Role of the COX-independent pathways in the ulcer-healing action of epigallocatechin gallate
Biplab
Adhikary†
a,
Sudhir K.
Yadav†
a,
Sandip K.
Bandyopadhyay
a and
Subrata
Chattopadhyay
*b
aIPGME & R, 244B, Acharya Jagadish Chandra Bose Road, Kolkata, 700 020, India
bBio-Organic Division, Bhabha Atomic Research Centre, Mumbai, 400 085, India. E-mail: schatt@barc.gov.in; Fax: +91-22-25505151; Tel: +91-22-25593703
First published on 27th May 2011
Abstract
The modulation of the cyclooxygenase-independent pathway by the green tea-derived polyphenol, epigallocatechin gallate (EGCG) during its healing action against indomethacin (IND)-induced stomach ulceration in mice was investigated. On the 3rd day of its administration, IND (18 mg kg−1) induced maximum stomach ulceration which was associated with increased myeloperoxidase (MPO) activity (2.1-fold, p < 0.001), and inducible nitric oxide synthase (iNOS) expression (2.5-fold, p < 0.001), along with augmented levels of serum nitrite (1.3-fold, p < 0.001), selectins and cell adhesion molecules (CAMs), as well as reduced endothelial nitric oxide synthase (eNOS) expression (53%, p < 0.001). Treatment with EGCG (2 mg kg−1) and omeprazole (3 mg kg−1) for 3 days reversed these parameters, and provided excellent (76–77%) ulcer healing.
1. Introduction
The nonsteroidal anti-inflammatory drugs (NSAIDs) are one of the most widely prescribed drugs in the world, and are extensively used to alleviate clinical cases of pain and inflammation,1 as well as prevention and treatment of ischemic heart disease2 and as antineoplastic agents.3 However, these drugs are well-known to induce stomach ulceration, and delay ulcer healing via inhibition of the cyclooxygenases (COXs).4–6 Hence, various synthetic anti-ulcer drugs are prescribed as adjuvants with the NSAID-therapy. However, efficient management of the NSAIDs-induced gastric ulceration remains a challenging medical problem, due to the various side effects and high cost of the existing drugs, and ulcer recurrence.7 Accumulated evidence suggests that, besides inhibition of prostaglandin (PG) synthesis,8 other factors such as oxidative stress,9 neutrophils infiltration,10 leukocyte-endothelium interaction,11,12 and cytokine imbalance13 also play key roles in the pathogenesis of the gastric damage induced by the NSAIDs.The activated neutrophils produce pro-oxidative and proinflammatory myeloperoxidase (MPO), which is considered a useful risk marker and diagnostic tool against oxidative stress even under clinical conditions.14,15 Because MPO is mainly produced by neutrophils, MPO activity is also considered as an index for the evaluation of neutrophil infiltration.16 Activated neutrophils produce many enzymes and free radicals including nitric oxide (NO), leading to oxidative burst,17 which inflicts endothelial damage.18,19 The interaction between adhesion molecules, expressed on the endothelial cell surface and their ligands on the leukocyte surface dictates the adhesion of neutrophilic polymorphonuclear leucocytes (PMNs) to endothelium that is a critical early step of NSAID-induced gastropathy.10 The NSAID-induced gastric mucosal injuries are significantly reduced in neutropenic rats, and by immunoneutralisation of CD18, intercellular adhesion molecule 1 (ICAM-1), and P- as well as E-selectins.11,12 The above inter-connected processes are critically governed by the imbalance of the pro- and anti-inflammatory (Th1vs. Th2) cytokines.
The physiologically important NO, produced during arginine catabolism by the nitric oxide synthases (NOSs), plays dual roles in gastric mucosal defense and injury. The low concentration of NO, produced by endothelial NOS (eNOS), one of the constitutive NOS isoforms, helps in wound healing by increasing blood flow20 and angiogenesis21 in the damaged gastric mucosa. However, its enhanced generation by inducible NOS (iNOS) may contribute to the pathogenesis of various gastroduodenal disorders including peptic ulcers.22 Thus, the status of the eNOS vs. iNOS expressions in the gastric tissues is very crucial for maintaining their integrity.
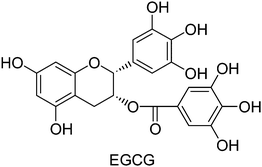
It is now well recognized that dietary adjustments may moderate the risk of gastritis or peptic ulcer. Hence there is a growing interest in dietary factors as supplements to prevent or treat symptoms of gastritis and ulceration. The natural phenolic, (−)-epigallocatechin-3-gallate (EGCG, Fig. 1), abundant in green tea is credited with anticancer, antidiabetic23 and cardioprotective24 activities. Research has indicated it to be a more potent antioxidant than vitamin C and vitamin E, and a promising anti-inflammatory agent.25 EGCG is also reported to control gastrointestinal toxicity induced by Helicobacter pylori,26 and reduce inflammation in spontaneously colitic IL-2-deficient mice.27
Very recently, we have found that oral administration of EGCG (2 mg kg−1) for three days could effectively heal the IND-induced (18 mg kg−1, p. o., single dose) stomach ulceration in mice. The healing activity of EGCG could be partly attributed to its antioxidant action as well as the ability to augment the COX isozymes that improved PG synthesis.28 Given the importance of the intrinsic COX-independent pathways in the IND-mediated gastric inflammation, presently we examined the inhibitory effect of EGCG on neutrophils infiltration and neutrophils-endothelial cells (ECs) interactions to promote healing of gastric ulcer in IND-treated mice. For this, we assessed the gastric mucosal MPO activity and the soluble forms of E- and P-selectins as well as ICAM-1 and vascular cell adhesion molecule 1 (VCAM-1) in the ulcerated and EGCG-treated mice. Because eNOS/iNOS ratio is crucial in ulcer healing,29,30 we also studied expressions of these enzymes in the gastric tissues of the different groups of mice. Further, the status of the pro- and anti-inflammatory cytokines that control the inflammatory response, during wound healing was also investigated.
