New insights on the interaction mechanism between tau protein and oleocanthal, an extra-virgin olive-oil bioactive component†
Maria Chiara
Monti
,
Luigi
Margarucci
,
Alessandra
Tosco
,
Raffaele
Riccio
and
Agostino
Casapullo
*
Dipartimento di Scienze Farmaceutiche e Biomediche, Università degli Studi di Salerno, Via Ponte don Melillo, 84084, Fisciano, Italy. E-mail: casapullo@unisa.it; Fax: +39 089 969602; Tel: +39 089 969243
First published on 7th July 2011
Abstract
Oleocanthal (OLC) is a phenolic component of extra-virgin olive oil, recently supposed to be involved in the modulation of some human diseases, such as inflammation and Alzheimer. In particular, OLC has been shown to abrogate fibrillization of tau protein, one of the main causes of Alzheimer neurodegeneration. A recent interpretation of this mechanism has been attempted on the basis of OLC reactivity with the fibrillogenic tau hexapeptide VQIVYK and SDS-PAGE of OLC/tau incubation mixtures, suggesting that covalent modification events modulate tau fibrillization. In this paper we report a detailed mass spectrometric investigation of the OLC reactive profile with both tau protein fibrillogenic fragment K18 and propylamine in biomimetic conditions. We show that K18 is prone to be covalently modified by OLC through Schiff base formation between the ε-amino group of lysine residues and OLC aldehyde carbonyls. Moreover, as expected from its de-structured conformation, K18 shows a non-selective modification profile, reacting with several lysine residues to give cyclic pyridinium-like stable adducts. These data give new insights on the mechanism of inhibition of tau fibrillization mediated by OLC.
Introduction
Extra virgin olive oil, the main source of fat in the Mediterranean diet, has long been associated with health benefits.1,2 For example, both the incidents of breast and colon cancers3–5 are remarkably low in the Mediterranean area compared to other geographical regions. In particular, olive oil phenolic compounds have attracted a wide attention since their characterization in the 1990s by Montedoro and co-workers,6–8 due to their relevant pharmacological properties and the involvement in some pathogenic processes, such as oxidative cell stress, inflammation, cancer and cardiovascular diseases.9–13Inside the secoiridoid family of olive oil constituents, oleocanthal (OLC, Fig. 1), the dialdehydic form of (−)-deacetoxy-ligstroside aglycon responsible for the bitter taste of olive oil, has been recently supposed to interfere within important pathways of relevant human diseases, such as inflammation and Alzheimer (AD).14,15
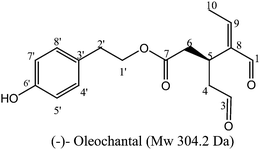 | ||
| Fig. 1 Chemical structure of (−)-oleocanthal. | ||
Recently, a role of OLC in the alteration of oligomeric structure of soluble Aβ peptides and tau protein, both involved in AD, has been reported.15 In particular, OLC increases the immunoreactivity of soluble Aβ species, when assayed with both sequence and conformation specific Aβ antibodies, protecting neurons from the synapto-pathological effects of Aβ assemblies.16 Moreover, since the aggregation of tau protein strongly correlates with the clinical progression of the AD, it seemed likely that inhibition or reversal of tau aggregation could protect the affected neurons.17
Tau is a microtubule-associated protein, particularly abundant in the neuronal axons, working as a stabilizer of the microtubules (MTs) by a direct interaction promoted by a microtubule-binding domain (MBD),18 thus modulating the plasticity of cytoskeleton. The MBD is located in the C-terminal half of the protein and is composed of three- or four-repeat structures (3RMBD or 4RMBD, respectively), with each repeat peptide (R1–R4) consisting of 31 or 32 amino acids; these four single-repeat structures have relatively similar and conserved amino acid sequences.19,20 Tau is a highly soluble protein with a random conformation in aqueous solution, and hardly shows any tendency to assemble under physiological conditions.21 In the AD patients, however, tau dissociates from axonal microtubules and abnormally aggregates to form an insoluble paired helical filament (PHF), which is implicated in neurodegeneration.22,23 Since the amount of tau aggregates has been correlated with neuron loss and the severity of dementia, the analysis of its self-assembly mechanism and the discovery of lead compounds capable of reducing the PHF formation24–28 could provide the needed information to develop an effective method to slow down the neurodegenerative process.
