DOI:
10.1039/C1FO10027K
(Paper)
Food Funct., 2011,
2, 328-337
Antioxidant, pro-oxidant and cytotoxic properties of parsley
Received
16th February 2011
, Accepted 2nd May 2011
First published on 18th May 2011
Abstract
Parsley (Petroselinum crispum) leaves were macerated with a mixture of methanol: water: acetic acid to produce a crude extract which was then defatted with (40°–60°) petrol. Antioxidant activity of the extract was evaluated using a battery of in vitro assays, viz., iron(III) reduction, iron(II) chelation and free radical scavenging assays. Evaluation of the pro-oxidant activity of the extract was based upon its effects upon DNA fragmentation and protein carbonylation. Cytotoxicity and apoptotic effects of the extract were determined in non-cancerous CV1-P fibroblast and cancerous A375 melanoma cells using MTT and LDH tests and caspase 3-like activity assay. The highest concentration, 2.0 mg ml−1, decreased the viability of both cell lines, however, the cancerous melanoma cells were slightly susceptible to the effects. The observed cytotoxicity was not due to the caspase 3 activity. In conclusion, the toxicity might be explained by the pro-oxidative activity of components within the extract against proteins and/or DNA but it is not related to caspase 3-dependent apoptosis within cells.
Introduction
Parsley (Petroselinum crispum) is a widely cultivated, light green biennial herb of the Apiaceae (Umbelliferae) family which is widely used for its organoleptic properties and essential oil. It is reported to contain a wide spectrum of components, viz., carotenoids,1 coumarins,2 flavonoids, tannins and triterpenes3 and volatile oils.4 Therefore, it should not be surprising that a number of health-promoting properties have been ascribed to parsley-derived preparations. The herb and root are often recommended in traditional herbal/folklore medicine for their alleged beneficial effects on gastric, menstrual and urinary disorders, on cough and myalgia.4 A cursory search through the scientific literature reveals that parsley extracts possess a variety of biological activities, including protection against diabetes-induced oxidative damage,5 antibiotic,6 antihypertensive,7 antioxidant,8 antitumorigenic,9 diuretic,7 enzyme inducing,10 gastroprotective3 and platelet normalizing11 actions amongst others.
Naturally occurring antioxidants have been shown to protect cells from oxidative stress,12 which is considered to have a major influence upon human health and wellbeing. Furthermore, they have been shown to possess antiproliferative or apoptosis inducing properties in tumor cell lines through various mechanisms, e.g. phosphatidylinositol-3-kinase inhibition,13 activation of mitogen-activated protein kinase14 and tumor suppression protein p53 accumulation,15 properties which may have major medicinal significance. Previously, we demonstrated that a water-soluble parsley extract possessed antioxidant-related activity in a battery of in vitro assays,8 however, the same extract did not exert any significant cytotoxic effect upon SH-SY5Y neuroblastoma cells and had no quantitative effect upon p53 levels, as determined by Western blotting.16 Accordingly, we decided to evaluate the cytotoxic, apoptotic and antioxidant (iron(III) reducing iron(II) chelating and free radical scavenging) potential of an aqueous methanol parsley extract to determine if another type of parsley extract would demonstrate better activity. Furthermore, as any cytotoxicity may be related to natural antioxidants' pro-oxidant tendencies,17–19 the parsley extract was evaluated for its effect upon oxidation-mediated DNA fragmentation and protein degradation.
Experimental
Plant material
Air-dried leaf material was obtained from Pimenta Oy (Finland). One hundred grams of leaves were placed in a screw-top bottle and macerated with 1000 ml (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:1, v/v/v) MeOH:H2O:HOAc with stirring for 24 h. The extractant was filtered and the extraction process was repeated a further 2 times. The resulting extracts were combined, filtered through cheese cloth, defatted using 40°–60° petrol, filtered through Whatman No. 4 filter paper, concentrated in vacuo (≤30 °C) and lyophilized.
:20:1, v/v/v) MeOH:H2O:HOAc with stirring for 24 h. The extractant was filtered and the extraction process was repeated a further 2 times. The resulting extracts were combined, filtered through cheese cloth, defatted using 40°–60° petrol, filtered through Whatman No. 4 filter paper, concentrated in vacuo (≤30 °C) and lyophilized.
Total phenol content
Total phenol content was estimated as gallic acid equivalents.20 To ca. 3.0 ml H2O, 50 μl of dissolved sample was transferred to a 5.0 ml volumetric flask to which was added 250 μl Folin-Ciocalteu reagent. After 1 min, 750 μl 20% (w/v) Na2CO3 solution was added and the volume was made up to 5.0 ml with H2O. After 60 min, the absorbance was measured at 760 nm.
Cell cultures and treatments
Human A375 melanoma cells were cultured in Dulbecco's modified eagle medium (DMEM) containing GlutaMAX I, 4500 mg l−1 of glucose and 110 mg l−1 of sodium pyruvate and supplemented with 10% (v/v) heat-inactivated foetal bovine serum, 100 U/ml penicillin and 100 μg ml−1 streptomycin. Green monkey CV1-P fibroblast cells were cultured in DMEM containing L-glutamine, 1000 mg l−1 of D-glucose and sodium pyruvate and supplemented with 10% (v/v) heat-inactivated foetal bovine serum, 100 U/ml penicillin and 100 μg ml−1 streptomycin. Both cell lines were cultured at 37 °C in a humidified atmosphere containing 5% CO2. Cells were plated in 6 cm cell culture plates for caspase 3 assays and in 96 well plates for cytotoxicity assays. After culturing cells over night in plates/wells, the medium was replaced with the medium containing different concentrations of the parsley extract and the cells were incubated for 24 h. The final concentration of MeOH was 1% (v/v) in all experiments including control cells.