2. Results
In our previous study, the dose of EGCG for effective ulcer healing was optimized by carrying out the treatment with different doses of EGCG (0.5–5 mg kg−1) up to seven days. Peak ulceration in mice was observed on the 3rd day after IND (18 mg kg−1, single dose) administration, and a three-day treatment with EGCG (2 mg kg−1) and omeprazole (OMEZ) (3 mg kg−1) provided optimal ulcer healing.28 Hence, most of the present experiments were carried out under the above conditions. However, the effective dose of EGCG for ulcer healing was reaffirmed by conducting the MPO assay using different doses of EGCG. This also provided the status of neutrophils infiltration after ulceration and during treatment.Regulation of MPO activity
Compared to the control, the MPO activity in the 3rd day ulcerated mice was significantly higher (2.1-fold). EGCG reduced it dose-dependently (Fig. 2). The effect of EGCG (2 mg kg−1) was significantly better than that of its lower doses, but comparable with that of EGCG (3 and 5 mg kg−1). Overall, treatment with EGCG (2 mg kg−1) and OMEZ (3 mg kg−1) brought down the MPO activity by 43.1% and 28.2%, respectively, compared to the untreated mice. These results corroborated our previous results.28 On its own, EGCG did not show any effect on the MPO level in control mice.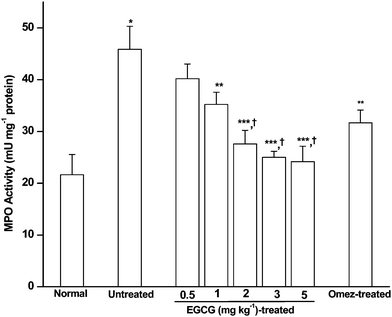 | ||
| Fig. 2 Effect of EGCG in modulating the mucosal MPO level in the IND-induced ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out for 3 days with different doses of EGCG. The supernatant of the gastric tissue homogenate was incubated with TMB and H2O2 in a suitable buffer, and the MPO activity assayed from the absorbance at 450 nm against HRPO as the standard. The values are mean ± S.E.M. of three independent experiments, each with five mice per group. * p < 0.001 compared to control; ** p < 0.05, *** p < 0.01, compared to untreated mice; † p < 0.05, compared to EGCG (0.5 and 1 mg kg−1) treatment. | ||
Compared to the ulcerated mice, those treated with L-NIL and L-NAME showed reduced MPO activity (39.7% and 28.8%, p < 0.05). On the other hand, compared to the EGCG (2 mg kg−1)-treated mice, those receiving EGCG and L-NAME showed significantly increased MPO activity (69.9%, p < 0.001). However, L-NIL did not show any effect.
Quantitative histology
Treatment with EGCG (2 mg kg−1) and OMEZ (3 mg kg−1) reduced the damage score (DS) by 77.6%, and 75.8%, respectively, compared to the ulcerated mice. Likewise, the inflammatory score (IS) was also reduced by EGCG (75%) and OMEZ (60.7%), compared to untreated mice (Table 1).| Group | DS value | DS reduction (%) | IS value | IS reduction (%) |
|---|---|---|---|---|
| a Stomach ulceration in mice was induced by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The assays were carried out 4 h after the last dose of the drugs and the values are mean ± S.E.M. of three independent experiments, each with 5 mice per group. b p < 0.001, compared to ulcerated mice. | ||||
| Untreated | 3.3 | 0 | 2.8 | 0 |
| EGCG (2 mg kg−1) | 0.74 | 77.6b | 0.7 | 75b |
| OMEZ (3 mg kg−1) | 0.81 | 75.8b | 1.1 | 60.7b |
Regulation of the serum nitrite level
In aqueous medium, cellular NO is rapidly converted to nitrite and nitrate. However, their ratio varies substantially depending on the environment. Hence in order to investigate the effect of EGCG on NO production in the ulcerated mice, we assayed the total nitrite concentration, after reducing the nitrate into nitrite. At peak ulceration (day 3), there was a significant increase (1.3-fold; p < 0.001) in the serum nitrite level in IND-treated compared to the control mice. Treatment with EGCG or OMEZ reduced such increase by 49% and 43%, respectively, compared to that in the untreated mice. (Fig. 3).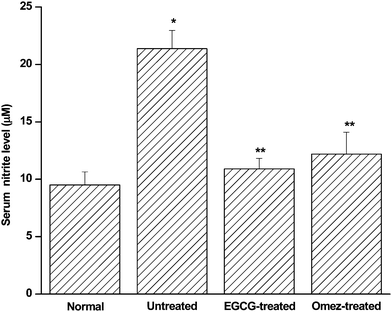 | ||
| Fig. 3 Effect of EGCG and OMEZ in regulating serum nitrite level in the IND-induced ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The NO level was measured using a colorimetric kit. The values are mean ± S.E.M. of three independent experiments, each with five mice per group. * p < 0.001 compared to control; ** p < 0.01 compared to untreated mice. | ||
Regulation of NOS activity
Compared to the control, ulceration increased the serum NOS activity (Fig. 4) 4.6-fold. EGCG and OMEZ suppressed it by 67.8%, and 65.5%, respectively, compared to the untreated mice. The effect of OMEZ was significantly less than that of EGCG.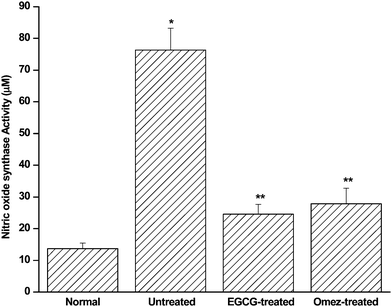 | ||
| Fig. 4 Effect of EGCG and OMEZ in regulating serum NOS activity in the IND-induced ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The NOS activity was measured using a colorimetric kit. The values are mean ± S.E.M. of three independent experiments, each with five mice per group. * p < 0.001 compared to control; ** p < 0.001 compared to untreated mice. | ||
Modulation of the mucosal iNOS and eNOS expressions
The Western blots of the iNOS and eNOS expressions in the gastric mucosa of the normal, ulcerated and drug (EGCG or OMEZ)-treated mice are shown in Fig. 5. The iNOS expression was very high in the ulcerated tissues, but insignificant in normal gastric tissues. Quantification of the bands revealed that stomach ulceration increased the expressions of iNOS (2.5-fold), but reduced the eNOS expression (53%), compared to that in control mice. Treatment with EGCG and OMEZ reversed the changes, the effect of OMEZ being less on the expressions of both iNOS and eNOS.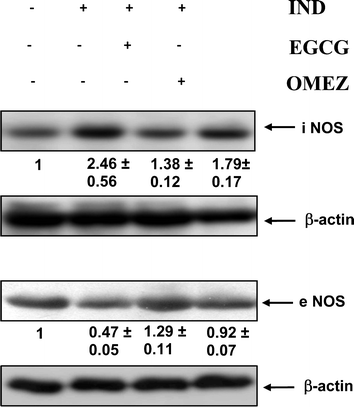 | ||
| Fig. 5 Immunoblots of the iNOS and eNOS expressions in the stomach tissues of normal, ulcerated and treated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The bands were quantified relative to that of β-actin bands of the corresponding lanes, using a Kodak Gelquant software. The individual bands of the ulcerated control and treatment groups were subsequently normalized, considering that of control as 1. The values (arbitary unit, mean ± S.E.M.) are the density scanning results of three independent experiments. | ||
Modulation of soluble E-selectin (sE-selectin) and P-selectin (sP-selectin) levels
