and finally the leaf extract (Table 1). HPLC analysis was subsequently performed to compare their chemical profiles. It was found that the chemical profile (Fig. 1) of the leaf extract displayed only one major peak and the profile of the root extract was similar to that of the stem extract. By using oxyresveratrol as a reference standard, it was found that oxyresveratrol, a well-known tyrosinase inhibitor was present in the twig, stem and root. Its content was very low in the twig and root, while it was one of the main components in the stem, suggesting it might be a major compound contributing to the tyrosinase inhibitory activity of the stem extract. In addition, the stem extract also contained other phenolics at high concentrations, and chromatographic and spectral methods were further introduced to study the chemical components of the stem extract.
Table 1
Tyrosinase inhibitory activity of different parts of C. cochinchinensis
| Plant |
Extracts |
IC50 (μg mL−1) |
|
C. cochinchinensis
|
Stem (crude extract) |
36.3 |
| Root (crude extract) |
56.2 |
| Twig (crude extract) |
>400 |
| Leaf (crude extract) |
>400 |
|
Kojic acid
|
|
10.2 |
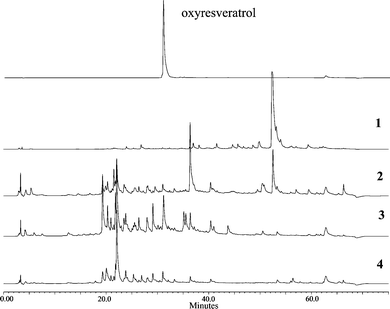 |
| | Fig. 1
HPLC
chromatograms of leaf (1), twig (2), stem (3), and root (4) extracts of C. cochinchinensis. | |
Structural elucidation of the new compound
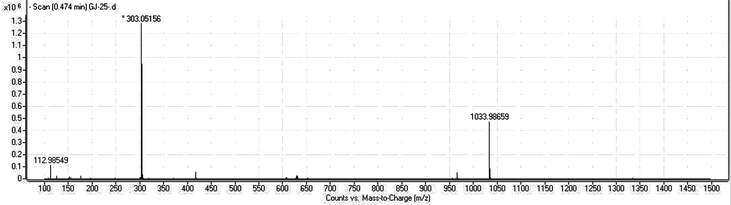
Compound 1 was found to be a new compound and its structure was elucidated by interpretation of HRESI-MS and NMR data. Its HRESI-MS showed a molecular ion peak [M − H]− at m/z 303.0516 (Fig. 2), and its 13C NMR spectrum showed the signals for fifteen carbons, consistent with a molecular formula of C15H12O7. In its 1H NMR spectrum (Table 2), five aromatic proton signals, including an ABX aromatic spin system at δ 7.22 (1H, d, J = 9.0 Hz), 6.36 (2H, overlap) and two meta-coupled aromatic protons at δ 5.96 (1H, d, J = 1.9 Hz), 5.91 (1H, d, J = 1.9 Hz), were observed, together with two doublets at δ 5.55 (J = 1.8 Hz) and δ 4.13 (J = 1.8 Hz), suggesting that compound 1 has a dihydroflavonol skeleton. This was supported by 13C NMR data (Table 1), which totally showed 15 carbon signals. Carbons at δ 79.4 (CH) and 72.1 (CH) were assignable to carbon at 2 and 3 positions. Moreover, five oxygenated carbons at δ 168.9, 166.5, 165.3, 159.8, and 156.7 were also observed in the down field of the 13C NMR spectrum, implying B ring of the dihydroflavonol was hydroxylated at C-2′ and C-4′.11 This was also consistent with the 13C NMR spectra of other compounds isolated from the same plant. For example, 2,3-trans-dihydromorin has carbons at δ 160.3 at position C-4′ and 158.8 at position C-2′ in the B ring, and morin-7-O-β-D-glucoside also has carbons at δ 162.8 at position C-4′ and 158.7 at position C-2′. Carefully comparing the 1H NMR and 13C NMR of compound 1 with that of 2,3-trans-dihydromorin, it was found that these two compounds might have similar structures except for the relative configuration of C-2 and C-3 position. For 2,3-trans-dihydromorin, its 