Mechanism of Hericium erinaceus (Yamabushitake) mushroom-induced apoptosis of U937 human monocytic leukemia cells
Sung Phil
Kim
a,
Mi Young
Kang
b,
Yong Hee
Choi
c,
Jae Ho
Kim
a,
Seok Hyun
Nam
*d and
Mendel
Friedman
*e
aDepartment of Molecular Science and Technology, Ajou University, Suwon, 443-749, Republic of Korea
bDepartment of Food Science and Nutrition, Kyoungpook National University, Daegu, 702-701, Republic of Korea
cDepartment of Food Science and Technology, Kyoungpook National University, Daegu, 702-701, Republic of Korea
dDepartment of Biological Science, Ajou University, Suwon, 443-749, Republic of Korea. E-mail: shnam@ajou.ac.kr; Fax: +82-31-219-1615; Tel: +82-31-219-2619
eWestern Regional Research Center, Agricultural Research Service, U.S Department of Agriculture, 800 Buchanan St., Albany, CA 94710, USA. E-mail: Mendel.Friedman@ars.usda.gov; Fax: +1-510-559-6162; Tel: +1-510-559-5615
First published on 8th June 2011
Abstract
Phytochemicals in some foods are a potential source of bioactive safe compounds for cancer chemoprevention and suppression of tumor initiation, promotion, and metastasis. In the present study, we evaluated hot water (HWE), microwaved 50% ethanol (MWE), acidic (ACE), and alkaline (AKE) extracts of the fruitbody (sporocarp) of Hericium erinaceus (Yamabushitake, Lion's Mane) mushrooms for their ability to induce apoptosis (programmed cell death) in U937 human monocytic leukemia cells. Cell culture, cell viability, cytotoxicity, flow cytometry, chromosomal DNA integrity, mitochondrial membrane potential, expression of pro- and anti-apoptotic proteins, and activation and inhibition of caspase assays were carried out to help define the mechanism of observed apoptosis. The aqueous and aqueous/ethanolic extracts were active in all assays, whereas the acidic and alkaline extracts with the similar proximate compositions were both inactive. The results of the bioassays with the active extracts are consistent with an apoptosis mechanism governing suppression of the cell proliferation pathway that involves activation of mitochondria-mediated caspase-3 and caspase-9 but not caspase-8. Proximate analysis of the freeze-dried mushroom powder showed that it contains high amounts of proteins, carbohydrates, and minerals. The results indicate that H. erinaceus mushrooms may have therapeutic potential against human leukemia.
Introduction
Hericium erinaceus, called “Yamabushitake” in Japan or “Houtou” in China, respectively, is an edible and medicinal mushroom of the Aphyllophorales, Hydnaceae (Hericiaceae) class. In a previous study, we showed that two extracts prepared from fruit of H. erinaceus upregulated nitrogen oxide (NO) production and phagocytosis in mouse macrophage RAW264.7 cells,1 suggesting a possible anti-cancer activity via augmentation of host immunity. To place findings described below in proper perspective, we will first briefly mention selected reported observations on anticarcinogenic and therapeutic aspects of mushrooms in chronological order.An aqueous extract of shiitake mushrooms (Lentinus edodes) induced apoptosis in U937 cells associated with decreased production of IL-1.2 Ethanol extracts of from Lentinula edodes mycelia significantly decreased the proliferation and induced apoptosis in murine skin carcinoma (CH72) cells via a transient G1 arrest.3 A mushroom lectin induced apoptosis in U937 cells via G2/M cell cycle arrest associated with upregulation of p21/Waf1 expression, decrease in expression of Bcl-2, enhanced release of cytochrome-c from mitochondria to cytoplasm, and activation of caspase-9 in treated cells.4 A polysaccharide isolated form H. erinaceus upregulated functional events mediated by macrophages in RAW264.7 cells such as production of nitric oxide (NO) and expression of cytokines (IL-1β and TNF-α).5 A purified polysaccharide from H. erinaceus fruitbody enhanced doxorubicin-induced apoptosis in human hepatocarcinoma cells, via sensitization of doxorubicin-mediated apoptotic signaling by reducing c-FLIP expression.6 A ubiquinone-9 isolated from chloroform extract from the mushroom Pleurotus eryngii induced apoptosis in U937 cells via fragmentation of DNA into an apoptotic DNA ladder and inhibition of topoisomerase activity.7 Feeding H. erinaceus mycelia added to mouse diet induced anti-inflammatory effects via elevation of plasma glutathione (GSH) level and amelioration of hepatic oxidative stress.8 Japanese men and women aged from 50 to 80 years fed 250 mg capsules containing H. erinaceus experienced improvement in mild cognitive impairment, suggesting that mushrooms can help prevent dementia.9 Processing, but not cultivation conditions, affects the antimicrobial and antioxidative potential of H. erinaceus.10
The main objective of this study was to determine the proximate composition of fruitbody extracts of H. erinaceus mushrooms and to investigate in more detail mechanistic aspects associated with the suppression of proliferation of U937 cells by induction of apoptosis through activation of the mitochondria-mediated intrinsic caspase pathway.