On the 3rd day after IND administration, there was an increase (1.6-fold) in the serum sE-selectin level, compared to the normal value. Treatment with EGCG and OMEZ reduced it by 28.1% and 25.8% respectively, compared to the ulcerated mice. Ulceration also increased (2.5-fold) the serum sP-selectin level, compared to that in the control mice. Treatment with EGCG and OMEZ reduced it by 30% and 27.5% respectively compared to that in the ulcerated mice. The effect of OMEZ on both the parameters was significantly less than that of EGCG. The results are presented in Fig. 6.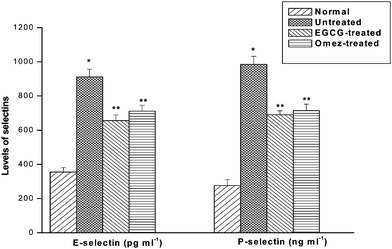 | ||
| Fig. 6 Effect of EGCG and OMEZ in regulation of serum sE- and sP-selectins in the IND-induced ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The serum sE- and sP-selectins were measured by ELISA. The values are mean ± S.E.M. of three independent experiments, each, with five mice per group. * p < 0.001 compared to control; ** p < 0.01 compared to ulcerated mice. | ||
Modulation of soluble ICAM-1 (sICAM-1) and VCAM-1 (sVCAM-1) levels
The levels of sICAM-1 and sVCAM-1 in the ulcerated mice were augmented by 2.6-fold and 65.4%, respectively, compared to the control group. Treatment with EGCG and OMEZ reduced sICAM-1 level by 34.2% and 32.5% respectively. Likewise, EGCG and OMEZ reduced the sVCAM-1 level by 24.1% and 13.4%, respectively, compared to the ulcerated mice. The effect of OMEZ on sVCAM-1 level was significantly less than that of EGCG. The combined results are shown in Fig. 7.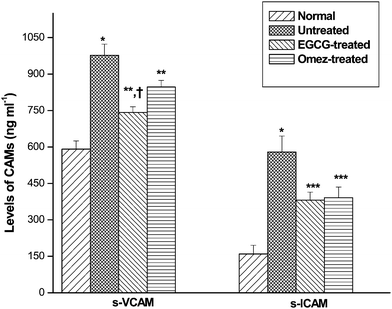 | ||
| Fig. 7 Abilities of EGCG and OMEZ in regulating serum sICAM-1 and sVCAM-1 in the IND-induced ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The serum sICAM-1 and sVCAM-1 levels were measured by ELISA. The values are mean ± S.E.M. of three independent experiments, each with five mice per group. * p < 0.001 compared to control; ** p < 0.05, *** p < 0.01 compared to ulcerated mice; † P < 0.05 compared to OMEZ treatment. | ||
Regulation of the serum Th1 (TNF-α and IL-6) and Th2 (IL-4 and IL-10) cytokines
Compared to the normal value, ulceration markedly increased the serum TNF-α and IL-6 levels by 1.7-fold and 2.5-fold, respectively. EGCG suppressed the TNF-α level by 44.3%, while OMEZ reduced it by 33.3%, compared to that in the untreated mice (Fig. 8). Likewise, EGCG and OMEZ suppressed the serum IL-6 level by 48.1% and 27.9% respectively, compared to that in the untreated mice. The effect of EGCG on both the cytokines was significantly better than that of OMEZ.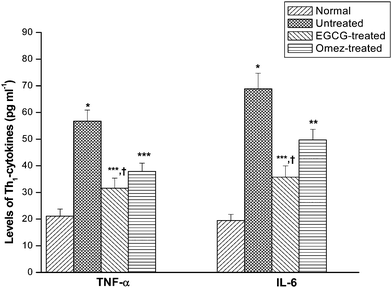 | ||
| Fig. 8 Modulation of the serum levels of the Th1 cytokines (TNF-α and IL-6) by EGCG and OMEZ in the ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The serum cytokine levels were assayed by ELISA. The values are mean ± S.E.M. of three independent experiments, each with 5 mice per group. * p < 0.001 compared to control; ** p < 0.05, *** p < 0.01 compared to ulcerated mice; † p < 0.01 compared to OMEZ treatment. | ||
The serum IL-10 and IL-4 levels in the ulcerated mice were reduced by 34.3% and 32.8% respectively, compared to the control group. Treatment with EGCG and OMEZ increased the serum IL-10 level by 33.7% and 29.4%, respectively, compared to the untreated group. Likewise, EGCG and OMEZ increased the serum IL-4 level by 20.8% and 18.6% respectively, compared to the untreated group. The results are shown in Fig. 9.
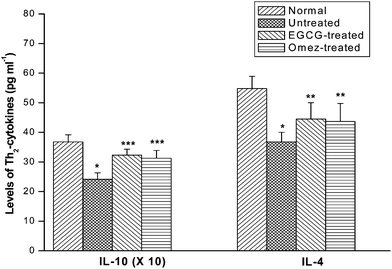 | ||
| Fig. 9 Modulation of the serum levels of the Th2 cytokines (IL-4 and IL-10) by EGCG and OMEZ in the ulcerated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The serum cytokine levels were assayed by ELISA. The values are mean ± S.E.M. of three independent experiments, each with 5 mice per group. * p < 0.01 compared to control; ** p < 0.05, *** p < 0.01 compared to ulcerated mice. | ||
Modulation of mucosal TNF-α expressions
The Western blots of TNF-α expressions in the gastric mucosa of the normal, ulcerated and drug (EGCG or OMEZ)-treated mice are shown in Fig. 10. IND administration increased (4.3-fold, p < 0.001) the mucosal TNF-α, compared to that in the control group. Treatment with EGCG and OMEZ reduced such increase by 54% (p < 0.01) and 26% (p < 0.05), respectively. The effect of EGCG was significantly better than that of OMEZ (p < 0.01). | ||
| Fig. 10 Immunoblots of TNF-α expressions in the stomach tissues of normal, ulcerated and treated mice. The mice were ulcerated by IND (18 mg kg−1, p. o.). Treatment was carried out with EGCG (2 mg kg−1, p. o.) and OMEZ (3 mg kg−1, p. o.) for 3 days. The bands were quantified relative to that of β-actin bands of the corresponding lanes, using a Kodak Gelquant software. The individual bands of the ulcerated control and treatment groups were subsequently normalized, considering that of control as 1. The values (arbitary unit, mean ± S.E.M.) are the density scanning results of three independent experiments. | ||
3. Discussion
Several factors such as enzymes, cytokines, and soluble mediators, liberated due to the inflammatory response play crucial roles in the IND-mediated gastric ulceration and the delayed ulcer healing.31 Thus, controlling these factors provides an opportunity to develop improved anti-ulcer medications. However, this aspect is often ignored while reporting gastric ulcer-healing activity of the test compounds. The impressive healing capacity of EGCG (IC50 = 2.94 ± 0.18 mg kg−1) against the IND-induced gastric ulceration in mice28 encouraged us to investigate its probable modulatory effect on the COX-independent pro-inflammatory parameters, because these are additional key contributors in wound healing.32 Oxidative stress and inflammation adversely effect endothelial function and gastrointestinal anomalies.There is compelling evidence that neutrophils infiltration plays key role in the initiation and progression of injury and inflammation in a variety of tissues including gastric mucosal damage by IND.33 Besides, infiltration of neutrophils also delays gastric ulcer healing.34 Reduction of neutrophils infiltration and inhibition of TNF-α expression was shown to accelerate ulcer healing.35 The oxygen free radicals produced by the activated neutrophils, delay gastric ulcer healing in rats.36 Furthermore, neutrophils infiltration induces microcirculatory abnormalities37 and its suppression promotes healing.38 Another important factor for ulcer healing is the gastric mucosal NO that shows biphasic behaviour, depending on its mode of generation and concentration. Therefore, all these parameters were the foci of the present study.