1H NMR showed two doublets at δ 5.38 (1H, d, J = 11.5 Hz, H-2), 4.78 (1H, d, J = 11.5 Hz, H-3), while for compound 1, the 1H NMR showed two doublets at δ 5.55 and 4.13 with coupling constant only 1.8 Hz, suggesting compound 1 is a 2,3-cis-isomer of 2,3-trans-dihydromorin. The coupling constant difference also agreed with what reported for 2,3-cis-dihydroquercetin and 2,3-trans-dihydroquercetin.13 Kiehlmann and Slade14 synthesized a series of didydroquercetin derivatives and compared the chemical shifts of H-2 and H-3 in 1H NMR and J23 coupling constant of cis- and trans-configuration, suggesting that the resonance of cis-configuration was usually 0.40 ppm lower at H-2 and 0.34–0.41 ppm higher at H-3 than that of trans-configuration, furthermore, its J23 coupling constant (usually 2–3 Hz) was much smaller than that of trans-isomer (usually 11–12 Hz). They hence summarized some rules to differentiate the cis- and trans-dihydroflavonols. These two compounds from our study followed these rules very well. Generally, the absolute configuration can be determined by circular dichroism (CD) data.15 However, a CD test showed that no cotton effect was detected in this study, suggesting that compound 1 should be a racemic mixture. On the other hand, the cis- and trans-configuration of dihydroflavonols can be differentiated not only by their 1H NMR data and CD, but also by the HPLC chromatogram. The trans-configuration was eluted a little bit later (different about 1 min) than that of the cis-configuration in the current HPLC condition (Fig. 3). Taken together, compound 1 was elucidated as (±)2,3-cis-dihydromorin, a racemic mixture with its type of 2,3-cis-configuration rarely found in nature.
Table 2
1H NMR (500 MHz) and 13C NMR (125 MHz) data of compounds 1 and 10 in CD3OD
| Positions |
Compound 1 |
2,3-trans-Dihydromorin (10) |
|
δ
H
|
δ
C
|
δ
H
|
δ
C
|
| 2 |
5.55 (1H, d, J = 1.8 Hz) |
79.4 (d) |
5.38 (1H, d, J = 11.5 Hz) |
80.2 (d) |
| 3 |
4.13 (1H, d, J = 1.8 Hz) |
72.1 (d) |
4.78 (1H, d, J = 11.5 Hz) |
72.7 (d) |
| 4 |
|
196.9 (s) |
|
199.1 (s) |
| 5 |
|
168.9 (s) |
|
168.7 (s) |
| 6 |
5.91 (1H, d, J = 1.9 Hz) |
97.5 (d) |
5.87 (1H, d, J = 2.0 Hz) |
97.2 (d) |
| 7 |
|
165.3 (s) |
|
165.1 (s) |
| 8 |
5.96 (1H, d, J = 1.9 Hz) |
96.5 (d) |
5.91 (1H, d, J = 2.0 Hz) |
96.5 (d) |
| 9 |
|
166.5 (s) |
|
165.5 (s) |
| 10 |
|
102.1 (s) |
|
102.1 (s) |
| 1′ |
|
115.2 (s) |
|
115.8 (s) |
| 2′ |
|
156.7 (s) |
|
158.8 (s) |
| 3′ |
6.36 (1H, overlapped) |
103.5 (d) |
6.34 (1H, overlapped) |
103.9 (d) |
| 4′ |
|
159.8 (s) |
|
160.3 (s) |
| 5′ |
6.36 (1H, overlapped) |
107.8 (d) |
6.34 (1H, overlapped) |
108.2 (d) |
| 6′ |
7.22 (1H, d, J = 9.0 Hz) |
131.0 (d) |
7.33 (1H, d, J = 8.0 Hz) |
131.1 (d) |
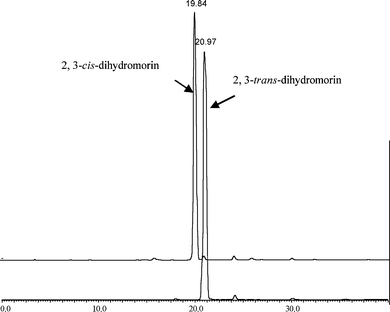 |
| | Fig. 3
HPLC
chromatograms of compound 1 (2,3-cis-dihydromorin) and 10 (2,3-trans-dihydromorin). | |
(±)2,3-cis-Dihydromorin (1): pale yellow powder; 1H NMR (CD3OD, 500 MHz) and 13C NMR (CD3OD, 125 MHz), see Table 2. HRESI-MS m/z 303.0516 [M − H]− (calcd for C15H11O7, 303.0510) (Fig. 4).