Experimental
Materials
RPMI 1640 medium, Hanks' balances salt solution (HBSS), fetal bovine serum (FBS) and other cell culture reagents were purchased from Hyclone Laboratories (Logan, Utah). Fluorescein isothiocyanate (FITC)-conjugated Annexin V and propidium iodide (PI) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). All reagents of analytical grade were purchased from Sigma (St. Louis, MO) and used without further purification.Preparation of H. erinaceus extracts
Dried fruitbody of H. erinaceus was obtained from Forest Environment Science Laboratory, College of Agriculture and Life Science, Kyungpook National University (Daegu, Korea). Fruitbody was ground into powder and then passed through a 40-mesh sieve. The powder was extracted with 20-fold weight of the following solvents as previously described:1 hot-boiling in pyrogen-free water for 3 h (HWE); microwaving in ethanol (50% v/v) at 60 W for 3 min using focused microwave-associated Soxhlet extractor (Prolabo, Paris, France) MWE); acidic extract (ACE) and alkaline extracts (AKE) by boiling in 1% HCl and 3% NaOH for 2 h, respectively (negative controls). After extraction, solid materials were removed by centrifuge at 3,000 × g for 30 min. To the recovered supernatants was added 4-fold volume of ethanol to precipitate polysaccharides at 4 °C for 24 h. The precipitates were dissolved in deionized water and dialyzed in a tube (Sigma) with cut-off molecular weight of 12 kDa against the same solvent. All extracts were lyophilized to powders.Protein, carbohydrate, ash (mineral), fat, fiber, and moisture of the four extracts were determined by standard methods.11
Cell culture and viability assay
Normal and leukemia cell lines were obtained from the American Type Tissue Culture Collection (ATCC, Manassas, VA) and cultured in RPMI 1640 medium supplemented with heat-inactivated FBS (10%), glutamine (2 mM), penicillin (100 U mL−1), and streptomycin (100 μg mL−1) at 37 °C in a humidified atmosphere with 5% CO2. Cell viability was assessed by MTT staining as described by Mosmann12 using a non-radioactive cell proliferation kit (Promega, Madison, WI). Briefly, cells were seeded into a 96-well plate at a density of 1 × 105 cells per well and cultivated for 24 h at 37 °C humidified air with 5% CO2. Cells were then treated with H. erinaceus extraction fractions for 48 h followed by staining with tetrazolium salts and subsequent solubilization of the intracellular chromogen formazan product according to the manufacturer's instructions. Absorbance was read in the microplate reader (Bio-Rad, Hercules, CA) at 570 nm and a reference wavelength of 655 nm. Viability was expressed as a percentage of control group treated with vehicle alone.Apoptosis assay by flow cytometry
Flow cytometry was carried out according to the method described by Vermes et al.13 Briefly, U937 cells (1 × 106) were seeded into culture dish and cultivated for 3 h in serum-free RPMI 1640 medium. We adopted the 3 h serum starvation time to rule out experimental noise because longer serum starvation leads to cell apoptosis. Cells were then treated with different H. erinaceus extracts at 500 μg mL−1. After incubation for 48 h, cells were collected by centrifugation at 1,000 × g for 5 min. Cell pellets were resuspended in a FACs binding buffer (0.1 mL; 10 mM HEPES, 150 mM NaCl, 2.5 mM CaCl2, pH 7.4) with Annexin V-FITC (1 μg) and propidium iodide (PI) (1 μg). After incubation for 15 min in dark, flow cytometry was carried out in a FACSCaliber™ (Becton, Dickinson, and Co. San Jose, CA).Cell cycle distribution assay
Sub-G1 phase cell population was measured by the method of Herrmann et al.14 Briefly, U937 cells (1 × 106) were seeded into culture dish and cultivated for 3 h in serum-free RPMI1640 medium to synchronize the cell cycle. Cells were then treated with H. erinaceus extracts at 500 μg mL−1 for 12 h, collected, washed, and fixed with ice-cold ethanol at 4 °C overnight. Ethanol-fixed cells were suspended in cold PBS (1 mL) containing RNase A (1 mg mL−1), and PI (50 μg mL−1). Cell suspensions were incubated at 37 °C for 1 h in the dark followed by a flow cytometry and analysis of histograms by CellQuest software.DNA fragmentation assay
U937 cells (1 × 106) with synchronized cell cycle were treated with H. erinaceus extracts at 500 μg mL−1 for 48 h, washed, and lysed in a buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, 0.5% SDS, and 1 mg mL−1 proteinase K) at 56 °C for 3 h. RNase A (0.5 mg mL−1) was added and incubation continued for 1 h at 56 °C. DNA was extracted with neutralized phenol/chloroform/isoamyl alcohol (25/24/1, v/v/v) and purified by ethanol precipitation by the method of Shen et al.15 Purified DNA was dissolved in Tris/EDTA buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA), and analyzed by electrophoresis on 1% agarose gel containing (0.5 μg mL−1) ethidium bromide (EtBr) (0.5 mg mL−1). Gels were photographed under UV light.Western blot assay
U937 cells (1 × 106) treated with H. erinaceus extracts for 12 h were harvested and washed twice with PBS. Cell pellets were lysed in a lysis buffer (50 mM Tris-Cl at pH 7.4, 150 mM NaCl, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, protease inhibitor cocktail) on ice for 20 min. Lysates were clarified by micro-centrifugation at 14,000 × g for 20 min. Protein concentration of lysates was determined by the Bradford method using a Protein Kit (Bio-Rad, Hercules, CA) with BSA as a standard. Lysates containing proteins (30 μg) were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a nitrocellulose membrane. The membrane was blocked in 5% skim milk in PBS containing Tween 20 (0.1%) at 4 °C overnight and incubated with primary antibodies diluted in an antibody buffer (5% skim milk and 0.1% Tween 20 in PBS) at room temperature for 3 h. Membranes were then washed thoroughly with PBS containing Tween 20 (0.1%) and incubated for 1 h with a peroxidase-conjugated secondary antibody 1,000-fold diluted in the antibody buffer. After subsequent washing with PBS containing Tween 20 (0.1%), membrane-bound antibodies were visualized by ECL western blotting detection reagent (Amersham, Newark, NJ) according to the manufacturer's instruction. Protein band intensity was determined in a luminescent image analyzer (Model Las-1000CH, Fuji, Tokyo, Japan).Primary antibodies used were as follows: rabbit polyclonal against mouse Bid, Bcl-2, cIAP-1, cIAP-2, XIAP (Millipore, Billerica, MA), caspase-8, -9, HSP90 (Santa Cruz Biotechnology, Santa Cruz, CA), and caspase-3 (Abcam, Cambridge, MA); mouse monoclonal against mouse Bad, BAX, Bcl-XL/XS, cytochrome-c, and β-actin (Millipore, Billerica, MA); and mouse polyclonal against PARP (Cell Signaling Technology, Danvers, MA).