The activated neutrophils produce MPO, which possesses powerful pro-oxidative and proinflammatory properties. Hence, it is considered a useful risk marker and diagnostic tool for severity of gastric ulcer. MPO activity is also considered as an index for the evaluation of neutrophils infiltration.19 A positive correlation between MPO activity and neutrophils infiltration in intestinal inflammation models has been reported.39 Earlier, we have established the healing action of EGCG by histology. Quantification of the histological slides in terms of damage score and inflammatory score provided a better assessment of the quality of healing. In the present study, IND administration led to mucosal damage and augmented the MPO activity in the ulcerated area of the gastric wall, suggesting the involvement of neutrophils infiltration in gastric ulceration. Treatment with EGCG could sufficiently restore the normal gastric mucosal integrity, while reducing the MPO activity. These results suggested a close relationship between the MPO activity and state of the gastric inflammation as well as ulcer healing.
Neutrophils inflict endothelial damage by generating various free radicals including NO that profoundly influences oxidative burst.16 It is now well-recognized that the enhanced generation of NO by the iNOS may contribute to the pathogenesis of various gastroduodenal disorders including peptic ulcer.22 High concentrations of NO may be detrimental by promoting inflammation via mucosal swelling and epithelial damage.30 An increase in iNOS activity and a decrease in eNOS activity in the gastric mucosa are closely related to the development of gastric mucosal lesions. Currently we confirmed that the IND-induced gastric ulceration increased the mucosal iNOS expression, while reducing the expression of eNOS in mice. The elevated expressions of iNOS accounted for the increased total NOS activity as well as serum nitrite level due to ulceration. The augmented neutrophil counts due to IND treatment would generate more iNOS-derived NO and superoxide radicals enhancing oxidative mucosal damage. The enhanced levels of NO add to the free radical load by generating secondary oxidizing and nitrating species. This would affect their growth, differentiation, and apoptosis, delaying ulcer healing. Consistent with this, our results also showed that both iNOS-specific inhibitor, L-NIL and the non-selective NOS inhibitor, L-NAME reduced the MPO activity of the ulcerated mice. However, the protective effect of L-NIL was more than that of L-NAME. This suggested that the iNOS-mediated NO was primarily responsible for IND-induced gastric ulceration. The activated neutrophils, observed in the ulcerated mice would also scavenge the beneficial vasodilatory NO through NADPH oxidase or MPO catalyzed reactions.41 Piotrowski et al. showed a 12-fold increase in gastric epithelial expression of iNOS activity in the IND-administered animals, compared to controls, and the increase correlated positively with the epithelium damage.40 Our results showed only 1.5-fold increased iNOS expression after ulceration. This may possibly be due to the fact that we assayed it on the 3rd day of ulceration.
Treatment with EGCG raised the eNOS/iNOS ratio to a level favorable for efficient ulcer-healing. The reduction of the total NOS activity and nitrite level by EGCG was primarily due to suppression of the iNOS expression. Earlier, using eNOS deficient mice, the importance of eNOS and eNOS-derived NO in regulating microvascular structure during acute inflammation has been demonstrated.29 Our results revealed that L-NAME, but not L-NIL adversely affected the healing activity of EGCG. Because, EGCG itself reduced the iNOS expression, L-NIL did not show any effect on its healing activity. However, L-NAME would negate the positive influence of EGCG on the eNOS expression, reducing the healing by EGCG. Taken together, these results suggested that the eNOS-derived NO contributed maximum to the ulcer healing property of EGCG, although a role for neuronal NOS-derived NO cannot be excluded. Overall, the ability of EGCG to reduce the neutrophils infiltration and enhance the eNOS expression at the gastric tissues might be instrumental in its ulcer-healing action.
Leukocyte capture by and rolling on ECs, followed by more firm adhesion to the endothelium is crucial at various stages of acute inflammation. Neutrophil transendothelial migration is staged into a series of events: initial rolling (primarily selectin mediated), activation (primarily chemoattractant dependent), firm adhesion (integrin mediated), and transmigration (utilising integrins and immunoglobulin superfamily molecules, for example, ICAM-1, VCAM-1 etc.). It has been already reported that soluble forms of adhesion molecules are good markers of an inflammatory disease.42 The circulating form of E-selectin (sE-selectin) is released by enzymatic cleavage or by shedding of damaged or activated ECs, and its concentration often correlates well with its expression on the surface of ECs. Likewise, sICAM-1 concentration is considered to reflect the expression of mICAM-1 on ECs. Therefore, plasma concentrations of these soluble inflammatory mediators might be a marker of endothelial cell damage or activation, and was assayed in the present investigation.