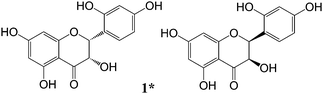 |
| | Fig. 4 Structures of new compounds from the stems of C. cochinchinensis. | |
Identification of known compounds
The structures for the 15 known compounds were identified by comparing ESI-MS, 1H NMR and 13C NMR data with the literature, as dihydrokaempferol 7-O-β-D-qlucopyranoside (2),16 skimmin (3),17quercetin-7-O-β-D-glucoside (4),182,3-dihydroquercetin 7-O-β-D-glucoside (5),19kaempferol-7-O-β-glucopyranoside (6),20quercetin-3,7-di-O-β-D-glucoside (7),21morin-7-O-β-D-glucoside (8),221,3,5,8-tetrahydroxyxanthen-9-one (9),232,3-trans-dihydromorin (10),24 aromadendrin (11),25oxyresveratrol (12),24genistin (13),26 protocatechuic acid (14),27kaempferol 3,7-di-O-β-glucopyranoside (15),28 and naringenin (16).29
Tyrosinase inhibition activities of purified compounds
These compounds were further tested for their inhibitory activities against mushroom tyrosinase, and the results showed three of them (oxyresveratrol, 2,3-trans-dihydromorin, and 2,3-cis-dihydromorin) exhibited better tyrosinase inhibitory activities than that of kojic acid (Table 3). Some of them (quercetin-7-O-β-D-glucoside, kaempferol-7-O-β-glucopyranoside and morin-7-O-β-D-glucoside) exhibited moderate inhibitory activities (Table 3). These results also provided information to establish the preliminary structure–activity relationships of these compounds. It was shown that both the number and the location of hydroxyl groups affected the tyrosinase inhibitory activities of flavonoids with the later parameter playing a more important role. In particular, simultaneous occurrence of a hydroxyl group at 2′ and 4′ position of the B ring would greatly enhance the resultant tyrosinase inhibitory activities of the compounds. When some of these hydroxyl groups were associated with other groups or substitute, activities of the compounds would be significantly affected. As an example, the glycosidation of the hydroxyl groups greatly reduced the tyrosinase inhibitory activities of the compounds, especially when the glycosidation of the hydroxyl group was occurred at C-3 position. This phenomenon could be observed by comparing the tyrosinase inhibitory activities of the following compounds, such as quercetin-7-O-β-D-glucoside and quercetin-3,7-di-O-β-D-glucoside; kaempferol-7-O-β-glucopyranoside and kaempferol-3,7-di-O-β-glucopyranoside; dihydrokaempferol 7-O-β-D-qlucopyranoside and aromadendrin. The tyrosinase inhibitory activity of quercetin-7-O-β-D-glucoside (IC50 = 143.0 μM) is at least seven-times stronger than that of quercetin-3,7-di-O-β-D-glucoside (IC50 > 1000 μM). Meanwhile, their inhibitory activities are much lower than that of quercetin.30 In addition, it was also deduced that the steric configuration of compounds might be an influencing factor on the tyrosinase inhibitory activity, such effect could be observed between the compounds 2,3-trans-dihydromorin and 2,3-cis-dihydromorin. The trans-configuration exhibited slightly stronger tyrosinase inhibitory activity than that of cis-configuration.
Table 3
Tyrosinase inhibitory activity of compounds from the stem of C. cochinchinensis
| Compounds (compound number, yield × 103 %) |
IC50 (μM) ± SD |
|
NT, not test.