Caspase-3, -8 and -9 activity assays
Caspase activity was measured using the caspase-8 activity (Chemicon, Temecula, CA) and caspase-9 activity assay kits (Millipore, Billerica, MA) according to the manufacturer's instructions. Briefly, treated U937 cells (1 × 106) were lysed in a lysis buffer on ice for 30 min. Lysates were clarified by micro-centrifugation at 14,000 × g for 10 min. Supernatants (cytosolic extract) containing proteins (50 μg) were incubated in an assay buffer (100 μL) containing the colorimetric substrates, Ac-Ile-Gle-Thr-Asp (IETD)-p-nitroanilline (pNA) for caspase-8 activity and Ac-Leu-Glu-His-Asp (LEHD)-pNA for caspase-9 activity assays at 37 °C for 2 h. Absorbance at 405 nm was quantified in a microplate reader.Mitochondrial membrane potential (MMP) assay
MMP was assessed using the lipophilic cationic probe 3,3-dihexyloxacarbocyanine iodide (DiOC6) (Sigma). U937 cells (1 × 105) were treated with H. erinaceus extracts for 48 h followed by staining with fluorescence probes by incubation in dark at 37 °C for 20 min in RPMI 1640 medium containing DiOC6 dye (10 μM). Changes in intensity were analyzed by flow cytometry using a FACS Caliber. In addition, JC-1-stained cells suspended in PBS were mounted on confocal dish (SPL Inc., Pocheon, Korea) and MMP was determined in a confocal microscope (LSM510, Carl Zeiss, Munich, Germany).Statistical analysis
Results are expressed as the means ± SD of three independent experiments. Significant differences between means were determined using Statistical Analysis System software package (SAS, Cary, NC). P < 0.05 is regarded as statistically significant.Results
Mushroom composition
To stimulate interest in the dietary use of H. erinaceus, we determined the proximate composition of the freeze-dried mushroom powder (Table 1).Growth inhibition of U937 cells
To determine whether HWE and MWE are cytotoxic, we evaluated cell viability by the MTT assay. As an internal control, we also tested two extracts (ACE and AKE) which did not induce NO production. Fig. 1 shows that treatment with HWE or MWE decreased cell viability in a concentration-dependent manner, whereas ACE or AKE were not cytotoxic. These data implied possible involvement of HWE or MWE in apoptosis of U937 cells.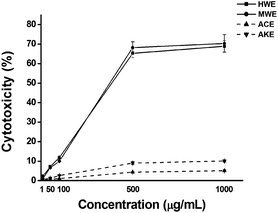 | ||
| Fig. 1 Cytotoxic and apoptotic effects of each H. erinaceus extract including HWE and MWE on human leukemia U937 cells. The cells were plated at a density of 1 × 105 cells/well in 96-well microplate, and then treated with various concentrations of each extracts for 48h. After incubation at 37 °C, cell death was determined by MTT assay. ACE and AKE were used as the internal negative controls selected among the previously reported H. erinaceus extracts. Cytotoxicity (%) = (Absorbance of extract-treated cells/Absorbance of PBS-treated control) × 100. | ||
Induction of apoptosis in U937 cells
Additional experiments were carried out to find out whether growth inhibition by HWE and MWE resulted from apoptotic cell death. First, the fraction of stained cell populations in different quadrants was analyzed by flow cytometry (Fig. 2). The proportion of single (FITC+, PI−) plus double positive (FITC+, PI+) cells, comprised of early apoptotic cells (the lower right quadrant) and late apoptotic/necrotic cells (the upper right quadrant), increased to 54.5% and 54.7% by the treatment with HWE and MWE, respectively. Induction levels were near to those of etoposide-treated control used to demonstrate apoptosis.16 Next, we analyzed the integrity of genomic DNA and the ratio of hypodiploid cells. Treatment with HWE or MWE caused appearance of chromosomal condensation and apoptotic bodies by staining with DAPI (Fig. 3A). Digestion of genomic DNA into ladder of internucleosomal fragmentation was also observed in cells treated with HWE and MWE. Digestion patterns were similar to those induced by etoposide (Fig. 3B).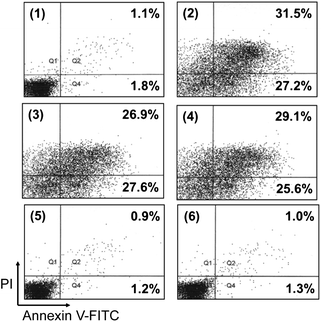 | ||
| Fig. 2 Contour diagram of Annexin V-FITC/PI flow cytometry of U937 cells after being treated with different H. erinaceus extracts including HWE and MWE. After being treatment with 500 μg mL−1 of each extracts for 48 h, cells were treated with 1 μg of Annexin V-FITC together with 1 μg of PI for 15 min. The lower left quadrants of each panels show the viable cells, which exclude PI and are negative for Annexin V-FITC binding. The upper right quadrants contain the late apoptotic/necrotic cells, positive for Annexin V-FITC binding and for PI uptake. The lower right quadrants represent the early apoptotic cells, Annexin V-FITC positive and PI negative. Treatments: panel 1, untreated control; panel 2, etoposide positive control; panel 3, WHE; panel 4, MWE; panel 5, ACE negative control; panel 6, AKE negative control. The figure represents three independent experiments. | ||