Our results are indicative of prominent role of sE-selectin at the early stages of tethering and recruitment of leukocytes in gastric injury by IND. The elevated levels of sP-selectin, sICAM-1 and sVCAM-1 help to maintain the high MPO activity in gastric mucosa as compared to the normal group, since ICAM-1 binds to CD11a/CD18 and CD11b/CD18 on leukocytes, and VCAM-1 binds to the α4β1 integrin, located at lymphocytes, monocytes and basophils.13 Because P-selectin and E-selectin are critical adhesion molecules for the capture and rolling of leukocytes in the microvasculature while ICAM-1 mediates firm adhesion, the up-regulation of these CAMs by indomehacin may result in more efficient leukocyte emigration into the surrounding tissues. Treatment with all the test samples reduced the levels of soluble selectins and CAMs. These results demonstrate that EGCG may interrupt the interaction of neutrophils and ECs both at the early rolling phase mediated by the selectins and at the late firm adhesion phase mediated by the CAMs. The effect was more prominent on sE-selectin, suggesting that the test samples primarily prevented the early loose tethering of leukocytes. The relative potency of the test samples was EGCG > OMEZ. Interestingly, we found that IND raised sE-selectin more than sP-selectin, and EGCG showed slightly better ability to reduce the former than the latter. This differs from the earlier observation where P-selectin was reported to have a more prominent role in the IND-mediated ulceration.11,12 Collectively, our results suggested that the ulcer-healing effect EGCG may be due to its anti-inflammatory property coupled with its ability to generate the beneficial eNOS-derived NO that helps in wound healing. This is consistent with several previous reports where drugs such as bombesin were found to promote ulcer healing by restricting the activity of neutrophil-derived inflammatory processes.43
Stimulation of inflammatory cytokines is extremely important in mucosal defense. One of the most prominent modes of mediation of IND-induced gastropathy is the increased expression of the pro-inflammatory cytokines,44 which also correlates with the extent of ulceration. The activation of ECs for expression of CAMs is mediated in part by inflammatory cytokines, such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ). There is consistent evidence in the literature that during acute and chronic colitis the sustained production of pro-inflammatory cytokines leads to the up-regulation of adhesion molecules. TNF-α promotes leukocyte adhesion by enhancing the transcription-dependent expression of endothelial cell adhesion molecules that can extend and further increase the leukocyte rolling and adherence/emigration responses.13 Enhancement of iNOS expression and the levels of soluble inflammatory modulators by IND reflected a pro-inflammatory trigger. In view of this, the immune response due to ulceration, and its modulation by EGCG and OMEZ was monitored. This enabled us to associate the inflammatory response with a better prognosis.
IND administration raised the serum levels of pro-inflammatory Th1 cytokines (TNF-α and IL-6) and reduced the anti-inflammatory Th2 cytokines (IL-4 and IL-10). We selected TNF-α for this study, as increased plasma level of TNF-α has been reported to increase leukocyte adherence after IND administration.45 Likewise, IL-4 that remains under the influence of NO, controls the expression of growth factors that is responsible for ulcer onset and healing. The cytokine imbalance, created by IND is presumed to trigger up-regulation of adhesion molecules, and augment the iNOS/NO pathway to produce excess NO, which is likely to promote oxidative stress and result in ulceration.46 Treatment with EGCG, however, reversed the imbalance by reducing the Th1 cytokines drastically, and restoring the levels of IL-4 and IL-10 to normalcy. In particular, our results clearly showed that EGCG could reduce the mucosal TNF-α expression as well as the circulating TNF-α level. Suppression of TNF-α would facilitate ulcer healing by enhancing epithelial cell proliferation and gastric blood flow, and decreasing epithelial apoptosis. The upregulation of the anti-inflammatory cytokines by EGCG is likely to inhibit the stimulatory effect of IND on the level of pro-inflammatory cytokines release in blood and gastric mucosa. In the previous paper, we have reported that EGCG augmented the PGE level in the IND-treated mice. The increase PGE might stimulate IL-10 release, which, in turn, controls PGE and Th1 cytokines via a negative feedback.
4. Conclusions
Overall, the data presented in the present study demonstrates that treatment with EGCG could accelerate healing of IND-induced gastric ulceration in mice. This healing can be associated to different mechanisms: (a) reducing leukocyte infiltration by moderating and/or abolishing the expression of various inflammation modulators including adhesion molecules, NO and Th1/Th2 cytokines; and (b) switching the gastric mucosal eNOS/iNOS ratio. These effects, along with EGCG ability to strengthen mucosal defense system by augmenting antioxidant defences, gastric mucin expression, and PGE, as disclosed in our previous studies, might be contributing to the ulcer healing action of EGCG.5. Experimental
5.1 Chemicals and reagents
Indomethacin, EGCG, Bradford reagent, triton X-100, leupeptin, phenylmethylsulfonyl fluoride (PMSF), glycine, sodium dodecyl sulfate (SDS), acrylamide, bis-acrylamide, tween 20, ethylenediaminetetraacetic acid (EDTA), 3,3′,5,5′-tetramethylbenzidine (TMB), omeprazole (OMEZ), Trizma base, cetyltrimethylammonium bromide (CTAB), and nitrocellulose membrane were procured from Sigma Chemicals (St. Louis, MO). Other reagents used were disodium hydrogen phosphate and sodium dihydrogen phosphate (BDH, Pool Dorset, U. K.), sulphuric acid, hydrochloric acid, phosphoric acid, sodium chloride (Thomas Becker, Mumbai, India), horseradish peroxidase (HRPO, Sisco Research Laboratory, Mumbai, India), rabbit polyclonal iNOS and eNOS antibodies (Santacruz Biotechnology, Delaware, USA), TNF-α antibodies (Cell Signaling Technology Inc., Danvers, MA, USA), peroxidase conjugated anti-rabbit IgG antibody, enhanced chemiluminescence detection kit (Roche, Mannheim, Germany), NOS and nitrite assay kits (Calbiochem, California, USA), and cytokine ELISA kits (Pierce Biotechnology, Rockford, USA).5.2 Instrumentation
The absorption spectrophotometry was carried out at 25 °C using an ELISA reader (Biotech Instruments, USA). The bands obtained from the western blots were quantified using the Gelquant software (DNR Bioimaging System, version 2.7.0, Israel).5.3 Preparation of the test samples
The test samples (EGCG and OMEZ) were prepared as aqueous suspensions in 2% gum acacia as the vehicle, and administered to the mice orally.5.4 Animals
Male Swiss albino mice, bred at BARC Laboratory Animal House Facility, Mumbai, India, were procured after obtaining clearance from the BARC Animal Ethics Committee (BAEC). All the experiments were conducted with strict adherence to the ethical guidelines laid down by European Convention for the Protection of Vertebrate Animals used for Experimental and Other Scientific Purposes. In addition, the ethical guidelines, laid down by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), constituted by the Animal Welfare Division, Government of India on the use of animals in scientific research was followed. The mice (6–8 weeks old, 25–30 g) were reared on a balanced laboratory diet as per National Institute of Nutrition, Hyderabad, India and given tap water ad libitum. They were kept at 20 ± 2 °C, 65–70% humidity, and 12 h day/12 h night cycles. The experiments were performed by two investigators blinded to the group and treatment of animals, which were identified by typical notches in the ear and limbs [performed at a pre-weaning stage to minimize the pain to the animals], and then randomized.5.5 Protocol for ulceration and biochemical studies
The mice were divided into several groups (each containing five mice), and each experiment was repeated three times. Except for the normal control, ulceration in the other mice was induced by IND (18 mg kg−1, p. o., single dose), dissolved in distilled water and suspended in 2% gum acacia as the vehicle. For the standardization of doses, EGCG (10–50 mg kg−1, p. o.) was given to the mice once daily up to 3 days, starting the first dose 6 h after the IND-administration. In the subsequent days, the test samples were given at 9 AM on each day. OMEZ (3.0 mg kg−1, p. o.) was used as the positive control. The doses of IND and OMEZ were standardized in our earlier study.47 The normal and ulcerated control groups of mice were given the vehicle (0.2 ml) during the entire period of study. Four hours after the last dose of the test samples, the mice were sacrificed on the 3rd day under anesthesia with thiopental, the stomachs were opened along the greater curvature, thoroughly rinsed with normal saline, and the wet weights of the tissues were recorded. In separate experiments, treatments were also carried out with L-NAME (10 mg kg−1, once daily), and L-NIL (3 mg kg−1, twice daily) alone or in conjunction with EGCG (at its optimized dose) for 3 days.5.6 Ulcer healing assessment
The ulcerated portions of the stomach were sectioned after fixing in 10% formol saline solution. After 24 h of fixation followed by embedding in a paraffin block, it was cut into sections of 5 micron onto a glass slide, stained with haematoxylene-eosin and the histology examined under a light microscope. One centimetre length of each histological section was divided into three fields. The damage score (DS)48 was assessed by scoring each field on a 0–4 scale: 0 – normal mucosa, 1 – epithelial cell damage, 2 – glandular disruption, vasocongestion or edema in the upper mucosa, 3 – mucosal disruption, vasocongestion or edema in the mid-lower mucosa, and 4 – extensive mucosal disruption involving the full thickness of the mucosa. The overall mean value of the damage scores (DS) for each of the fields was taken as the histological ulcer index for that section.Likewise, the inflammatory scores (IS)49 were assigned after reviewing all slides to assess the range of inflammation as follows: 0 – normal mucosa, 1 – minimal inflammatory cells, 2 – moderate number of inflammatory cells, and 3 – large number of inflammatory cells.