Cytotoxicity assays
Cytotoxicity was measured by two different methods. Metabolic activity of cells was determined by the reduction of yellowish methylthiazolyldiphenyl-tetrazolium bromide (MTT) to dark blue formazan by viable and metabolically active cells, membrane integrity of cells was measured by the leakage of lactate dehydrogenase (LDH) from cells using a commercial CytoTox-ONE Homogeneous Membrane Integrity Assay kit (Promega). Both assays were performed as previously described with minor modifications.16 In brief, after 24 h treatment with the parsley extract, 50 μl of medium from treated cells was mixed with the same amount of CytoTox-ONE reagent and incubated for 30 min at RT with shaking during the first 10 min. The reaction was stopped by adding CytoTox-ONE Stop Solution and the fluorescence was measured with excitation at 560 nm and emission at 590 nm using a fluorescence microplate reader (Varioskan, Thermo Scientific, USA). The percentage membrane integrities were calculated using an eqn (1) from the values of 3–4 independent experiments. The metabolic activity of cells was determined by adding the MTT dissolved in PBS to the cells and remaining medium at a final concentration of 0.5 mg/ml. Both cell lines were incubated with the MTT for 2.5 h at 37 °C in 5% CO2. Then the medium was removed and the formazan crystals were dissolved in DMSO. The absorbance was measured at 550 nm, subtracting the background at 655 nm using a microplate reader. The percentage metabolic activities were calculated using eqn (2) from the values of 4–5 independent experiments.| | | Percentage membrane integrity = Fluorescence treated cells-blank/Fluorescence untreated cells-blank × 100 | (Eq. 1) |
| | | Percentage metabolic activity = Absorbance treated cells − blank/Absorbance untreated cells-blank × 100 | (Eq. 2) |
Caspase 3 activity
The caspase 3 like activity assay was performed as previously described.21,22 In brief, the cells were collected by scraping and centrifuged for 5 min at 3000 g at 4 °C. Cell pellets were stored at −80 °C until they were lysed with an ice-cold lysis buffer (10 mM Tris-HCl, 10 mM Na2H2PO4, 10 mM Na2HPO4, 1% Triton X-100, pH 7.5) and centrifuged for 15 min at 20 000 g at 4 °C. Supernatants were collected and protein amounts were defined by BCA protein assay. The caspase 3 like activity was measured by mixing the equal amounts of supernatant and assay buffer (20 mM HEPES, 10% glycerol, pH 7.5) containing 120 μM Ac-DEVD-AMC and 2 mM DTT in 96-well plate. The reaction was incubated for 2 h at 37 °C and the cleavage of Ac-DEVD-AMC measured with excitation at 370 nm and emission at 445 nm using a fluorescence microplate reader. The results were calculated as release of AMC in pmol min−1 mg−1 of protein.
Iron(III) reductive capacity was assessed as previously described.23 A 0.5 ml aliquot of dissolved sample was mixed with 0.75 ml phosphate buffer (0.2 M, pH 6.6) and 0.75 ml of a 1% (w/v) potassium hexacyanoferrate solution. After 30 min at 50 °C, 0.75 ml 10% (w/v) aq. trichloroacetic acid (TCA) solution was added and the mixture was centrifuged. A 0.75 ml aliquot was mixed with 0.75 ml H2O and 0.25 ml 0.1% (w/v) aq. FeCl3·6H2O solution and the absorbance was measured at 700 nm. Data are presented as ascorbic acid equivalent (AscAE) values expressed as μmol ascorbic acid/g sample (dry wt.) of the sample.
Iron(II) chelation was assessed as previously described.24 A 675 μl aliquot of sample dissolved in acetate buffer (10 mM, pH 3.6) was added to 75 μl of a 1 mM FeSO4·7H2O (in MeOH) solution. After 20 min, 750 μl of 1.0 mM ferrozine (in acetate buffer) solution was added. After 10 min, the absorbance was measured at 562 nm. Data are presented as EDTA equivalent (EDTAE) values expressed as μmol EDTA/g sample (dry wt.).
1,1-Diphenyl-2-picrylhydrazyl (DPPH•) free radical scavenging
DPPH• scavenging was assessed as previously described.25 A 50 μl aliquot of dissolved sample was mixed with 450 μl Tris-HCl buffer (50 mM, pH 7.4) and 1.0 ml of 0.1 mM DPPH• (in MeOH) solution. After 30 min, the absorbance was measured at 517 nm. The percentage inhibition was calculated using eqn (3).| | | Percentage inhibitory activity = (Absorbance DPPH• − Absorbance sample)/Absorbance DPPH• x 100 | (Eq. 3) |
2,2′-Azinobis(3-ethylbenzothiazoline)-6-sulfonic acid (ABTS•+) free radical scavenging
ABTS•+ scavenging was assessed as previously described.26 The ABTS•+ was generated by reacting a 7 mM ABTS solution with K2S2O8 (2.45 mM, final concentration) in the dark for ca. 12–16 h and thereafter adjusting the Abs734nm to 0.700 (±0.030) at room temperature. A 15 μl aliquot of dissolved sample was added to 1.485 ml ABTS•+ free radical and the absorbance at 734 nm was measured at 10 min after initial mixing. The percentage inhibition was plotted as a function of concentration and the Trolox equivalent (TE) values were calculated against a Trolox calibration curve.