It has been recently reported that two VQIXXK motifs in the MT binding region, named PHF6 (from V306 to K311) and PHF6* (from V275 to K280), are responsible for the development of β-sheet structure and tau fibril formation.21
In 2009, Li and co-workers demonstrated that OLC and several analogues abrogate tau fibrillization, by means of ThT fluorescence, sedimentation analyses and EM experiments, freezing the protein in its unfolded state.15 They also assessed, on the basis of the OLC reactivity with N-Bz-lys-OMe and the fibrillogenic hexapeptide PHF6, the key role of the two OLC aldehyde groups in the covalent modification of the PHF6 lysine (K133), mediated by a Schiff base formation, and consequently their potential relevance in the inhibitory activity of OLC. However, only a few suggestions on the exact molecular mechanism of interaction between the native protein and OLC were reported, essentially based on the SDS-PAGE analysis of OLC-K18 mixtures and low resolution MALDI-MS analysis.15 These data, in our opinion, are not fully adequate to infer the exact mechanism of action of OLC and the punctual reaction site on the entire native tau protein, since Li and co-workers pointed out OLC reaction pathway using the short PHF6 peptide which cannot be representative of a complex protein system. Thus, following our previous studies on bioactive aldehyde-containing natural products,29–33 we investigated OLC-tau protein interaction in pseudo-physiological conditions by high resolution MS approach. Since the longest tau isoform T40 was not suitable for MS evaluations, we used its K18 fragment that, as previously discussed, contains all four MT-binding domains representing the integral core element of the pathological tau filament responsible for the fibrillization process.34 Our work was based on the MS characterization of the reaction profile of OLC with K18 and the identification of residues involved in the covalent modification of the polypeptide. The following analysis of OLC reactivity with propylamine in relevant bio-mimetic conditions, by 1D/2D NMR, LC-ESIMS and tandem MS in real time, gave additional support to the proposed mechanism of ligand-protein interaction. Our results point towards a non-selective covalent modification of K18 in which the two OLC carbonyl groups react with different tau lysine residues to give cyclic pyridinium-like stable adducts.
Materials and methods
OLC isolation and structural characterization
The samples of extra-virgin olive oil were kindly provided by Dr Guido Prattico from Tenuta Acquamara, Rocca d'Evandro, Caserta, Italy. The extraction and purification procedure of OLC was carried out essentially as reported by Impellizeri and Owen.35 Briefly, 45 ml of olive oil were extracted with 8 ml of CH3OH under vacuum for three times. The atmosphere in the extraction flask was saturated by nitrogen to prevent OLC oxidation. The falcon flask was centrifuged for 30 min at 5000 rpm to separate the oil from the organic phase. The methanol extract was then dried and the residue was dissolved in CH3CN. An equal volume of n-hexane was added to remove oil traces. After the extraction procedure, the CH3CN phase was dried again and the sample was purified by reverse phase-HPLC on a semi-preparative column C-18 Phenomenex (250 × 10 mm) by means an isocratic gradient at 25% of aqueous CH3CN at 4 ml min−1. The chromatographic profile was monitored at λ = 264 nm on HP 1100 binary pump system (Agilent Technologies, Palo Alto CA, USA). 190 mg of OLC were purified from each kg of oil (yield of 0.019%). NMR spectra were recorded on a Bruker DRX 600 [600 MHz (1H) and 150 MHz (13C)]. The 1H and 13C chemical shifts were referenced to the solvent peaks (δH = 7.26 ppm and δC = 77.0 ppm in CDCl3); IR spectra were recorded on a Shimadzu FTIR-8101M spectrometer; UV spectra were obtained on a Beckman DU-70 spectrophotometer; optical rotations were measured with a JASCO DIP-1000 polarimeter. MS spectra were recorded on a Q-ToF Premiere instrument (Waters, Co.) equipped with an ESI source and a Waters 6975 pump systems. Alternatively, LC-MS runs were also performed on a Finnigan LCQ Deca mass spectrometer equipped with P4000 Spectra System quaternary pumps (ThermoQuest, Co). OLC spectroscopic characterization is reported in supplementary information.Expression and purification of K18