Histological sections were coded to eliminate an observer bias. Data for the histological analyses are presented as the mean ± S. E. M. from the review of a minimum of three sections per animal and five animals per group.
5.7 MPO assay
The MPO activity was assayed following a reported method50 with slight modifications. The glandular portions of the stomach tissues were homogenized for 30 s in a 50 mM phosphate buffer (pH 6.0), containing 0.5% CTAB and 10 mM EDTA, followed by freeze thawing three times. The homogenate was centrifuged at 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 20 min at 4 °C. The supernatant was collected, and the protein content determined. The supernatant (50 μl) was added to 80 mM phosphate buffer, pH 5.4 (250 μl), 0.03 M TMB (150 μl) and 0.3 M H2O2 (50 μl). After incubating the mixture at 25 °C for 25 min, the reaction was terminated by adding 0.5 M H2SO4 (2.5 ml). The absorbance of the mixture at 450 nm was recorded using HRPO as the standard. The MPO activity is expressed as mU mg−1 protein.
000 × g for 20 min at 4 °C. The supernatant was collected, and the protein content determined. The supernatant (50 μl) was added to 80 mM phosphate buffer, pH 5.4 (250 μl), 0.03 M TMB (150 μl) and 0.3 M H2O2 (50 μl). After incubating the mixture at 25 °C for 25 min, the reaction was terminated by adding 0.5 M H2SO4 (2.5 ml). The absorbance of the mixture at 450 nm was recorded using HRPO as the standard. The MPO activity is expressed as mU mg−1 protein.
5.8 Total NOS assay
The NOS activity in the serum was measured using a commercially available colorimetric kit following manufacturer's protocol.5.9 Immunoblots of iNOS, eNOS and TNF-α
The glandular part of the stomach tissue was washed with PBS containing protease inhibitors, minced and homogenized in a lysis buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100) containing aprotinin (2 μg ml−1), leupeptin (10 μg ml−1) and PMSF (0.4 μM). Following centrifugation at 15000 × g for 30 min at 4 °C, the supernatant was collected, and the protein concentration measured. The proteins (80 μg) were resolved by 10% SDS-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane. The membrane was blocked for 2 h in TBST buffer (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, 0.02% Tween 20) containing 99% fat-free milk powder and incubated overnight at 4 °C with rabbit polyclonal iNOS, eNOS or TNF-α antibodies. The membrane was washed for 1 h with TBST and incubated with peroxidase conjugated anti-rabbit IgG antibody (1:2500 dilution). The bands were detected using an enhanced chemiluminescence detection kit and quantified.
5.10 Estimation of nitrite
Following manufacturer's instruction, the serum nitrite concentration was measured using a commercially available colorimetric kit that measures the total nitrite concentration of the sample.5.11 Cytokines assays
The TNF-α, IL-6, IL-4 and IL-10 levels in the serum and glandular part of the stomach tissue were estimated using commercially available ELISA kits following manufacturer's protocols.5.12 Assays of soluble levels of selectins and CAMs
The serum levels of P-selectin, E-selectin, VCAM-1 and ICAM-1 were estimated using ELISA kits according to manufacturer's protocol.5.13 Statistical analysis
All the data are presented as mean ± S.E.M. The parametric data, which included all the biochemical parameters, were analyzed using a paired Student's t test for the paired data, or one-way analysis of variance (ANOVA) followed by Dunnet's multiple comparisons post hoc test. Non-parametric data (histology scoring) were analyzed using the Kruskal–Wallis test (non-parametric ANOVA) followed by Dunn's multiple comparison post hoc test.References
- P. Brooks, P. Emery, J. F. Evans, H. Fenner, C. J. Hawkey, C. Patrono, J. Smolen, F. Breedveld, R. Day, M. Dougados, E. W. Ehrich, J. Gijon-Banos, T. K. Kvien, M. H. Van Rijswijk, T. Warner and H. Zeidler, Interpreting the clinical significance of the differential inhibition of COX-1 and COX-2, Rheumatology, 1999, 38, 779–788 Search PubMed.
- P. A. Gladding, M. W. Webster, H. B. Farrell, I. S. Zeng, R. Park and N. Ruijne, The antiplatelet effect of six non-steroidal anti-inflammatory drugs and their pharmacodynamic interaction with aspirin in healthy volunteers, Am. J. Cardiol., 2008, 101, 1060–1063 Search PubMed.
- G. A. Piazza, A. B. Keeton, H. N. Tinsley, J. D. Whitt, B. D. Gary, B. Mathew, R. Singh, W. E. Grizzle and R. C. Reynolds, NSAIDs: Old drugs reveal new anticancer targets, Pharmaceuticals, 2010, 3, 1652–1667 Search PubMed.
- A. F. Barrison and M. M. Wolfe, Management of NSAID-related gastrointestinal mucosal injury, Inflammopharmacology, 1999, 7, 277–286 Search PubMed.