Alkylperoxyl (LOO•) free radical scavenging
LOO• radical scavenging was assessed using the β-carotene-18:2(n–6) auto-oxidation method as previously described.27 To a 50 μl aliquot of dissolved sample was added 5 ml β-carotene-linoleic acid aq. emulsion (4.0 μg ml−1 β-carotene, 0.4 mg ml−1 linoleic acid and 4 mg ml−1 Tween 20). After careful mixing the absorbance (Abs0) at 470 nm was measured. The remaining samples were placed in a water bath at 50 °C for a period of 60 min. Thereafter, the absorbance (Abs60) was measured. Data are presented as percentage antioxidant activity (AA %) values, calculated using eqn (4).| | | Percentage antioxidant activity = [1 − (Absorbance samplet = 0 − Absorbance samplet = 60)/(Absorbance controlt=0 − Absorbance controlt = 60)] × 100 | (Eq. 4) |
Superoxide anion (O2•−) free radical scavenging
O2•− free radical scavenging was assessed as previously described.28 The reaction mixture contained 2.4 ml of NaH2PO4-NaOH buffer (50 mM, pH 7.4), 100 μl of (5.0 mg ml−1) dissolved extract, 100 μl of 30 mM of Na2EDTA solution, 100 μl of 3 mM hypoxanthine solution and 200 μl of 1.42 mM nitroblue tetrazolium solution. After 3 min incubation at room temperature, the reaction was initiated by the addition of 100 μl of a 0.50 U/ml xanthine oxidase solution. The absorbance at 560 nm was measured after 20 min. To confirm the O2•− scavenging, uric acid formation was confirmed. Data are presented as Abs560 nm values.
Nitric oxide (NO•) free radical scavenging
NO• scavenging was assessed as previously described.29 A 0.5 ml aliquot (0.5 mg ml−1) of extract dissolved in KH2PO4-KOH (50 mM, pH 7.4) was mixed with 0.5 ml (10 mM) sodium nitroprusside solution. The mixture was incubated at 37 °C for 2.5 h under normal light condition. After incubation the sample was placed in the dark for 20 min. Thereafter, 1 ml of Griess reagent (1 g l−1N-(1-naphtyl) ethylenediamine and 10 g l−1 sulphanilamide dissolved in 20 ml l−1 aqueous H3PO4) was added and the absorbance at 546 nm was measured after 40 min. Data are presented as quercetin equivalent (QuE) values expressed as mmol quercetin/g (dry wt.) of the sample.
•OH-mediated oxidative degradation of bovine phospholipid-derived liposomes at physiological pH was assessed as previously described.30 Bovine brain phospholipid extract was mixed with 10 mM phosphate-buffered saline (PBS, pH 7.4) and sonicated until an opalescent suspension was obtained containing 5 mg ml−1 phospholipid liposomes. A 0.2 ml aliquot of liposome solution was mixed with 0.5 ml of PBS, 0.1 ml of 1 mM FeCl3·6H2O solution and 0.1 ml of (1 mg ml−1) dissolved extract. Peroxidation was initiated by adding 0.1 ml of 1 mM ascorbate solution. After 60 min incubation at 37 °C, 50 μl of 2% (w/v) BHT was added followed by 1 ml of 2.8% (w/v) TCA and 1 ml of 1% (w/v) 2-thiobarbituric acid (TBA) solution. The resulting solution was then heated at 100 °C for 20 min and the resulting chromogen was extracted into 2 ml of n-BuOH and the absorbance was measured at 532 nm.
Inhibition of non-site-specific •OH-mediated degradation was assessed as previously described.31 The reaction mixture contained 400 μl of NaH2PO4-NaOH buffer (50 mM, pH 7.4), 100 μl dissolved extract, 100 μl of 30 mM 2-deoxy-D-ribose solution, 200 μl of 100 μM FeCl3 and 104 mM EDTA (1:1 v/v) premixed solution, 100 μL of 1.0 mM H2O2 and 100 μl of 1.0 mM ascorbic acid solution. Tubes were vortexed and incubated at 37 °C for 60 min. Thereafter, 50 μl of 2.0% (w/v) BHT was added to each tube followed by 1 mL of 2.8% (w/v) TCA and 1 mL of 1.0% (w/v) TBA solution. The samples were vortexed and heated in a water bath at 100 °C for 20 min. The reaction was stopped by placing the test tubes in an ice-H2O bath for a 5 min period. To each tube was added 2 ml of n-BuOH, and the mixture was vigorously vortexed. After centrifugation, the extent of oxidation was estimated from the absorbance of the organic layer at 532 nm. Data are presented as percentage inhibition values.
The ability to inhibit site-specific •OH-mediated degradation was assessed as described above except the EDTA solution was substituted by buffer.
The ability of the parsley extract to induce DNA oxidation mediated by •OH was assessed as previously described.32 The reaction mixture contained 100 μl (1.8 mM) 1,10-phenanthroline solution, 480 μl (250 μM) CuCl2·2H2O solution, 300 μl (1.68 mg ml−1) DNA solution 220 μl (100 mM, pH 7.4) KH2PO4-KOH buffer and 100 μl (0.1–5.0 mM) aq. ascorbic acid or (0.1–5.0 mg mL−1) dissolved extract. The reaction mixture was then incubated at 37 °C for 60 min. Thereafter, 100 μl (0.1 M) EDTA solution was added followed by 1 ml (1%, w/v) TBA solution (in 50 mM NaOH) and 1 ml (25%, v/v) HCl solution. Once vortexed, the samples were heated at 100 °C for 20 min. After a 5 min immersion in an ice-H2O bath, 2 ml n-BuOH was added to each tube and the mixture was vigorously vortexed. After centrifugation, the absorbance of the organic layer was measured at 532 nm. The data are presented as mean Abs532 nm values.
The ability of the parsley extract to induce protein oxidation mediated by •OH was assessed as previously described.33 The reaction mixture contained 2.4 ml (1.25 mg ml−1) bovine serum albumin protein solution, 0.3 ml dissolved sample at 1.0 mg ml−1 or 0.3 ml (100 mM) 2,2′-azobis(2-amidinopropane) hydrochloride solution. The reaction mixture was then incubated at 50 °C for 90 min. The degree of protein oxidation was then determined using the 2,4-dinitrophenylhydrazine (DNPH) method as previously described.34 A 0.5 ml aliquot of 20 mM DNPH solution (in 2.5 M HCl) was added to 1.0 ml aliquot of the reaction mixture. After 1 h incubation at ambient temperature, 0.5 ml of 20% (w/v) TCA was added and the reaction mixture was left for 10 min in an ice-H2O bath. Thereafter it was centrifuged at 3000 g for 10 min. The supernatant was carefully discarded and the protein pellet was carefully washed 3 times with 3 ml (1:1, v/v) EtOAc:EtOH solution then dissolved in 1.5 ml of 6 M guanidine solution (in 2 M HCl). The absorbance was then measured at 370 nm. The date are presented as mean Abs370 nm values.