The expression plasmid of K18 (starting from Q244 and ending with E372 of tau-441) cloned into the bacterial expression vector pRK172 has been kindly provided by Prof. Virginia Lee (University of Pennsylvania). The protein was expressed in Escherichia coli BL21(DE3) RIL strain (Agilent Techn.) and the sample was treated as reported by W. Li34 with some modification briefly described. Supernatants were dialyzed into FPLC buffer A [20 mM piperazine-N,N-bis(2-ethanesulfonic acid), 10 mM NaCl, 1 mM EGTA, 1 mM MgSO4, 2 mM DTT, 0.1 mM PMSF, pH 6.5], applied onto a HiTrap SP FF cation-exchange column (Amersham Pharmacia Biotech, Inc., Piscataway, NJ), and eluted with a 0–1 M NaCl gradient using an AKTA Purifier FPLC system (Amersham Pharmacia Biotech, Inc., Piscataway, NJ). K-18 was eluted at 0.7 M NaCl. The fractions were checked for the presence of the tau proteins by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie Blue G-250 staining. Those containing the desired tau profile were pooled together and dialyzed against 100 mM sodium acetate buffer, pH 7.0. Proteins were chromatographed by LC-MS procedure to check the purity and the Mw of the species on an Atlantis dC18 NanoAcquity column (100 mm × 75 μm) with a 5 μm Symmetry C18 precolumn (180μm × 20 mm) and eluted by means of a linear gradient from 15% to 70% aqueous acetonitrile containing 0.05% TFA and 1% formic acid, over 35 min (See supplementary information Figure S3). Mass spectra were collected in a m/z range of 1500–3000 on a Q-ToF PremierTM (Waters Co.) equipped with a nanoAcquity Ultraperformance LC system and a nanospray source. Protein concentration was determined using Biorad assay and 2 ml of 60 μM K18 were collected. A MALDI-MS mapping of the tryptic digest of K–18 has been carried out on a MALDI Micro MX™ Waters to check the sequence of the protein, finally covered at 98%Reaction mechanism of OLC on K–18 by MS
OLC was dissolved in acetonitrile (ACN; 1 mg mL−1) and then aliquots of this mother solution were added to K–18 (20 μM in PBS buffer, pH 7) for different times (from 5 min to 5 h) at 4 °C or 37 °C, with molar excesses of inhibitor ranging from 3 to 5 times (60 μM, 100 μM final OLC concentration). The final volume of ACN in the reaction mixture was always kept lower than 6%. When required, the mixture was diluted with an equal volume of NaBH4 or NaBD4 (molar ratio NaBH4 or NaBD4 : K18 = 150![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in Na2B4O7 (20 mM) for 10 min at 4 °C and the reaction was quenched adding 3 μl of a HCl 6M solution. The mixture was loaded on an Atlantis dC18 NanoAcquity column (100 mm × 75 μm) with a 5 μm Symmetry C18 precolumn (180μm × 20 mm) and eluted by means of a linear gradient from 15% to 70% aqueous acetonitrile containing 0.05% TFA and 1% formic acid, over 35 min. Mass spectra were collected in a m/z range of 1500–3000 on a Q-ToF PremierTM (Waters Co.) equipped with a nanoAcquity Ultraperformance LC system and a nanospray source.
:1) in Na2B4O7 (20 mM) for 10 min at 4 °C and the reaction was quenched adding 3 μl of a HCl 6M solution. The mixture was loaded on an Atlantis dC18 NanoAcquity column (100 mm × 75 μm) with a 5 μm Symmetry C18 precolumn (180μm × 20 mm) and eluted by means of a linear gradient from 15% to 70% aqueous acetonitrile containing 0.05% TFA and 1% formic acid, over 35 min. Mass spectra were collected in a m/z range of 1500–3000 on a Q-ToF PremierTM (Waters Co.) equipped with a nanoAcquity Ultraperformance LC system and a nanospray source.
The mixture of un-reacted and modified protein upon incubation at 4 °C and NaBH4 reduction was analyzed by RP-HPLC on a Phenomenex Proteo column (250 × 2.0 mm) by means of a linear gradient from 20% to 70% aqueous acetonitrile containing 0.1% TFA, over 50 min at 0.150 ml min−1. The elution profile was monitored at 220 and 280 nm. The fractions were collected and analyzed on a MALDI micro MX™ (Waters Co, Milford Massachusetts, USA) in reflectron positive ion mode, using α-cyano-4-hydroxycinnamic acid (10 mg ml−1) dissolved in H2O–CH3CN (50/50 v/v) 0.1% TFA as matrix, in a m/z range of 10000–30000.
Identification of reactive sites of OLC on K-18
The mixtures of un-reacted and modified proteins upon incubation at 4 °C or 37 °C and NaBH4 reduction were purified as reported above, lyophilized and diluted in sodium bicarbonate (50 mM, pH 8). Then, the samples were digested with trypsin at 37 °C for 4 h with a 1:50 (w/w) enzyme : substrate ratio. Proteolytic fragments were analyzed by MALDI-MS. Mass spectra were acquired in an m/z interval of 600–1800.