- A. Lanas, M. A. Perez-Aisa, F. Feu and J. Ponce, A nationwide study of mortality associated with hospital admission due to severe gastrointestinal events and those associated with non-steroidal anti-inflammatory drug use, Am. J. Gastroenterol., 2005, 100, 1685–1693 Search PubMed.
- C. Hawkins and G. W. Hanks, The gastroduodenal toxicity of nonsteroidal anti-inflammatory drugs. A review of the literature, J. Pain Symptom Manage., 2000, 20, 140–151 Search PubMed.
- F. K. L. Chan, Primer: managing NSAID-induced ulcer complications—balancing gastrointestinal and cardiovascular risks, Nat. Clin. Pract. Gastroenterol. Hepatol., 2006, 3, 563–573 Search PubMed.
- R. Langenbach, S. G. Morham, H. F. Tiano, C. D. Loftin, B. I. Ghanayem, P. C. Chulada, J. F. Mahler, C. A. Lee, E. H. Goulding, K. D. Kluckman, H. S. Kim and O. Smithies, Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and IND-induced gastric ulceration, Cell, 1995, 83, 483–492 Search PubMed.
- T. Yoshikawa, Y. Naito, A. Kishi, T. Tomii, T. Kaneko, S. Iinuma, H. Ichikawa, M. Yasuda, S. Takahashi and M. Kondo, Role of active oxygen, lipid peroxidation, and antioxidants in the pathogenesis of gastric mucosal injury induced by indomethacin in rats, Gut, 1993, 34, 732–737 Search PubMed.
- L. Osborn, Leukocyte adhesion to endothelium in inflammation, Cell, 1990, 62, 3–6 Search PubMed.
- J. L. Wallace, K. E. Arfors and G. W. McKnight, A monoclonal antibody against the CD18 leukocyte adhesion molecule prevents indomethacin-induced gastric damage in the rabbit, Gastroenterology, 1991, 100, 878–883 Search PubMed.
- J. L. Wallace, W. McKnight, M. Miyasaka, T. Tamatani, J. Paulson, D. C. Anderson, D. N. Granger and P. Kubes, Role of endothelial adhesion molecules in NSAID-induced gastric mucosal injury, Am. J. Physiol., 1993, 265(Gastrointest. Liver Physiol. 28), G993–G998 Search PubMed.
- J. Panés, M. Perry and D. N. Granger, Leukocyte-endothelial cell adhesion: avenues for therapeutic intervention, Br. J. Pharmacol., 1999, 126, 537–550 Search PubMed.
- V. Loria, I. Dato, F. Graziani and L. M. Biasucci, A new biomarker of inflammation in ischemic heart disease and acute coronary syndromes, Mediators Inflammation, 2008, 2008, 135625, DOI:10.1155/2008/135625.
- C. C. Wu, J. S. Chen, W. M. Wu, T. N. Liao, P. Chu, S. H. Lin, C. H. Chuang and Y. F. Lin, Myeloperoxidase serves as a marker of oxidative stress during single haemodialysis session using two different biocompatible dialysis membranes, Nephrol., Dial., Transplant., 2005, 20, 1134–1139 Search PubMed.
- K. Nishida, Y. Ohta, T. Kobayashi and I. Ishiguro, Involvement of the xanthine-xanthine oxidase system and neutrophils in the development of acute gastric mucosal lesions in rats with water immersion restraint stress, Digestion, 1997, 58, 340–351 Search PubMed.
- M. Chatterjee, R. Saluja, S. Kanneganti, S. Chinta and M. Dikshit, Biochemical and molecular evaluation of neutrophil NOS in spontaneously hypertensive rats, Cell Mol. Biol., 2007, 53, 84–92 Search PubMed.
- D. Salvemini, G. de Nucci, R. J. Gryglewski and J. R. Vane, Human neutrophils and mononuclear cells inhibit platelet aggregation by releasing a nitric oxide-like factor, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 6328–6332 CAS.
- B. Kristal, R. Shurtz-Swirski, J. Chezar, J. Manaster, R. Levy, G. Shapiro, I. Weissman, S. M. Shasha and S. Sela, Participation of peripheral polymorphonuclear leukocytes in the oxidative stress and inflammation in patients with essential hypertension, Am. J. Hypertens., 1998, 11, 921–928 Search PubMed.
- B. J. R. Whittle, Nitric oxide in gastrointestinal physiology and pathology, in L. R. Johnson (ed.), The physiology of the gastrointestinal tract, Raven Press, New York, USA, 1994, pp. 267–294 Search PubMed.
- M. Ziche, L. Morbidelli, E. Masini, S. Amerini, H. J. Granger and C. A. Maggi, Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance P, J. Clin. Invest., 1994, 94, 2036–2044 CrossRef CAS.
- M. H. L. P. Souza, H. P. Lemos, R. B. Oliveira and F. Q. Cunha, Gastric damage and granulocyte infiltration induced by indomethacin in tumour necrosis factor receptor 1 (TNF-R1) or inducible nitric oxide synthase (iNOS) deficient mice, Gut, 2004, 53, 791–796 Search PubMed.
- A. L. Brown, J. Lane, J. Coverly, J. Stocks, S. Jackson, A. Stephen, L. Bluck, A. Coward and H. Hendrickx, Effects of dietary supplementation with the green tea polyphenol epigallocatechin-3-gallate on insulin resistance and associated metabolic risk factors: randomized controlled trial, Br. J. Nutr., 2008, 101, 886–894 Search PubMed.
- P. T. Devika and P. S. M. Prince, Protective effect of (−)-epigallocatechin-gallate (EGCG) on lipid peroxide metabolism in isoproterenol induced myocardial infarction in male Wistar rats: A histopathological study, Biomed. Pharmacother., 2008, 62, 701–708 Search PubMed.
- J. B. Calixto, M. M. Campos, M. F. Otuki and A. R. Santos, Anti-inflammatory compounds of plant origin. Part II. Modulation of pro-inflammatory cytokines, chemokines and adhesion molecules, Planta Med., 2004, 70, 93–103 Search PubMed.
- K. M. Lee, M. Yeo, J. S. Choue, J. H. Jin, S. J. Park, J. Y. Cheong, K. J. Lee, J. H. Kim and K. B. Hahm, Protective mechanism of epigallocatechin–3–gallate against Helicobacter pylori–induced gastric epithelial cytotoxicity via the blockage of TLR–4 signaling, Helicobacter, 2004, 9, 632–642 Search PubMed.
- G. W. Varilek, F. Yang, E. Y. Lee, W. J. deVilliers, J. Zhong, H. S. Oz, K. F. Westberry and C. J. McClain, Green tea polyphenol extract attenuates inflammation in interleukin-2-deficient mice, a model of autoimmunity, J. Nutr., 2001, 131, 2034–2039 Search PubMed.