Statistical analysis
Data are presented as mean values ±95% confidence intervals. Analysis of variance was performed by ANOVA procedures. Significant differences between means were determined by Tukey's pairwise comparison test at a level of p < 0.05 whilst significant differences for cell-based assays were assessed by the Dunnett's multiple comparison test at a level of p < 0.05.
Results
Extract characteristics
The extraction procedure yielded a 23% (w/w) extract with a total phenol content, estimated by the Folin-Ciocalteu reagent method, equivalent to 18.25 ± 1.21 mg gallic acid/g (dry wt.) extract.
Evaluation of cytotoxic and apoptotic-related activities
Certain plant extracts have shown an increased toxicity against cancerous cells compared to non-cancerous cells.35,36 To determine whether the parsley extract possesses this trait, the metabolic activity and membrane integrity of cells were measured to evaluate and compare the possible cytotoxic effects of the extract in cancerous (A375) and non-cancerous (CV1-P) cells (Fig. 1). Cancerous cells were more susceptible to the 24 h treatment with different concentrations of the extract than non-cancerous cells. The metabolic activity of A375 cells decreased 30% and 50% after treatments with the concentrations of 1.5 and 2.0 mg ml−1, respectively. CV1-P cells responded to the treatment with the concentration of 2.0 mg ml−1 but the metabolic activity of cells did not change significantly. The membrane integrity decreased significantly in both cell lines after 2.0 mg ml−1 of treatment with the parsley extract. The leakage of LDH increased 210% from A375 cells and 179% from CV1-P cells. The slight increase in the LDH leakage from A375 cells was detected after the treatment with the concentration of 1.5 mg/ml. Both assays indicate the decreased viability of cells but do not reveal the mechanisms of toxicity. Therefore the activity of caspase 3 was determined in the cells to show whether declined cell viability is due to apoptosis or not. The concentrations 0.5, 1.0 or 2.0 mg ml−1 of the parsley extract had no effects on the caspase 3 activity after 24 h in both cell lines (data not shown). Our results imply the absence of caspase 3-dependent apoptotic mechanisms behind the cytotoxic effects observed with the parsley extract.
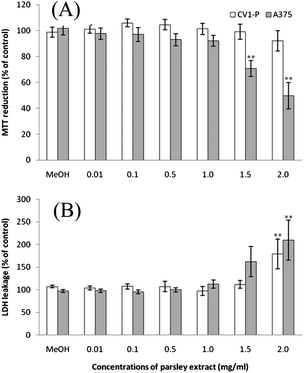 |
| | Fig. 1 The cytotoxic effects of parsley extract in fibroblast (CV1-P) and melanoma (A375) cells: (A) MTT reduction and (B) LDH leakage after 24 h treatment with different parsley extract concentrations. Data are presented as mean values ± SEM. Statistical significance was determined by one-way ANOVA, followed by Dunnett's multiple comparison test; **p < 0.01. | |
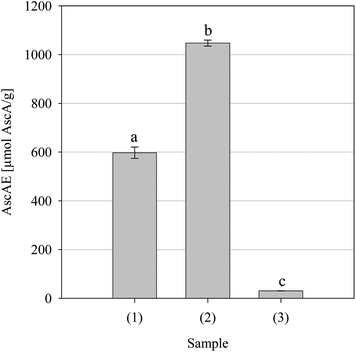 |
| | Fig. 2 Iron(III) to iron(II) reductive activity for the parsley extract and positive controls BHT and Trolox. Key: (1) BHT, (2) Trolox and (3) parsley extract. Data are presented as mean values ± SD (n = 4). Bars with the same lowercase letters (a–c) are not significantly (p > 0.05) different. | |
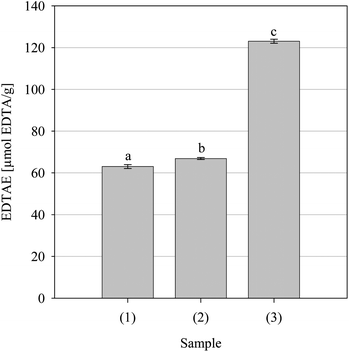 |
| | Fig. 3 Iron(II) chelating activity of the positive controls Pycnogenol (1) and Trolox (2) and the parsley extract. Data are presented as mean values ± SD (n = 4). Bars with the same lowercase letters (a–c) are not significantly (p > 0.05) different. | |
DPPH• free radical scavenging
Iron(III) reduction is regularly used to indicate the ET ability but it cannot demonstrate reactivity against free radicals or indicate HAT-type activity. Natural products recognized as possessing potent antioxidant activity are strong scavengers of the synthetic nitrogen-centered free radical DPPH•.40 Accordingly, the DPPH• scavenging ability of the parsley extract was evaluated. As can be seen from the dose-response curve in Fig. 4, the extract was capable of scavenging DPPH• and did so in a concentration-dependent fashion. From the estimated fifty percent scavenging concentration (SC50) values, the concentration required to scavenge 50% of the available DPPH•, it can be seen that the extract (10.95 ± 1.07 mg ml−1) was significantly less potent than either BHT (0.057 ± 0.002 mg ml−1) or Trolox (0.161 ± 0.003 mg ml−1). Despite this, the data suggests that the extract may afford protection to susceptible components by interfering with the propagation phase of free radical-mediated chain reactions by scavenging free radicals via an ET- or HAT-type mechanism.