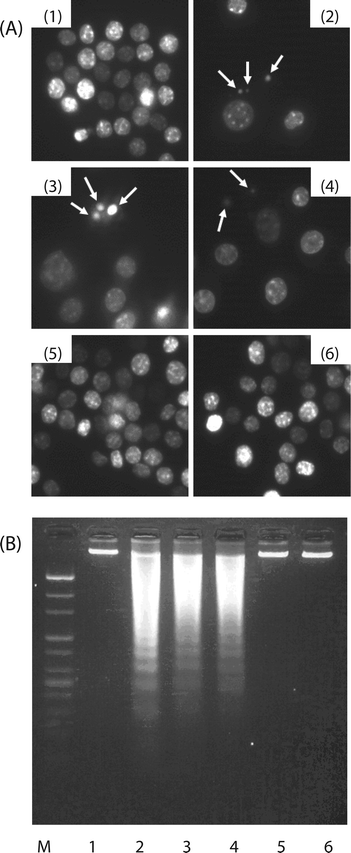 | ||
| Fig. 3 Appearance of apoptotic bodies and loss of chromosomal DNA integrity in U937 cells treated with different H. erinaceus extracts including HWE and MWE. U937 cells were treated with each extract at 500 μg mL−1 for 48 h. (A) H. erinaceus extract-treated cells were fixed and stained with DAPI. Stained nuclei with DAPI solution were photographed with a fluorescent microscope (×400). Arrows indicate apoptotic bodies. (B) U937 cells were treated with different H. erinaceus extracts as described. Chromosomal DNA in cells was then extracted and electrophoresed through 1% agarose gel and visualized by staining with ethidium bromide (EtBr). Treatments: lane 1, untreated control; lane 2, etoposide positive control; lane 3, WHE; lane 4, MWE; lane 5, ACE negative control; lane 6, AKE negative control. The figure represents three independent experiments. | ||
We also measured changes in ratio of hypodiploid cells in treated cells by flow cytometry. As shown in Fig. 4, the sub-G1 DNA contents of U937 cells at 12 h with at 500 μg mL−1 of HWE or MWE were 56.0% and 59.4%, respectively. Hypodiploid cells resulting from HWE and MWE treatments increased in a time-dependent manner. These data demonstrate that HWE and MWE, but not ACE and AKE, are acting as apoptosis inducers in U937 cells.
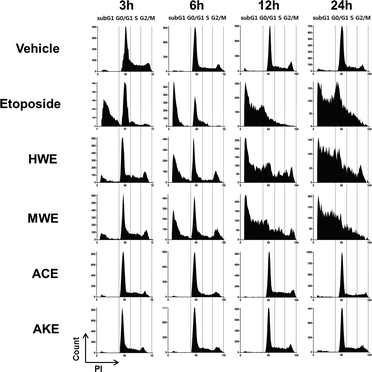 | ||
| Fig. 4 Induction of hypodiploid cells in U937 cells treated with different H. erinaceus extracts including HWE and MWE. U937 cells (1 × 106) were treated with each extract at 500 μg mL−1 for indicated time periods, followed by the treatment with 50 μg mL−1 PI for 5 min. The appearance of hypodiploid cells was detected by flow cytometry at various time intervals after treatment. ACE and AKE were used as the internal negative controls. The figure represents three independent experiments. | ||
Expression of pro-and anti-apoptotic proteins in U937 cells
To examine the apoptotic pathway activated by HWE and MWE, expression of the numerous pro-and anti-apoptotic proteins were analyzed by Western blot assay. Fig. 5 shows that both HWE and MWE markedly inhibited expression of the anti-apoptotic proteins including Bcl-2, Bcl-xL(S), XIAP, and cIAPs, whereas the expression levels of pro-apoptotic, but not XIAP Bad, Bid and Bax remained virtually unchanged. These data suggest that down-regulation of anti-apoptotic proteins, but not up-regulation of pro-apoptotic proteins, is involved in HWE or MWE-induced apoptosis through intrinsic signal pathway.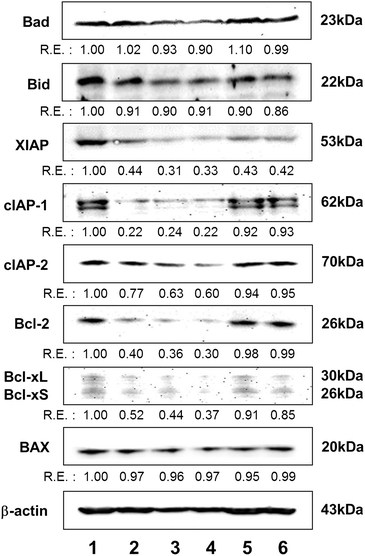 | ||
| Fig. 5 Effects of each H. erinaceus extracts including HWE and MWE on the expression levels of pro- and anti-apoptotic protein members in U937 cells. After incubation with each extract for 48 h, cells were lysed and total cellular proteins (40 μg) were resolved in SDS-polyacrylamide gels, after which they were transferred onto nitrocellulose membranes. The membranes were then probed with the indicated antibodies. Protein bands were visualized using ECL detection system. Each protein was expressed as relative expression (R.E.) value calculated from the target gene/β-actin expression. β-Actin was used as a control for constitutively expressed protein. Treatments: lane 1, untreated control; lane 2, etoposide positive control; lane 3, WHE; lane 4, MWE; lane 5, ACE negative control; lane 6, AKE negative control. The figure represents three independent experiments. | ||
Reduction of MMP and release of cytochrome-c
Fig. 6A shows that treatment of the cells with HWE and MWE led to a significant decline of pro-apoptotic cytochrome-c in mitochondria and concurrent appearance of cytochrome-c in cytosol, suggesting release of cytochrome-c from mitochondria into cytosol. The treatments also significantly reduced their MMP by 62.3 and 59.8%, respectively, levels comparable to those induced by etoposide (Fig. 6B). These data suggest that HWE and MWE induce U937 cell apoptosis through disruption of mitochondrial membrane integrity.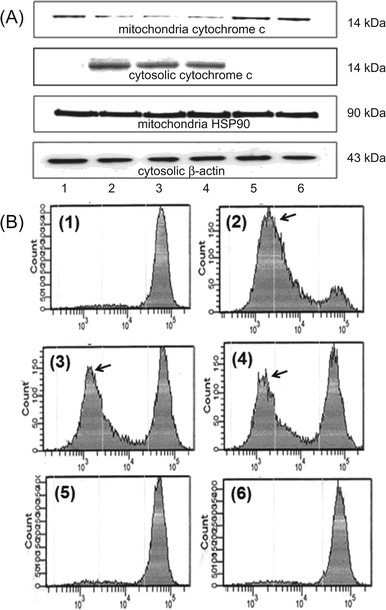 | ||