- B. Adhikari, S. K. Yadav, S. K. Bandyopadhyay and S. Chattopadhyay, Epigallocatechin gallate accelerates healing of indomethacin-induced stomach ulceration in mice, Pharmacol. Rep., 2011, 63, 527–536 Search PubMed.
- J. C. Luo, V. Y. Shin, E. S. L. Liu, W. H. L. So, Y. N. Ye, F. Y. Chang and C. H. Cho, Non-ulcerogenic dose of dexamethasone delays gastric ulcer healing in rats, J. Pharmacol. Exp. Ther., 2003, 307, 692–698 Search PubMed.
- H. Meurs, H. Maarsingh and J. Zaagsma, Response to Ricciardolo: The functional significance of arginase in asthma is supported by gene expression, Trends Pharmacol. Sci., 2003, 24, 562–563 Search PubMed.
- S. Fiorucci, E. Antonelli and A. Morelli, Mechanism of non-steroidal anti-inflammatory drug-gastropathy, Dig. Liver Dis., 2001, 31(suppl 2), S35–S43 Search PubMed.
- I. Tegeder, J. Pfeilschifter and G. Geisslinger, Cyclooxygenase-independent actions of cyclooxygenase inhibitors, FASEB J., 2001, 15, 2057–2072 Search PubMed.
- J. L. Wallace, C. M. Keenan and D. N. Granger, Gastric ulceration induced by nonsteroidal anti-inflammatory drugs is a neutrophil-dependent process, Am. J. Physiol., 1990, 259, G462–G467 Search PubMed.
- H. Fujita, S. Takahashi and S. Okabe, Mechanism by which indomethacin delays the healing of acetic acid-induced ulcer in rats. Role of neutrophils antichemotactic and chemotactic activities, J. Physiol. Pharmacol., 1998, 49, 71–82 Search PubMed.
- N. Shimizu, T. Watanabe, T. Arakawa, Y. Fujiwara, K. Higuchi and T. Kuroki, Pentoxifylline accelerates gastric ulcer healing in rats: roles of tumor necrosis factor alpha and neutrophils during the early phase of ulcer healing, Digestion, 2000, 61, 157–164 Search PubMed.
- Y. Suzuki, M. Ishihara and M. Ito, Anti-ulcer effects of antioxidants, quercetin, α-tocopherol, nifedipine and tetracycline in rats, Jpn. J. Pharmacol., 1998, 78, 435–441 Search PubMed.
- C. F. Bou-Abboud, H. Wayland, G. Panlsen and P. H. Guth, Microcirculatory stasis precedes tissue necrosis in ethanol-induced gastric mucosal injury in rat, Dig. Dis. Sci., 1988, 33, 872–877 Search PubMed.
- Y. Tsukimi, C. Nozue and S. Okabe, Effects of teminoprazole, omeprazole and sucralfate on indomethacin-induced delayed healing of kissing gastric ulcers in rats, J. Gastroenterol. Hepatol., 1996, 11, 335–340 Search PubMed.
- J. E. Krawisz, P. Sharon and W. F. Stenson, Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity, Gastroenterology, 1984, 87, 1344–1350 Search PubMed.
- J. Piotrowski, A. Slomiany and B. L. Slomiany, Activation of apoptotic caspase-3 and nitric oxide synthase-2 in gastric mucosal injury induced by indomethacin, Scand. J. Gastroenterol., 1999, 34, 129–134 Search PubMed.
- J. Morton, B. Coles, K. Wright, A. Gallimore, J. D. Morrow, E. S. Terry, P. B. Anning, B. P. Morgan, V. Dioszeghy, H. Kiihn, P. Chaitidis, A. J. Hobbs, S. A. Jones and V. B. O'Donnell, Circulating neutrophils maintain physiological blood pressure by suppressing bacteria and IFN gamma-dependent iNOS expression in the vasculature of healthy mice, Blood, 2008, 111, 5187–5194 Search PubMed.
- B. Glowinska, M. Urban, J. Peczynska and B. Florys, Soluble adhesion molecules (sICAM-1, sVCAM-1) and selectins (sE selectin, sP selectin, sL selectin) levels in children and adolescents with obesity, hypertension, and diabetes, Metab., Clin. Exp., 2005, 54, 1020–1026 Search PubMed.
- O. Günal, B. K. Oktar, E. Özçınar, D. Tansuker, S. Arbak and B. Ç. Yeğen, Healing–promoting effect of bombesin treatment on chronic gastric ulcer in rats, Regul. Pept., 2002, 106, 81–88 Search PubMed.
- T. Brzozowski, P. C. Konturek, S. J. Konturek, Z. Sliwowski, R. Pajdo, D. Drozdowicz, A. Ptak and E. G. Hahn, Classic NSAID and selective cyclooxygenase (COX)-1 and COX-2 inhibitors in healing of chronic gastric ulcers, Microsc. Res. Tech., 2001, 53, 343–353 Search PubMed.
- C. B. Appleyard, D. M. McCafferty, A. W. Tigley, M. G. Swain and J. L. Wallace, Tumor necrosis factor mediation of NSAID-induced gastric damage: role of leukocyte adherence, Am. J. Physiol., 1996, 270, G42–G48 Search PubMed.
- S. Chatterjee, S. Premachandran, R. S. Bagewadikar, S. Bhattacharya, S. Chattopadhyay and T. B. Poduval, Arginine metabolic pathways determine its therapeutic benefit in experimental heat stroke: Role of Th1/Th2 cytokine balance, Nitric Oxide, 2006, 15, 408–416 Search PubMed.
- D. Banerjee, B. Maity, A. K. Bauri, S. K. Bandyopadhyay and S. Chattopadhyay, Gastroprotective property of Myristica malabarica against indomethacin-induced stomach ulceration: A mechanistic exploration, J. Pharm. Pharmacol., 2007, 59, 1555–1565 CrossRef.
- D. Dokmeci, M. Akpolat, N. Aydogu, L. Doganay and F. N. Turan, L-Carntine inhibits ethanol-induced gastric mucosal injury in rats, Pharmacol. Rep., 2005, 57, 481–488 Search PubMed.
- W. L. Beck and R. Xavier, Mechanism of NSAID induced gastrointestinal injury defined using mutant mice, Gastroenterology, 2000, 119, 699–705 CrossRef.
- K. Suzuki, H. Ota, S. Sasagawa, T. Sakatani and T. Fujikura, Assay method for myeloperoxidase in human polymorphonuclear leukocytes, Anal. Biochem., 1983, 132, 345–352 CAS.
Footnote |
| † Equal contributions from these authors. |
| This journal is © The Royal Society of Chemistry 2011 |