LC-MS and NMR analysis of OLC-propylamine complexes
OLC was incubated with propylamine (Sigma-Aldrich) in the following conditions: 50 μM of OLC diluted in CH3CN was added to a solution of sodium acetate 20 mM at pH = 7, containing 10-fold molar excess of propylamine. The reaction temperature was set at 37 °C. When required, the sample was treated with an equal volume of a NaBH4 solution (molar excess NaBH4 : OLC = 10:1) diluted in NaOH 15 mM, and the reaction was stopped after 30 min at room temperature with HCl 6 M. The incubation mixture was analyzed by RP-HPLC-MS on a Phenomenex C18 narrow-bore column by means a linear gradient from 10% to 80% aqueous acetonitrile containing 0.05% TFA, over 30 min. Alternatively, aliquots were diluted with CH3CN–H2O 1:1 with 0.1% TFA and immediately analyzed by a direct injection in the mass spectrometer ion source. A time course NMR analysis of the reaction was realized by adding 3 mM of propylamine to an equivalent CD3CN solution of OLC, in a NMR tube. 1H spectra were registered every 10 min. Structural characterization of the HPLC purified compounds was done by 1D and 2D NMR (CDCl3).
A3: ESIMSm/z 326.3 [M]+; ESIMS/MSm/z 121.5;.1H NMR δ (CDCl3, 600.13 MHz) 8.73 (H–1, bs), 8.46 (H–3, d, J = 6.3.Hz), 7.79 (H–4, d, J = 6.3 Hz), 3.92 (H–6, bs), 6.80 (H–9, dd, J = 17.4 and 11.5 Hz), 5.73 (H–10a, d, J = 11.5 Hz), 5.94 (H–10b, d, J = 17.4 Hz), 4.18 (H–1′a, m), 4.22 (H–1′b, m), 2.80 (H–2′, t, J = 7.0 Hz), 7.05 (H–4′/8′, d, J = 8.0 Hz), 6.76 (H–5′/7′, d, J = 8.0 Hz); 4.46 (H–1′′, t, J = 7.3 Hz), 1.99 (H–2′′, sext., J = 7.3 Hz), 0.98 (H–3′′, t, J = 7.3 Hz); 13C NMR δ (CDCI3, 150.9 MHz) 140,1 (C–1), 141,4 (C–3), 128,0 (C–4), 150.0 (C–5), 37.5 (C–6), 171.9 (C–7), 127.4 (C–8), 138.4 (C–9), 122.3 (C–10), 65.2 (C–1′), 34.3 (C–2′), 129.5 (C–3′), 130.1 (C–4′/8′), 115.4 (C–5′/7′), 154.1 (C–6′), 61.9 (C–1′′), 23.4 (C–2′′), 11.0 (C–3′′)
Results and discussion
OLC was isolated from an extra-virgin olive oil sample, according to the method reported in literature35 with some minor changes, and fully characterized by 1 and 2D NMR, ESIMS and MS/MS (see ESI†).The assessment of the mechanism of OLC-K18 interaction between OLC and K18 consisted of the following steps: (a) structural analysis of the ligand-protein complexes and (b) characterization of the OLC reaction profile with propylamine in bio-mimetic conditions. To this end, we employed an experimental protocol based on the combination of NMR spectroscopy, mass spectrometry and classical protein chemistry (proteolytic digestion, RP-HPLC).
Structural analysis of OLC-K18 complexes
K18 fragment of tau, expressed and purified as reported in the experimental section, was treated with a 5-fold molar excess of OLC and the reaction was carried out either at 4 °C or 37 °C, monitoring its progress by LC-MS at different time intervals (5 min, 15 min, 1h, 3h and 5h) with or without NaBH4 (and NaBD4) as reducing agent.The time course LCMS analysis of the reaction at 4 °C (see spectra in Fig. 2A) showed the presence of two main peaks, already after 5 min, corresponding to the unmodified K18 and mono-modified K18 species with a mass increment of 268.5 ± 1.3 Da, that increased at 272 ± 0.8 Da and 274 ± 0.6 Da after reduction with NaBH4 and NaBD4 respectively (Table 1). Mechanistic considerations, attempted on the basis of this data set, pointed towards two possible reaction pathways, both mediated by Schiff base intermediates formed between lysine ε-amino group(s) on K18 and the OLC aldehyde carbonyls.