| Fig. 6 Loss of MMP and release of cytochrome-c induced by treatment with H. erinaceus extracts including HWE and MWE in U937 cells. (A) After incubation for 48 h, equal amounts of cytosolic and mitochondrial proteins were extracted. The proteins were separated by SDS-polyacrylamide gels, and transferred onto nitrocellulose membrane. The membranes were probed with anti-cytochrome-c antibody. β-Actin and HSP90 were included as controls as indicators of markers of cytosolic and mitochondrial protein, respectively, for protein loading. (B) U937 cells were treated with H. erinaceus extracts and stained with 10 μM DiOC6. The mean fluorescence intensity was measured by flow cytometry. Arrows indicate subpopulation of cells with loss of MMP. Treatments: panel 1, untreated control; panel 2, etoposide positive control; panel 3, WHE; panel 4, MWE; panel 5, ACE negative control; panel 6, AKE, negative control. The figure represents three independent experiments. | ||
Activation of caspases
Fig. 7B shows that both HWE and MWE treatments markedly increased activities of caspase-9 (11.8- and 13.0-fold increase, respectively) and caspase-3 (14.8- and 18.3-fold increase, respectively) compared to vehicle-treated group. Caspase-8 activity was also increased. However, the induction ratio of caspase-8 activity was much lower than of caspase-3 and -9 (3.1- and 3.9-fold increase, respectively). Although antigen-binding specificities of anti-caspase-8 and -9 antibodies failed to detect cognate pro-forms, Western blot analysis indicates that degradation of their pro-forms is critical for expression of enzymatic activity of caspase-8, and -9 (initiator caspases) and caspase-3 (effector caspases). Activation of caspase-3 occurred in parallel with proteolytic cleavage of PARP from 116 kDa intact molecules into 85 kDa fragments (Fig. 7A). The induction of HWE- or MWE-mediated apoptosis can be largely attributed to triggering of ‘initiator caspase’ caspase-9.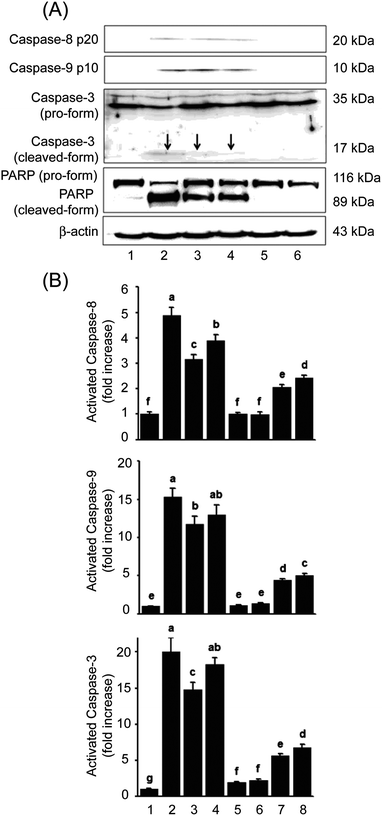 | ||
| Fig. 7 Activation of caspases by treatment with H. erinaceus extracts including HWE and MWE in U937 cells. (A) After incubation for 48 h, equal amounts of total cell proteins were separated on SDS-polyacrylamide gels and transferred onto nitrocellulose membrane. The membranes were probed with antibodies against each protein. β-Actin was included as a control of constitutively expressed protein marker. (B) The enzymatic activities of caspase-3, -8, and -9 in the cell lysates were determined by incubation with specific colorigenic substrates DEVD-pNA, IETD-pNA, and LEHD-pNA. Caspase-3, -8, and -9 peptide inhibitors, DEVD-CHO, IETD- CHO and LEHD- CHO, were incubated with the cell lysates prior to being challenged with HWE or MWE. Treatments: lane 1, untreated control; lane 2, ectopocide positive control; lane 3, WHE; lane 4, MWE; lane 5, ACE negative control; lane 6, AKE negative control; lane 7, WHE in the presence of inhibitor; lane 8, MWE in the presence of inhibitor. The figures are the representative of three independent experiments. Bars not sharing a common letter are significantly different between groups at P < 0.05. | ||
Suppression of induced apoptosis by caspase-3 inhibitor
To confirm that HWE- or MWE-induced apoptosis occurred with effector function of caspase-3 activated mainly by initiator function of caspase-9, cells were pre-incubated with DEVD-CHO, IETD-CHO and LEHD-CHO, inhibitors for caspase-3, -8, and -9, respectively, before the treatment. Fig. 7B shows that pre-treatment with caspase-3 or -9 inhibitors decreased cognate enzymatic activities more effectively than with caspase-8 inhibitor. Activation of caspase-3 via degradation of its pro-form mainly by caspase-9 plays a key role in HWE- and MWE-induced apoptosis.Discussion
Mushroom composition and nutrition
Mushrooms are a good source of good quality proteins, containing 19 to 40% protein on dry wt basis.17 The essential amino acids of most species are found in the same proportion as in eggs. The vitamin content is also high, similar to that of yeast except for thiamin. We found that for the four H. erinaceus extracts, protein, carbohydrate, and mineral (ash) content were all high, ranging (in % of dry wt) from 35.5 to 38.5; 33.8 to 39.5, and 14.6 to 19.0, respectively (Table 1). Moisture content was about 8% and fiber and fat content <1%. Low-fat mushrooms are a good source of essential nutrients and micronutrients.The fact that the water and aqueous ethanol extracts were highly active in all bioassays, whereas the corresponding acid and alkaline extracts with similar proximate composition were inactive suggests that the apoptotic compound(s) is not stable to strong acid or alkaline conditions.18,19