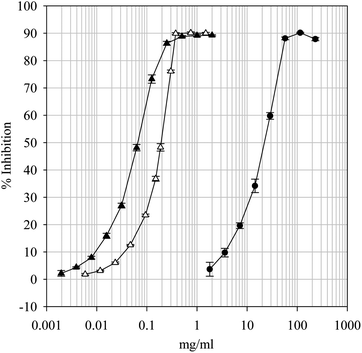 |
| | Fig. 4 1,1-Diphenyl-2-picrylhydrazyl free radical scavenging dose-response curves for the parsley extract and positive controls BHT and Trolox. Key: BHT (—▲—), Trolox (—△—) and Parsley extract (—●—). Data are presented as mean values ± SD (n = 4). | |
ABTS•+ free radical scavenging
The ABTS•+ cation is a reactive nitrogen species regularly used to screen natural products for free radical scavenging activity and is often used to complement DPPH•-based studies when solubility issues or colorimetric interference may arise.41 Accordingly, the ABTS•+ scavenging ability of the parsley extract was evaluated. As can be seen from Fig. 5, the extract was capable of scavenging ABTS•+ and did so in a concentration-dependent fashion (data not shown). The extract was significantly less potent than either ascorbic acid or BHT which is in accord with the DPPH• scavenging data presented in Fig. 4. Thus, similarly, the extract may interfere with the propagation phase of free radical-mediated chain reactions via an ET- or HAT-type mechanism.
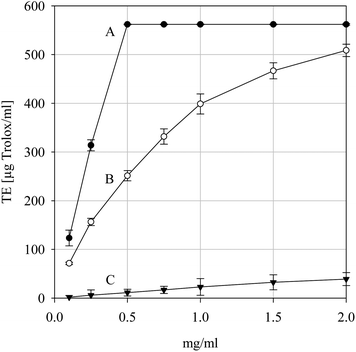 |
| | Fig. 5 2,2′-Azinobis(3-ethylbenzothiazoline)-6-sulfonic acid cation free radical scavenging activity the positive controls ascorbic acid (A) and BHT (B) and for the parsley extract (C). Data are presented as mean values ± SD (n = 5). | |
LOO• free radical scavenging
Alkylperoxyl free radicals are generated during enzymatic and non-enzymatic lipid peroxidation.42 Whilst LOO• are deactivated to less toxic hydroperoxides during enzymatic metabolism, LOO• from non-enzymatic sources react with their immediate surroundings to produce a range of cytotoxic by-products.42 Accordingly, the LOO• scavenging ability of the parsley extract was evaluated. As can be seen from Fig. 6, the extract was capable of scavenging LOO•, though BHT and Trolox were significantly more potent. Despite this, the data suggests that the extract may afford protection to susceptible components by interfering with LOO•-mediated free radical reactions via a HAT-type mechanism.
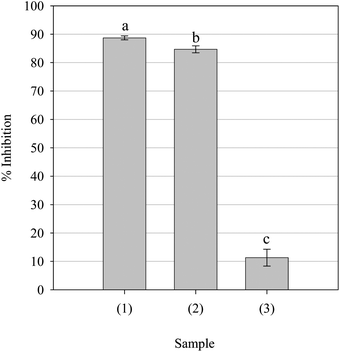 |
| | Fig. 6 Alkylperoxyl free radical scavenging activity of the parsley extract and positive controls BHT and Trolox. Key: (1) BHT (0.5 mg ml−1), (2) Trolox (0.5 mg ml−1) and (3) parsley extract (1.0 mg ml−1). Data are presented as mean values ± SD (n = 5). Bars with the same lowercase letters (a–c) are not significantly (p > 0.05) different. | |
O2•− free radical scavenging
Superoxide anion free radicals are generated during normal cellular metabolism; however, sustained over expression can occur during malignancy.43,44 Although O2•− is not particularly reactive per se, its toxicology is related to its generation of far more toxic species, viz., peroxynitrite (ONOO−) and LOO•/•OH free radicals. Accordingly, the O2•− scavenging ability of the parsley extract was evaluated. As can be seen from Fig. 7, the extract was as capable of scavenging O2•− with potency statistically indistinguishable from that of ascorbic acid but not equivalent to that demonstrated by gallic acid. As phenolic antioxidants are known to inhibit xanthine oxidase,45 the scavenging data was confirmed by the presence of uric acid, a by-product of xanthine oxidase-dependent O2•− generation. Thus the extract was able to scavenging O2•− at physiological pH and may be able to affect the creation of more reactive species and reduce their possible interaction with susceptible cellular components.
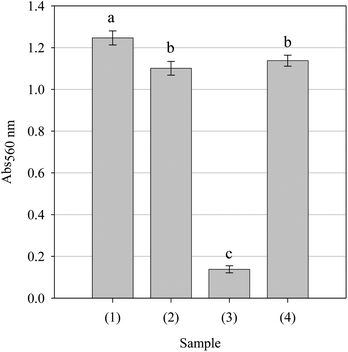 |
| | Fig. 7 Superoxide anion free radical scavenging activity of the parsley extract and positive controls ascorbic acid and gallic acid. All samples were assessed at a concentration of 5.0 mg/ml. Key: (1) Hypoxanthine + xanthine oxidase, (2) ascorbic acid, (3) gallic acid and (4) parsley extract. Data are presented as mean values ± SD (n = 4). Bars with the same lowercase letters (a–c) are not significantly (p > 0.05) different. | |
NO• free radical scavenging
NO• is a regulatory molecule in vivo,46 however, sustained or excessive production is thought to influence the pathogenesis of several diseases, viz., atherosclerosis, cancer and arthritis. As with O2•−, NO• is not a particularly reactive species per se but the powerful oxidant and nitrating agent ONOO- is formed when it reacts with O2•−.47 Accordingly, the NO• scavenging ability of the parsley extract was evaluated. As can be seen from the Fig. 8, the extract was capable of scavenging NO• and did so in a concentration-dependent fashion (data not shown); however, it was not as potent as either Pycnogenol or Trolox, which were statistically indistinguishable in potency.