| Mw after OLC treatment | |||||
|---|---|---|---|---|---|
| Measured K18 Mw | Measured Mw | ΔMw | Reducing Agent | Temperature | Time |
| 13812.2(±1.0) | 14080.7(±1.3) | 268.5 | none | 4 °C | 5′;15′,60′,180′,300′ |
| 13812.5(±0.9) | 14084.6(±0.8) | 272.1 | NaBH4 | 4 °C | 5′;15′,60′,180′,300′ |
| 13812.4(±1.5) | 14086.4(±0.6) | 274.0 | NaBD4 | 4 °C | 5′;15′,60′,180′,300′ |
| 13812.2(±1.0) | 14080.8(±1.3) | 268.6 | none | 37 °C | 5′; |
| 13812.5(±0.9) | 14084.8(±0.8) | 272.3 | NaBH4 | 37 °C | 5′; |
| 13812.4(±1.5) | 14086.7(±0.6) | 274.3 | NaBD4 | 37 °C | 5′; |
| 13812.7(±0.8) | 13901.8(±0.8) | 89.1 | none | 37 °C | 15′,60′,180′,300′ |
| 13812.3(±1.1) | 13904.3(±0.9) | 92.0 | NaBH4 | 37 °C | 15′,60′,180′,300′ |
| 13812.8(±1.2) | 13906.9(±0.6) | 94.1 | NaBD4 | 37 °C | 15′,60′,180′,300′ |
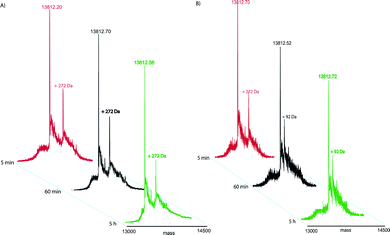 | ||
| Fig. 2 After addition of NaBH4 deconvoluted mass spectra of K18 incubated in presence of OLC. The spectra were recorded at different times (for convenience only three time points are shown) and different temperatures 4 °C (A) and 37 °C (B). | ||
In the first path (Scheme 1A), OLC cross-links two different K18 lysine residues, giving rise to a double imine reversible macrocyclic system (ΔMW of 268.5 Da) easily reduced in presence of NaBH4 or NaBD4 (Table 1). In a second hypothesis (Scheme 1B), a single lysine ε-amino group reacts with either of OLC carbonyls to give a stable iminium six-member ring adduct (ΔMW of 269 Da), that is converted to a cyclic amine15 with NaBH4.
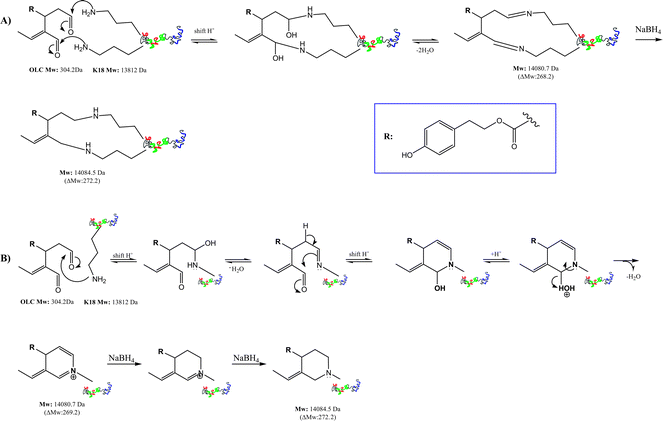 | ||
| Scheme 1 Possible reaction mechanisms for the covalent modification of K18 by OLC at 4 °C. Mass increments were measured by LC-ESIMS. Panel A shows the reaction mechanism between OLC and two different lysine residues on a unique K18-protein. Panel B reports the reaction between a single lysine ε-amino group and both OLC carbonyls to give an iminium six-member ring. | ||
When the reaction was carried out at 37 °C, a more complex pathway has been observed, as shown in Fig. 2B. As a matter of fact, we monitored the same K18 adduct observed at 4 °C, after 5 min (ΔMW of 268.5 Da), that evolved after 15 min to a species with a mass increment of 89.5 Da, subsequently reduced with NaBH4 (ΔMW of 92.1 Da) or NaBD4 (ΔMW of 94.2 Da), which remained unchanged even after 5 h (Fig. 2B and Table 1).
We suggested a plausible mechanism for this new evidence (Scheme 2), in which the early forming species with a mass increment of 268.5 Da turns into a more stable rearranged adduct giving rise to the loss of 4-hydroxy-phenylethyl acetate.
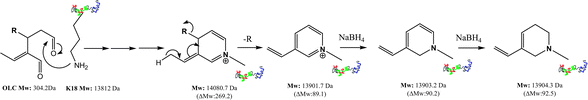 | ||
| Scheme 2 Proposed mechanism for the covalent modification of K18 protein by OLC at 37 °C. | ||
Finally, we determined the sites of covalent modifications on K18 in the reaction with OLC, either at 4 °C or 37 °C. The reaction mixtures, treated with NaBH4, were digested with trypsin and analyzed by MALDI-MS. This experiment enlightened on the occurrence of the mechanism of K18 covalent modification reported in Scheme 1.