Apoptosis mechanisms - general aspects
Apoptosis occurs according to a regulated process that involves activation of cellular events characterized by cell shrinkage, cell surface expression of phosphatidylserine, chromatin condensation, DNA fragmentation, and cellular fragmentation into apoptotic bodies.20 Phytochemicals can induce apoptosis by both intrinsic (mitochondrial) and extrinsic (death receptor) pathways.21 The intrinsic pathway involves mitochondrial outer-membrane permealization and release of pro-apoptotic factors, whereas the extrinsic pathway involves interactions between the plasma membrane death receptors and the ligand, which then activate the proteases leading to apoptotic cell death. For apoptosis induction, extrinsic and intrinsic pathways share cytochrome-c release and modulation of Bcl-2 family protein expressions in mitochondria.The fact that black rice bran extracts exhibited strong anti-oxidative, anti-inflammatory, and anti-carcinogenic properties22–24 suggests that carcinogenesis may also be caused by oxidative and inflammatory stress on cells mediated by integrated Nrf2, NF-kBm and AP-1-signalling pathways.21 The apoptosis signal is provided to the nucleus by interaction of FasL, tumor necrosis factor α (TNF-α), and tumor necrosis factor-related apoptosis-inducing ligand with their cognate receptors in the membrane and subsequent death receptor activation through adaptor molecules and caspase-8.25
In another pathway, mitochondrial release of cytochrome-c into the cytosol initiates the mitochondria-mediated apoptosis.26 Within the cytosol, cytochrome-c leads to activation of the apoptosis initiator caspase-9 by binding to APAF-1.27 Activated caspase-9 then cleaves an inactive pro-caspase-3 to an active executioner caspase-3.28 Activated caspase-3 then cleaves several substrates for caspase-3 such as poly (ADP-ribose) polymerase (PARP) and D4-GDI. This event is coordinated with DNA fragmentation and morphological changes of target cells.29 In addition, the Bcl-2 protein family participates as key regulators of cell death and functions either as activators (Bax, Bcl-Xs, Bid, and Bad) or as inhibitors [(Bcl-2 and Bcl-XL(S)] in the control of cell death30 leading to tumorigenesis.31
Apoptogenetic mechanisms of mushroom extracts
The present findings indicate that HWE or MWE exerted direct cytotoxic effects against cancer cell growth. Another human T-cell leukemia cell line (Molt-4) was also inhibited at 47% and 44%, respectively, by treatments with 500 μg mL−1 of HWE and MWE (Table 2), However, a murine leukemia cell line (L1210) and normal NIH-3T3 murine fibroblast and HGF-1 human fibroblast cells showed only 5 to 8% decreases following exposure to HWE or MWE (Table 2). Although, we do not know why HWE and MWE preferentially induced death of cells of human origin, one possibility is that selective cancer cell death might be associated with the induction of NO production in target cells.1,5 Several studies showed that selective cytotoxicity is due to low level of antioxidant enzymes in cancer cells.32,33 Such cells offer competitive advantage to formation of toxic reactive oxygen and nitrogen species. If the antioxidant capacity becomes fatally limiting, then oxidative or nitrosative stress may exert cytotoxic effects on cancer but not on normal cells.8| Treatment | Cytotoxicity (%) | ||||
|---|---|---|---|---|---|
| NIH-3T3 | HGF-1 | L1210 | MOLT-4 | U-937 | |
| Vehicle | 0.0 ± 0.7 | 0.0 ± 2.4 | 0.0 ± 1.8 | 0.0 ± 8.9 | 0.0 ± 4.2 |
| HWE | 8.6 ± 1.7 | 5.8 ± 1.6 | 8.8 ± 4.7 | 47.1 ± 4.8 | 67.9 ± 1.4 |
| MWE | 8.7 ± 2.0 | 4.5 ± 2.2 | 8.8 ± 4.0 | 44.2 ± 0.8 | 69.6 ± 1.3 |
| ACE | 1.3 ± 1.4 | 0.5 ± 0.7 | 1.7 ± 3.0 | 6.2 ± 8.6 | 5.4 ± 0.4 |
| AKE | 5.1 ± 2.9 | 2.5 ± 5.3 | 5.6 ± 0.6 | 12.9 ± 0.9 | 8.4 ± 0.6 |