|
|
(±)2,3-cis-dihydromorin (1, 0.88) |
31.14 ± 0.49 |
|
dihydrokaempferol 7-O-β-D-qlucopyranoside (2, 3.98) |
>1000 |
| skimmin (3, 0.32) |
NT
a |
|
quercetin-7-O-β-D-glucoside (4, 0.42) |
143.037 ± 2.16 |
|
2,3-dihydroquerctin 7-O-β-D-glucoside (5, 3.21) |
>500 |
|
kaempferol-7-O-β-glucopyranoside (6, 0.27) |
161.54 ± 4.12 |
|
quercetin-3,7-di-O-β-D-glucoside (7, 0.46) |
>1000 |
|
morin-7-O-β-D-glucoside (8, 0.27) |
196.33 ± 4.47 |
|
1,3,5,8-tetrahydroxyxanthen-9-one (9, 0.18) |
NT
a |
|
2,3-trans-dihydromorin (10, 0.25) |
21.09 ± 0.70 |
| aromadendrin (11, 1.54) |
>500 |
|
oxyresveratrol (12, 12.1) |
2.33 ± 0.24 |
|
genistin (13, 0.06) |
NT
a |
| protocatechuic acid (14, 0.52) |
>1000 |
|
kaempferol 3,7-di-O-β-glucopyranoside (15, 0.52) |
>1000 |
|
naringenin (16, 1.0) |
>800 |
|
kojic acid
|
50.79 ± 0.96 |
Conclusions
In this study, the phytochemical composition of the ethanol extract of the stem of C. cochinchinensis was systematically observed and sixteen compounds, including one new compound and fifteen known compounds were isolated. Thirteen compounds were tested on mushroom tyrosinase, three of them showed strong tyrosinase inhibitory activities indicating that the stem extract of C. cochinchinensis might be a promising candidate for application as a skin whitening ingredient in cosmetic products.
References
- C. W. G. Van Gelder, W. H. Flurkey and H. J. Wichers, Sequence and Structural Features of Plant and Fungal Tyrosinase, Phytochemistry, 1997, 45, 1309–1323 CrossRef CAS.
- M. I. Gil, E. Aguayo and A. A. Kader, Quality Changes and Nutrient Retention in Fresh-cut Versus Whole Fruits During Storage, J. Agric. Food Chem., 2006, 54, 4284–4296 CrossRef CAS.
- W. R. Cortez-Vega, A. M. Becerra-Prado, J. M. Soares and G. G. Fonseca, Effect of L-ascorbic Acid and Sodium Metabisulfite in the Inhibition of the Enzymatic Browning of Minimally Processed Apple, Inter. J. Agric. Res., 2008, 3, 196–201 Search PubMed.
- F. Solano, B. Stefania, M. Picardo and G. Ghanem, Hypopigmenting Agents: An Updated Review on Biological, Chemical and Clinical Aspects, Pigm. Cell Res., 2006, 19, 550–571 Search PubMed.
- Y. Xu, A. H. Stokes, W. M. Freeman, S. C. Kumer, B. A. Vogt and K. E. Vrana, Tyrosinase mRNA is Expressed in Human Substantia Nigra, Mol. Brain Res., 1997, 45, 159–162 CrossRef CAS.
- S. M. Son, K. D. Moon and C. Y. Lee, Inhibitory Effects of Various Aanti-browning Agents on Apple Slices, Food Chem., 2001, 73, 23–30 CrossRef CAS.
- K. Shimizu, R. Kondo and K. Sakai, Inhibition of Tyrosinase by Flavonoids, Stilbenes and Related 4-substituted Resorcinols: Structure–activity Investgations, Planta Med., 2000, 66, 11–15 CrossRef CAS.
- H. J. Kim, S. H. Seo, B. G. Lee and Y. S. Lee, Identification of Tyrosinase Inhibitors from Glycyrrhiza uralensis, Planta Med., 2005, 71, 785–787 CrossRef CAS.
- Z. P. Zheng, K. W. Cheng, J. T. K. To, H. Li and M. Wang, Isolation of Tyrosinase Inhibitors from Artocarpus heterophyllus and Use of Its Extract as Anti-browning Agent, Mol. Nutr. Food Res., 2008, 52, 1530–1538 CrossRef CAS.
- Z. P. Zheng, S. Chen, S. Wang, X. C. Wang, K. W. Cheng, J. J. Wu, D. J. Yang and M. Wang, Chemical Components and Tyrosinase Inhibitors from the Twigs of Artocarpus heterophyllus, J. Agric. Food Chem., 2009, 57, 6649–6655 CrossRef CAS.
- P. C. Zhang, Z. M. Feng and Y. H. Wang, Flavonoids, Including an Unusual Flavonoids-Mg2+ Salt, from Roots of Cudrania cochinchinensis, Phytochemistry, 2005, 66, 2759–2765 CrossRef CAS.
- N. J. Sun, C. J. Chang and J. M. Cassady, A Cototoxic Isoflavone from Cudrania cochinchinensis, Phytochemistry, 1988, 27, 951–952 CrossRef CAS.