Indeed, as expected for an intrinsically disordered polypeptide, we found several lysine residues modified by OLC both at 4 °C and 37 °C (see ESI, Fig. S1†). Besides, the early reaction at 4 °C evolved by a cross-link modification mechanism (Scheme 1A), since we detected in the tryptic mixture only fragments containing two peptides cross-linked to OLC. Increasing the time, the reaction equilibrium shifted to the mechanism depicted in Scheme 1B, as we monitored only single peptides modified by OLC on a single lysine residue.
Since the 1,5-dialdehyde moiety of OLC is prone to undergo attack by nitrogenous nucleophiles giving rise to a complex web of multiple chemical equilibria and an array of possible intermediates, as already suggested by Li and co-workers,15 we decided to give a firm evidence of the reaction pathways proposed above. On this basis, we analyzed the covalent reactivity of OLC towards the small nucleophile propylamine (PA), in relevant bio-mimetic conditions, following the time course of the reaction by 1D/2D NMR and MS. Thus, we applied two parallel sets of HPLC-ESIMS and NMR experiments: in the first step we followed the course of the reaction monitoring by LCMS the intermediates formation, in different conditions and after addition of NaBH4. Then, we applied 1D and 2D NMR to track the reaction course and characterize the final product(s).
The interpretation of the LCMS and NMR data supported the reaction pathway depicted in Scheme 3, in which two key intermediates (A1 and A2) and the final product A3 can be postulated. OLC promptly reacted with PA producing, after 3 min, a main species at m/z 346.3 (OLC + PA – H2O), matching a Schiff base formation between PA and one of the OLC aldehyde carbonyls. The most reactive aldehyde function was identified tracking the reaction course by NMR. Proton spectra were recorded after addition (every 10 min) of little amounts of PA to a CD3CN solution of OLC in the NMR tube (see Materials and methods for details). The reduction and final disappearance of the signal at δ 9.64 (H–3) (see ESI, Fig. S2†) supported the formation of the imine intermediate A1. This species, through the isobar intermediate A1*, evolved to the adduct A2 (m/z 328.3, 15 min incubation time), that slowly turned into two species at m/z 326.3 and 148.2 (30 min incubation time). The species at m/z 326.3 was isolated and unequivocally characterized by 1D and 2D NMR (see Materials and methods) as the aromatic N-alkyl pyridinium A3 and consequently the other compound as A4 species, arising from the rearrangement of A2 (Scheme 3). This picture was confirmed by LCMS analysis of the reaction after addition of NaBH4 (Scheme 4). The reduction of the imineA1 gave the amineB1 that, after cyclization and dehydration, evolved into the imonium species B2, rapidly reduced to the cyclic alkyl-amine B3.
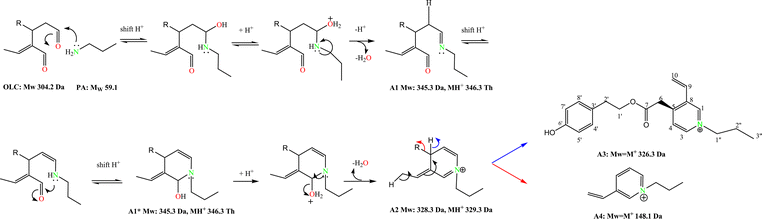 | ||
| Scheme 3 Reaction mechanism between OLC and propylamine. | ||
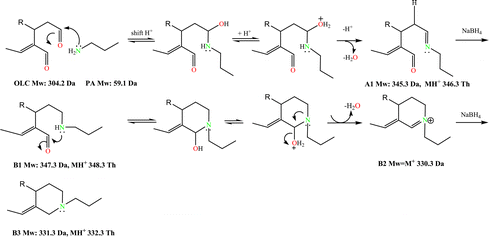 | ||
| Scheme 4 Reaction mechanism between OLC and propylamine in presence of NaBH4. | ||
Conclusions
The reduced risk of neurodegenerative pathologies as AD in Mediterranean area has been associated with high consumption of extra virgin olive oil, which also exerts beneficent effects on cancer and cardiovascular diseases. These healthy properties have been attributed mainly to minor phenolic compounds found in extra virgin olive oil,36 which also bear antioxidant, anti-inflammatory and anti-thrombic activities.14OLC represents about 10% of the total phenolic compounds in extra virgin olive oil, and has been found to reduce the fibrillization of tau protein already at 100 μM concentration.15 In this scenario, a detailed analysis of the reactive profile of OLC towards the fragment K18 of the tau protein in biologically relevant conditions has been performed, giving new insights into the mechanism of interaction at the molecular level. K18 is prone to be covalently modified by OLC with a 1:1 stoichiometry, through Schiff base formation between the ε-amino group of lysine residues and the OLC aldehyde carbonyls. Due to its mainly unstructured conformation, K18 is prone to undergo covalent modifications in an unspecific fashion. Moreover, the reaction course is strictly dependent on the temperature and time of contacts between the counterparts. The initial cross-link of OLC with two different lysine residues on K18 is rapidly converted in the modification of a single lysine, producing a cyclic adduct that evolves towards a more stable pyridinium-like complex by rearrangement of the skeleton. This picture, confirmed by a the analysis of reaction between OLC and propylamine, enlarges the knowledge on the interaction between OLC and tau protein, useful to give more lights into the mechanism of inhibition of tau fibrillization mediated by OLC.