The following observations reinforce the conclusion that cancer cell-specific cytotoxicity induced by HWE or MWE treatments operates through apoptosis. First, flow cytometry on Annexin V-labeled cells showed a rapid translocation and accumulation of membrane phosphatidylserine from the inner to the outer side of membrane leaflet. Second, the use of treated cells labeled with DAPI revealed the appearance of nuclei with condensed chromatin and apoptotic bodies. Third, the treatments caused digestion of genomic DNA into DNA ladders of ∼200 base-pairs. Fourth, flow cytometry of DNA profiles of treated cells showed increased accumulation of hypodiploid cells in sub-G1 phase. Finally, the cell cycle arrest pattern of these cells was similar to that of etoposide-treated control.
One objective of the present study was to find out whether HWE- or MWE-induced apoptosis is associated with down-regulation of Bcl-2, Bcl-xL and Bcl-xS as well as other anti-apoptotic proteins, including XIAP and cIAPs.21 As expected, the treatments induced marked down-regulation of anti-apoptotic Bcl-2 family proteins. By contrast, pro-apoptotic Bcl-2 family protein levels, including Bad, Bid and Bax, were only marginally altered. The treatments also increased the ratio of [pro-apoptotic protein]/[anti-apoptotic proteins] and induced the loss of MMP, leading to apoptosis due to mitochondrial dysfunction.30
Although chemical heterogeneity of each H. erinaceus extract examined makes it difficult to define the precise apoptotic signal pathway, the present data provides evidence for predominant involvement of mitochondrial pathway through activation of “initiator” caspase-9. The data rule out the possibility that both HWE and MHE induce death receptor-mediated apoptosis because of failure to detect cleaved product of Bid and substantially low levels of caspase-8 activity relative to that of caspase-9.34
Based on cited results, we propose that HWE- and MHE-triggered mitochondrial apoptotic pathway activates “initiator” caspase-9, which in turn activates caspase-3. The HWE and MHW induced translocation of cytochrome-c release from mitochondria into cytosol is probably due to reduced expression of anti-apoptotic proteins without any significant changes in the pro-apoptotic protein expressions. These events then lead to activation of caspase-9 and -3, together with concurrent cleavage of PARP, but without marked activation of caspase-8. This mechanism is also supported by the lower inhibitory effect of caspase-8 inhibitor on cognate enzyme action compared to inhibition observed with caspase-3, and -9 inhibitors.
Conclusions
The current findings indicate that aqueous and aqueous/ethanol extracts HWE and MHW of H. erinaceus that induced NO production in macrophage cells, strongly suppressed the proliferation of U937, but not normal cells by induction of apoptosis through activation of mitochondria-mediated caspase pathway. The two acidic and alkaline extracts (ACE and AKE), which did not induce NO production, were inactive in all bioassays, suggesting that the active mushroom ingredients are not stable to strong acidic or basic conditions. Our data indicate that both HWE and MWE extracts of H. erinaceus mushrooms increased levels of pro-apoptotic protein (Bax, Bad, and Bid) relative to apoptotic proteins {(Bcl-2, Bcl-xL(S) and cIAPs)}, thereby promoting apoptosis in U937 human leukemia cells. These considerations suggest that the whole mushrooms or active extracts merit further study for possible therapeutic potential against human leukemia. Finally, the present study complements previous observations on the anti-carcinogenic potential of other plant compounds, including tea ingredients35 and potato and tomato glycoalkaloids.36–39Acknowledgements
This work was supported by a grant (No. 107082-03-1-HD110) from Korean Institute of Planning and Evaluation for Technology in Food, Agriculture, Forestry, and Fisheries. We thank Carol E. Levin for assistance with the preparation of the manuscript.References
- S. P. Kim, Y. H. Choi, M. Y. Kang and S. H. Nam, Effects of the extracts by extraction procedures from Hericium erinaceus on activation of macrophage, J. Korean Soc. Appl. Biol. Chem., 2005, 48(3), 285–291 Search PubMed.
- G. M. Sia and J. K. Candlish, Effects of shiitake (Lentinus edodes) extract on human neutrophils and the U937 monocytic cell line, Phytother. Res., 1999, 13(2), 133–137 Search PubMed.
- Y.-H. Gu and M. A. Belury, Selective induction of apoptosis in murine skin carcinoma cells (CH72) by an ethanol extract of Lentinula edodes, Cancer Lett., 2005, 220(1), 21–28 Search PubMed.