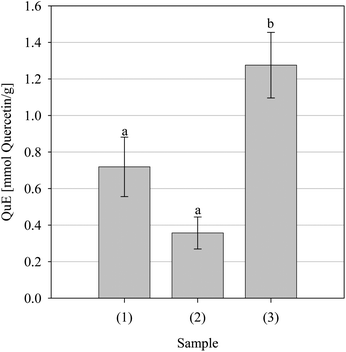 |
| | Fig. 8 Nitric oxide free radical scavenging activity of the positive controls Pycnogenol (1) and Trolox (2) and the parsley extract (3). Data are presented as mean values ± SD (n = 4). Bars with the same lowercase letters (a–b) are not significantly (p > 0.05) different. | |
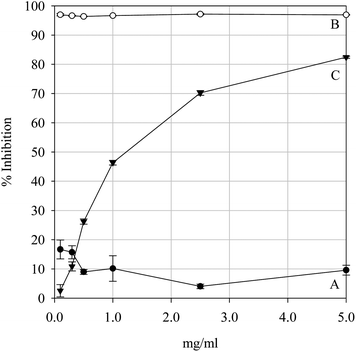 |
| | Fig. 10 Non-site-specific hydroxyl-mediated 2-deoxy-D-ribose degradation inhibition by the positive controls Pycnogenol (A) and Trolox (B) and the parsley extract (C). Data are presented as mean values ± SD (n = 4). | |
In the site specific assay, •OH is generated on the surface of the carbohydrate. The hypothesis that a test sample can intercept •OH before fragmentation occurs is not very plausible considering the reactivity of this oxygen species. A more credible premise would be that fragmentation is prevented by inhibiting •OH formation of via iron chelation. As can be seen from Fig. 11, the extract had no significant effect upon the degradation of 2-deoxy-D-ribose between 0.1–2.5 mg ml−1 concentration levels but did have a significant effect at 5.0 mg ml−1 – a profile mirrored by Pycnogenol – whilst Trolox had a significant effect upon fragmentation at 0.1 mg/ml. The extract was not capable of preventing the oxidative degradation of 2-deoxy-D-ribose by significantly interrupting the generation of •OH except at the highest concentration tested, thus it is unlikely that the extract would be able to afford protection to carbohydrates from oxidative degradation by Fenton chemistry-type reactions at realistic concentrations.
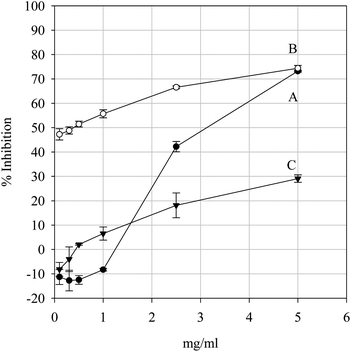 |
| | Fig. 11 Site-specific hydroxyl-mediated 2-deoxy-D-ribose degradation inhibition by the positive controls Pycnogenol (A) and Trolox (B) and the parsley extract (C). Data are presented as mean values ± SD (n = 4). | |
| Reaction mixture |
DNA damage (A532 nm) |
|
Data are presented as mean values ± SD (n = 5). Ph: 1,10-phenanthroline; Cu2+: cupric ions. See text for experimental details.
|
| DNA |
0.012 ± 0.005 |
| DNA (Ph/Cu2+) |
0.016 ± 0.005 |
| DNA (Ph/Cu2+) + Ascorbic acid (mM) |
| 0.10 |
0.083 ± 0.004 |
| 0.25 |
0.173 ± 0.008 |
| 0.50 |
0.272 ± 0.016 |
| 1.00 |
0.405 ± 0.008 |
| 5.00 |
0.557 ± 0.051 |
| DNA (Ph/Cu2+) + Extract (mg ml−1) |
| 0.10 |
0.016 ± 0.009 |
| 0.25 |
0.018 ± 0.004 |
| 0.50 |
0.025 ± 0.005 |
| 1.00 |
0.030 ± 0.004 |
| 5.00 |
0.063 ± 0.002 |
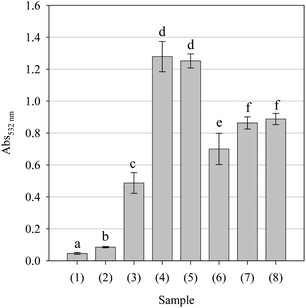
Protein polypeptide backbones and side chains are vulnerable to free radical-mediated fragmentation, resulting in enzyme and/or receptor inactivation and potentially cellular dysfunction and irreversible cell death.34 Accordingly, as natural antioxidants are known to catalyse pro-oxidative damage to cellular components such as proteins despite protecting lipids,51 the pro-oxidant potential of the parsley extract against bovine serum album was evaluated. As can be seen in Fig. 12, as expected, AAPH caused a significant increase in carbonyl formation when compared with the protein sample without AAPH. Ascorbic acid had no significant effect upon protein oxidation whilst gallic acid had a minor, though significant, pro-oxidative effect. Pycnogenol not only had a significant pro-oxidant effect but was significantly better at inducing protein carbonylation than the azo-polymerising agent AAPH. The extract demonstrated pro-oxidative activity, being more pro-oxidant than gallic acid but less oxidative than Pycnogenol. There appeared to be no difference between the extract and AAPH at inducing carbonylation of the bovine serum albumin substrate. Thus, it is likely that the extract may indeed cause damage to enzymes or receptors; however, whether the extract would cause damage in vivo cannot be determined with any authority from this data.