Acknowledgements
Financial support by the University of Salerno and by MIUR (Rome) is gratefully acknowledged. Authors also acknowledge the use of the instrumental facilities of the Centre of Competence in Diagnostics and Molecular Pharmaceutics supported by Regione Campania (Italy) through POR funds. Prof. V. Lee (University of Pennsylvania) is kindly acknowledged for providing K18 plasmid.References
- V. Fogliano and R. Sacchi, Mol. Nutr. Food Res., 2006, 50(1), 5–6 Search PubMed.
- F. Visioli, D. Caruso, S. Grande, R. Bosisio, M. Villa, G. Galli, C. Sirtori and C. Galli, Eur. J. Nutr., 2004, 44(2), 121–127 CrossRef.
- A. Keys, Am. J, Clin. Nutr., 1995, 61(6), 1321–1323 Search PubMed.
- J. Ruano, J. Lopez-Miranda, F. Fuentes, J. A. Moreno, C. Bellido, P. Perez-Martinez, A. Lozano, P. Gomez, Y. Jimenez and J. F. Perez, J. Am. Coll. Cardiol., 2005, 46(10), 1864–1868 CrossRef CAS.
- F. Visioli, A. Poli and C. Gall, Med. Res. Rev., 2002, 22(1), 65–75 CrossRef CAS.
- G. Montedoro, M. Servili, M. Baldioli and E. Miniati, J. Agric. Food Chem., 1992, 40(9), 1571–1576 CrossRef CAS.
- G. Montedoro, A. Sillen, M. Baldioli and E. Miniati, J. Agric. Food Chem., 1992, 40(9), 1576–1580 Search PubMed.
- G. Montedoro, M. Baldioli, M. Servili, R. Selvaggini and E. Miniati, J. Agric. Food Chem., 1993, 41(11), 2228–2234 CrossRef CAS.
- S. Brunelleschi, C. Bardelli, A. Amoruso, G. Gunella, F. Ieri, A. Romani, W. Malorni and F. Franconi, Pharmacol. Res., 2007, 56(6), 542–549 CrossRef CAS.
- Y. Z. Hashim, I. R. Rowland, H. McGlynn, M. Servili, R. Selvaggini, A. Taticchi, S. Esposto, G. Montedoro, L. Kaisalo, K. Wahala and C. I. Gill, Int. J. Cancer, 2008, 122(3), 495–500 CrossRef CAS.
- C. I. Gill, A. Boyd, E. McDermott, M. McCann, M. Servili, R. Selvaggini, A. Taticchi, S. Esposto, G. Montedoro, H. McGlynn and I. Rowland, Int. J. Cancer, 2005, 117(1), 1–7 CrossRef CAS.
- C. Manna, P. Galletti, V. Cucciolla, G. Montedoro and V. Zappia, J. Nutr. Biochem., 1999, 10(3), 159–165 CrossRef CAS.
- S. M. Monti, A. Ritieni, R. Sacchi, K. Skog, E. Borgen and V. Fogliano, J. Agric. Food Chem., 2001, 49(8), 3969–3975 CrossRef CAS.
- G. K. Beauchamp, R. S. Keast, D. Morel, J. Lin, J. Pika, Q. Han, C. H. Lee, A. B. Smith and P. A. Breslin, Nature, 2005, 437(7055), 45–46 CrossRef CAS.
- W. Li, J. B. Sperry, A. Crowe, J. Q. Trojanowski, A. B. Smith, III and V. M. Lee, J. Neurochem., 2009, 110(4), 1339–1351 CrossRef CAS.