- L. N. Lundgren and O. Theander,
cis and trans-Dihydroquercetin Glucosides from Needles of Pinus sylvestris, Phytochemistry, 1988, 27, 829–832 CrossRef CAS.
- E. Kiehlmann and P. W. Slade, Methylation of Dihydroquercetin Acetates: Synthesis of 5-O-methyldihydroquercetin, J. Nat. Prod., 2003, 66, 1562–1566 CrossRef CAS.
- D. Slade, D. Ferreira and J. P. J. Marais, Circular Dichroism, a Powerful Tool for the Assessment of Absolute Configuration of Flavonoids, Phytochemistry, 2005, 66, 2177–2215 CrossRef CAS.
- L. Y. Foo and J. J. Karchesy, Polyphenolic Glycosides from Douglas fir Inner Bark, Phytochemistry, 1989, 28, 1237–1240 CrossRef CAS.
- J. Reisch and S. H. Achenbach, A Furanocoumarin Glucoside from Stembark of Skimmia japonica, Phytochemistry, 1992, 31, 4376–4377 CrossRef CAS.
- K. R. Markham, B. Ternai, R. Stanley, H. Geiger and T. J. Mabry, Carbon-13 NMR Studies of Flavonoids. III. Naturally Occurring Flavonoid Glycosides and Their Acylated Derivatives, Tetrahedron, 1978, 34, 1389–1397 CrossRef CAS.
- H. F. Pan and L. N. Lundgren, Phenolics from Inner Bark of Pinus sylvestris, Phytochemistry, 1996, 42, 1185–1189 CrossRef CAS.
- J. Xu, X. Li, P. Zhang, Z. L. Li and Y. Wang, Anti-inflammatory Constituents from the Roots of Smilax bockii Warb, Arch. Pharmacal Res., 2005, 28, 395–399 Search PubMed.
- A. J. Dick and K. C. Smith, Quercetin Glucosides and Galactosides: Substrates and Inhibitors of Apple β-galactosidase, J. Agric. Food Chem., 1990, 38, 923–926 CrossRef CAS.
- S. Z. Zheng, L. P. Sun and X. W. Shen, Chemical Constituents of Mosla chinensis Maxim, Zhiwu Xuebao, 1996, 38, 156–160 Search PubMed.
- H. M. Zhou, Y. L. Liu, G. Blasko and G. A. Cordell, Swertiabisxanthone-I from Swertia macrosperma, Phytochemistry, 1989, 28, 3569–3571 CrossRef CAS.
- Z. P. Zheng, K. W. Cheng, Q. Zhu, X. C. Wang, Z. X. Lin and M. Wang, Tyrosinase Inhibitory Constituents from the Roots of Morus nigra: A Structure–Activity Relationship Study, J. Agric. Food Chem., 2010, 58, 5368–5373 CrossRef CAS.
- G. D. Monache, R. Scurria, A. Vitali, B. Botta, B. Monacelli, G. Pasqua, C. Palocci and E. Cernia, Two Isoflavones and a Flavone from the Fruits of Maclura pomifera, Phytochemistry, 1994, 37, 893–898 CrossRef.
- A. A. Drenin, E. K. Botirov and E. V. Petrulyak, New Genistein Monogalactoside from the Aerial Part of Trifolium pretense, Chem. Nat. Compd., 2008, 44, 178–181 CrossRef CAS.
- C. H. Yang, Q. F. Tang, J. H. Liu, Z. J. Zhang and W. Y. Liu, Preparative Isolation and Purification of Phenolic Acids from Smilax china by High-speed Counter-current Chromatography, Sep. Purif. Technol., 2008, 61, 474–478 CrossRef CAS.
- J. H. Lin and Y. Z. Lin, Flavonoids from the Leaves of Loranthus kaoi (Chao) Kiu, J. Food Drug Anal., 1999, 7, 185–190 Search PubMed.
- H. B. Wang, M. G. Nair, G. M. Strasburg, A. M. Booren and J. I. Gray, Antioxidant Polyphenols from Tart Cherries (Prunus cerasus), J. Agric. Food Chem., 1999, 47, 840–844 CrossRef CAS.
- I. Kubo and I. Kinst-Hori, Flavonols from Saffron Flower: Tyrosinase Inhibitory Activity and Inhibition Mechanism, J. Agric. Food Chem., 1999, 47, 4121–4125 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.