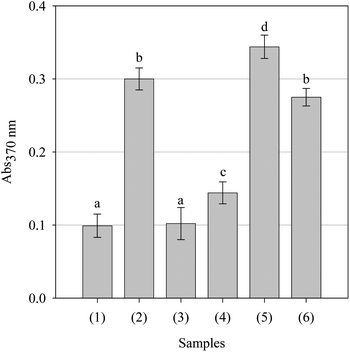 |
| | Fig. 12 Effect of the positive controls and parsley extract upon alkylperoxyl free radical-mediated protein (bovine serum albumin) degradation. All samples were assessed at a concentration of 1.0 mg/ml. Key: (1) Protein, (2) protein + AAPH, (3) ascorbic acid, (4) gallic acid, (5) Pycnogenol and (6) parsley extract. Data are presented as mean values ± SD (n = 5). Bars with the same lowercase letters (a–d) are not significantly (p > 0.05) different. | |
Conclusion
Parsley is extensively cultivated for its organoleptic properties, however, despite its dietary significance, scientific literature regarding its antioxidant properties are scant – especially considering the volume of work available regarding other, arguably, less important herbs – whilst cytotoxic-related reports are rarer still. To address this and because of the consensus regarding the potential beneficial effects of natural products with antioxidant and apoptotic properties we decided to screen a defatted acidified hydroalcoholic extract for antioxidant, pro-oxidant and cytotoxic properties. We found that the parsley extract demonstrated activity in both physiologically and non-physiologically-relevant antioxidant models and that the extract demonstrated pro-oxidant tendencies in a protein-based model but not in a DNA-based model. Our earlier studies with the water-soluble parsley extract showed no cytotoxic effects in cancerous neuroblastoma cells.16 The acidified aqueous methanol parsley extract used in this study had slight cytotoxic effects in non-cancerous and cancerous cells but only after the treatment with highest concentrations, i.e., 1.5 and 2.0 mg/ml. The toxicity might be explained by the pro-oxidative activity of components within the extract against proteins and/or DNA shown in our study but it is not related to caspase 3-dependent apoptosis within cells. It should be remembered, however, when interpreting the cell culture data that the observed cyctotoxicity may also be explained via the generation of H2O2 from components within the media reacting with the phenolic components within the extract and not through a direct effect of the extract components themselves on the intracellular components.52 Further works are required to determine whether this phenomenon occurred and to elucidate the underpinning mechanism(s) responsible of the observed cytotoxicity. Nevertheless, these results suggest that parsley may have a potential role as an adjunct in a pharmaceutical approach to tackle certain diseases, viz. cancer, by tipping the oxidative imbalance in favour of cell death–an idea gaining momentum in the chemotherapeutic treatment of human cancers. However, to make such a claim with any degree of authority, further investigations should be carried out. Nonetheless, the in vitro data at least supports the possibility of the concept though it does not confirm it.
References
- T. Daly, M. A. Jiwan, N. M. O'Brien and S. A. Aherne, Plant Foods Hum. Nutr., 2010, 65, 164–169 Search PubMed.
- R. C. Beier, G. W. Ivie and E. H. Oertli, Phytochem., 1994, 36, 869–872 CrossRef CAS.
- T. Al-Howiriny, M. Al-Sohaibani, K. El-Tahir and S. Rafatullah, Am. J. Chin. Med., 2003, 31, 699–711 Search PubMed.
-
C. A. Newall, L. A. Anderson, J. D. Phillipson, 1996. Herbal medicines, in A Guide for Health-care Professionals. Pharmaceutical Press, London, pp. 203–204 Search PubMed.
- G. Sener, Ö. Saçan, R. Yanardag and G. Ayanoglu-Dülger, Plant Foods Hum. Nutr., 2003, 58, 1–7 CrossRef.
- T. Ojala, S. Remes, P. Haansuu, H. Vuorela, R. Hiltunen, K. Haahtela and P. Vuorela, J. Ethnopharmacol., 2000, 73, 299–305 Search PubMed.
- K. E. de Campos, A. P. C. Balbi and M. J. Q. D. Alves, Rev. Bras. Farmacogn., 2009, 19, 41–45 Search PubMed.
- I. Hinneburg, H. J. D. Dorman and R. Hiltunen, Food Chem., 2006, 97, 122–129 Search PubMed.
- G. Q. Zheng, P. M. Kenney, J. Zhang and L. K. Lam, Carcinogenesis, 1992, 13, 1921–1923 Search PubMed.
- H. Ahmad, V. Valdivia, A. Cadena, E. Martinez, B. Robles, R. Patel, A. Zapata, A. Mancha, W. S. Lai, R. Gelman and M. Amro, Acta Hort., 2009, 841, 47–54 Search PubMed.
- D. Gadi, M. Bnouham, M. Aziz, A. Ziyyat, A. Legssyer, C. Legrand, F. F. Lafeve and H. Mekhfi, J. Ethnopharmacol., 2009, 125, 170–174 Search PubMed.
- S. J. Duthie, A. R. Collins, G. G. Duthie and V. L. Dodson, Mutat. Res., Genet. Toxicol. Environ. Mutagen., 1997, 393, 223–231 Search PubMed.
- G. Agullo, L. Gamet-Payrastre, S. Manenti, C. Viala, C. Rémésy, H. Chap and B. Payrastre, Biochem. Pharmacol., 1997, 53, 1649–1657 CrossRef CAS.
- W.-Y. Chen, Y.-A. Hsieh, C. I. Tsai, Y.-F. Kang, F.-R. Chang, Y.-C. Wu and C.-C. Wu, Invest. New Drugs, 2010 DOI:10.1007/s10637-010-9497-0 , in press.
- S.-H. Kook, Y.-O. Son, S.-W. Chung, S.-A. Lee, J.-G. Kim, Y.-M. Jeon and J.-C. Lee, Apoptosis, 2007, 12, 1289–1298 Search PubMed.
- T. A. Lantto, M. Colucci, V. Závadová, R. Hiltunen and A. Raasmaja, Food Chem., 2009, 117, 405–411 Search PubMed.
- M. S. Ahmad, F. Fazal, A. Rahman, S. M. Hadi and J. H. Parish, Carcinogenesis, 1992, 13, 605–608 CrossRef.