- J. Pitt, W. Roth, P. Lacor, A. B. Smith, III, M. Blankenship, P. Velasco, F. F. De, P. Breslin and W. L. Klein, Toxicol. Appl. Pharmacol., 2009, 240(2), 189–197 CrossRef CAS.
- A. Crowe, C. Ballatore, E. Hyde, J. Q. Trojanowski and V. M. Lee, Biochem. Biophys. Res. Commun., 2007, 358(1), 1–6 CrossRef CAS.
- M. D. Mukrasch, J. Biernat, B. M. von, C. Griesinger, E. Mandelkow and M. Zweckstetter, J. Biol. Chem., 2005, 280(26), 24978–24986 CrossRef CAS.
- J. Sevcik, R. Skrabana, R. Dvorsky, N. Csokova, K. Iqbal and M. Novak, FEBS Lett., 2007, 581(30), 5872–5878 CrossRef CAS.
- K. Okuyama, C. Nishiura, F. Mizushima, K. Minoura, M. Sumida, T. Taniguchi, K. Tomoo and T. Ishida, FEBS J., 2008, 275(7), 1529–1539 CrossRef CAS.
- C. Smet, A. Leroy, A. Sillen, J. M. Wieruszeski, I. Landrieu and G. Lippens, ChemBioChem, 2004, 5(12), 1639–1646 CrossRef CAS.
- M. Pickhardt, G. Larbig, I. Khlistunova, A. Coksezen, B. Meyer, E. M. Mandelkow, B. Schmidt and E. Mandelkow, Biochemistry, 2007, 46(35), 10016–10023 CrossRef CAS.
- M. Goedert, A. Klug and R. A. Crowther, J. Alzheimers. Dis., 2006, 9(3 Suppl), 195–207 CAS.
- I. Khlistunova, J. Biernat, Y. Wang, M. Pickhardt, B. M. von, Z. Gazova, E. Mandelkow and E. M. Mandelkow, J. Biol. Chem., 2005, 281(2), 1205–1214 CrossRef.
- M. Pickhardt, Z. Gazova, B. M. von, I. Khlistunova, Y. Wang, A. Hascher, E. M. Mandelkow, J. Biernat and E. Mandelkow, J. Biol. Chem., 2004, 280(5), 3628–3635 CrossRef.
- C. M. Wischik, P. C. Edwards, R. Y. Lai, M. Roth and C. R. Harrington, Proc. Natl. Acad. Sci. U. S. A., 1996, 93(20), 11213–11218 CrossRef CAS.
- A. Crowe, C. Ballatore, E. Hyde, J. Q. Trojanowski and V. M. Lee, Biochem. Biophys. Res. Commun., 2007, 358(1), 1–6 CrossRef CAS.
- B. Bulic, M. Pickhardt, I. Khlistunova, J. Biernat, E. M. Mandelkow, E. Mandelkow and H. Waldmann, Angew. Chem., Int. Ed., 2007, 46(48), 9215–9219 CrossRef.
- M. C. Monti, A. Casapullo, R. Riccio and L. Gomez-Paloma, FEBS Lett., 2004, 578(3), 269–274 CrossRef CAS.
- M. C. Monti, A. Casapullo, C. N. Cavasotto, A. Tosco, P. F. Dal, A. Ziemys, L. Margarucci and R. Riccio, Chem.–Eur. J., 2009, 15(5), 1155–1163 CrossRef CAS.
- M. C. Monti, A. Casapullo, C. Santomauro, M. V. D'Auria, R. Riccio and L. Gomez-Paloma, ChemBioChem, 2006, 7(6), 971–980 CrossRef CAS.
- M. C. Monti, A. Casapullo, R. Riccio and L. Gomez-Paloma, Rapid Commun. Mass Spectrom., 2005, 19(10), 1–6 Search PubMed.
- M. C. Monti, A. Casapullo, C. N. Cavasotto, A. Napolitano and R. Riccio, ChemBioChem, 2007, 8(13), 1585–1591 CrossRef CAS.
- W. Li and V. M. Lee, Biochemistry, 2006, 45(51), 15692–15701 CrossRef CAS.
- J. Impellizzeri and J. Lin, J. Agric. Food Chem., 2006, 54(9), 3204–3208 CrossRef CAS.
- A. Daccache, C. Lion, N. Sibille, M. Gerard, C. Slomianny, G. Lippens and P. Cotelle, Neurochem. Int., 2011, 58(6), 700–707 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1fo10064e |
| This journal is © The Royal Society of Chemistry 2011 |