- L. V. Jorgensen, C. Cornett, U. Justesen, L. H. Skibsted and L. O. Dragsted, Free Radical Res., 1998, 29, 339–350 Search PubMed.
- D. Metodiewa, A. K. Jaiswal, N. Cenas, E. Dickancaite and J. Segura-Aguilar, Free Radical Biol. Med., 1999, 26, 107–116 CrossRef CAS.
- V. L. Singleton, R. Orthofer and R. M. Lamuela-Raventós, Methods Enzymol., 1999, 299, 152–178.
- B. Ossola, T. M. Kääriäinen, A. Raasmaja and P. T. Männistö, Toxicology, 2008, 250, 1–8 Search PubMed.
- Š. Maňáková, A. Singh, T. Kääriäinen, H. Taari, S. K. Kulkarni and P. T. Männistö, Brain Res., 2005, 1038, 83–91 Search PubMed.
- M. Oyaizu, Jap. J. Nutr., 1986, 44, 307–315 Search PubMed.
- P. Carter, Anal. Biochem., 1971, 40, 450–458 CAS.
- M. A. Gyamfi, M. Yonamine and Y. Aniya, Gen. Pharmacol., 1999, 32, 661–667 Search PubMed.
- R. Re, N. Pellegrinni, A. Proteggente, A. Pannala, M. Yang and C. Rice-Evans, Free Radical Biol. Med., 1999, 26, 1231–1237 CrossRef CAS.
- I. I. Koleva, J. P. H. Linssen, T. A. van Beek, L. N. Evstatieva, V. Kortenska and N. Handjieva, J. Sci. Food Agric., 2003, 83, 809–819 Search PubMed.
- J.-C. Lee, H.-R. Kim, J. Kim and Y.-S. Jang, J. Agric. Food Chem., 2002, 50, 6490–6496 Search PubMed.
- O. I. Aruoma, J. Spencer, D. Warren, P. Jenner, J. Butler and B. Halliwell, Food Chem., 1997, 60, 149–156 CrossRef CAS.
- B. Halliwell, J. M. C. Gutteridge and O. I. Aruoma, Anal. Biochem., 1987, 165, 215–219 CrossRef CAS.
- O. I. Aruoma, P. J. Evans, H. Kaur, L. Sutcliffe and B. Halliwell, Free Radical Res., 1990, 10, 143–157 Search PubMed.
- K. Nakagawa, M. Kaku, T. Abukawa, K. Aratani, M. Yamaguchi and S. Uesato, J. Health Sci., 2007, 53, 591–595 Search PubMed.
- R. L. Levine, K. Garland, C. N. Oliver, A. Amici, I. Climent, A. G. Lenz, B. W. Ahn, S. Shaltiel and E. R. Stadtman, Methods Enzymol., 1990, 186, 464–478 CAS.
- E. Jaszewska, A. Kośmider, A. K. Kiss and M. Naruszewicz, J. Agric. Food Chem., 2009, 57, 8282–8289 Search PubMed; Y.-Q. Tang, I. B. Jaganath and S. D. Sekaran, PLoS One, 2010, 5, e12544 Search PubMed.
- D. Huang, B. Ou and R. Prior, J. Agric. Food Chem., 2005, 53, 1841–1856 CrossRef CAS.
-
M. H. Gordon, in ‘Food Antioxidants’, Ed. B. J. F. Hudson, Elsevier Applied Science, London, 1990, p. 1–18 Search PubMed.
- R. C. Hider, Z. D. Liu and H. H. Khodr, Methods Enzymol., 2001, 335, 190–203 CAS.
- A. Depkevicius, T. A. van Beek, G. P. Lelyveld, A. van Veldhuizen, A. de Groot, J. P. H. Linssen and R. Venskutonis, J. Nat. Prod., 2002, 65, 892–896 Search PubMed.
- M. B. Arnao, Trends Food Sci. Technol., 2000, 11, 419–421 Search PubMed.
- G. Spiteller, Free Radical Biol. Med., 2006, 41, 362–387 CrossRef CAS.
- B. A. Freeman and J. D. Crapo, J. Biol. Chem., 1981, 256, 10986–10992 Search PubMed.
- S. J. Weiss and A. F. LoBuglio, Ann. Rev. Pharm. Toxicol., 1982, 23, 239–257 Search PubMed.
- N. S. Ahmed, M. Farman, M. H. Najmi, K. B. Mian and A. Hasan, J. Basic Appl. Sci., 2006, 2, 1–6 Search PubMed.
- J.-N. Sheu, T.-H. Lin, C.-K. Lii, C.-C. Chen, H.-W. Chen and K.-L. Liu, Food Chem. Toxicol., 2006, 44, 409–416 Search PubMed.
- R. Radi, J. S. Beckman, K. M. Bush and B. A. Freeman, Arch. Biochem. Biophys., 1991, 288, 481–487 CrossRef CAS.
- B. Halliwell and J. M. C. Gutteridge, Methods Enzymol., 1990, 186, 1–85 CAS.
- S. N. Chatterjee and S. Agarwal, Free Radical Biol. Med., 1988, 4, 51–72 CrossRef CAS.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, J. Am. Soc. Exp. Biol., 2003, 17, 1195–1214 Search PubMed.
- M. J. Laughton, B. Halliwell, P. J. Evans and J. R. S. Hoult, Biochem. Pharmacol., 1989, 38, 2859–2865 CrossRef CAS.
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, J. Am. Soc. Exp. Biol., 2003, 17, 1195–1214 Search PubMed.
- M. J. Laughton, B. Halliwell, P. J. Evans and J. R. S. Hoult, Biochem. Pharmacol., 1989, 38, 2859–2865 CrossRef CAS.
- B. Halliwell, FEBS Lett., 2003, 540, 3–6 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2011 |
Click here to see how this site uses Cookies. View our privacy policy here.