- Y. Koyama, T. Suzuki, A. Kajiya and M. Isemura, Involvement of G2/M cell cycle arrest and the mitochondrial pathway in Boletopsis leucomelaena (Pers.) Fayod (agaricomycetideae) lectin-induced apoptosis of human leukemia U937 cells, Int. J. Med. Mushrooms, 2005, 7(1–2), 201–212 Search PubMed.
- J. S. Lee, J. Y. Cho and E. K. Hong, Study on macrophage activation and structural characteristics of purified polysaccharides from the liquid culture broth of Hericium erinaceus, Carbohydr. Polym., 2009, 78(1), 162–168 Search PubMed.
- J. S. Lee and E. K. Hong, Hericium erinaceus enhances doxorubicin-induced apoptosis in human hepatocellular carcinoma cells, Cancer Lett., 2010, 297(2), 144–154 Search PubMed.
- J.-S. Bae, J. W. Park, S. H. Park, J. B. Park, Y.-H. Rho, Y. B. Ryu, K.-S. Lee, K.-H. Park and Y.-S. Bae, Apoptotic cell death of human leukaemia U937 cells by ubiquinone-9 purified from Pleurotus eryngii, Nat. Prod. Res., 2009, 23(12), 1112–1119 Search PubMed.
- J.-H. Jang, K.-H. Noh, J.-N. Choi, K.-S. Jin, J.-H. Shin, J.-H. On, C.-W. Cho, W.-S. Jeong, M.-J. Kim and Y.-S. Song, Effect of Hericium erinaceus mycelia supplementation on the oxidative stress and inflammation processes stimulated by LPS and their mechanisms in BALB/C mice, Journal of the Korean Society of Food Science and Nutrition, 2010, 39(2), 227–236 Search PubMed.
- K. Mori, S. Inatomi, K. Ouchi, Y. Azumi and T. Tuchida, Improving effects of the mushroom Yamabushitake (Hericium erinaceus) on mild cognitive impairment: A double-blind placebo-controlled clinical trial, Phytother. Res., 2009, 23(3), 367–372 Search PubMed.
- K. H. Wong, V. Sabaratnam, N. Abdullah, U. R. Kuppusamy and M. Naidu, Effects of cultivation techniques and processing on antimicrobial and antioxidant activities of Hericium erinaceus (Bull.:Fr.) Pers. extracts, Food Technol. Biotechnol., 2009, 47(1), 47–55 Search PubMed.
- AOAC Official Methods of Analysis, 1990, Washington, DC: Association of Official Analytical Chemists Search PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65(1–2), 55–63 CrossRef CAS.
- I. Vermes, C. Haanen, H. Steffens-Nakken and C. Reutelingsperger, A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V, J. Immunol. Methods, 1995, 184(1), 39–51 CrossRef CAS.
- M. Herrmann, H.-M. Lorenz, R. Voll, M. Grunke, W. Woith and J. R. Kalden, A rapid and simple method for the isolation of apoptotic DNA fragments, Nucleic Acids Res., 1994, 22(24), 5506–5507 Search PubMed.
- S.-C. Shen, Y.-C. Chen, F.-L. Hsu and W.-R. Lee, Differential apoptosis-inducing effect of quercetin and its glycosides in human promyeloleukemic HL-60 cells by alternative activation of the caspase 3 cascade, J. Cell. Biochem., 2003, 89(5), 1044–1055 Search PubMed.
- E. Lee, R. Enomoto, C. Suzuki, M. Ohno, T. Ohashi, A. Miyauchi, E. Tanimoto, K. Maeda, H. Hirano, T. Yokoi and C. Sugahara, Wogonin, a plant flavone, potentiates etoposide-induced apoptosis in cancer cells, Ann. N. Y. Acad. Sci., 2007, 1095(1), 521–526 Search PubMed.
- R. Kurtzman, Mushrooms as a source of food protein, in Protein nutritional quality of foods and feeds. Part II. Quality factors - plant breeding, composition, processing, and antinutrients, M. Friedman, Editor. 1975, Marcel Dekker: New York. p. 305–318 Search PubMed.
- P. Manzi, S. Marconi, A. Aguzzi and L. Pizzoferrato, Commercial mushrooms: Nutritional quality and effect of cooking, Food Chem., 2004, 84(2), 201–206 Search PubMed.
- A. Omarini, C. Henning, J. Ringuelet, J. A. Zygadlo and E. Albertó, Volatile composition and nutritional quality of the edible mushroom Polyporus tenuiculus grown on different agro-industrial waste, Int. J. Food Sci. Technol., 2010, 45(8), 1603–1609 Search PubMed.
- H. Okada and T. W. Mak, Pathways of apoptotic and non-apoptotic death in tumour cells, Nat. Rev. Cancer, 2004, 4(8), 592–603 CrossRef CAS.
- L. Shu, K.-L. Cheung, T. O. Khor, C. Chen and A.-N. Kong, Phytochemicals: Cancer chemoprevention and suppression of tumor onset and metastasis, Cancer Metastasis Rev., 2010, 29(3), 483–502 Search PubMed.
- S. H. Nam, S. P. Choi, M. Y. Kang, N. Kozukue and M. Friedman, Antioxidative, antimutagenic, and anticarcinogenic activities of rice bran extracts in chemical and cell assays, J. Agric. Food Chem., 2005, 53(3), 816–822 Search PubMed.
- S. P. Choi, M. Y. Kang, H. J. Koh, S. H. Nam and M. Friedman, Antiallergic activities of pigmented rice bran extracts in cell assays, J. Food Sci., 2007, 72(9), S719–726 Search PubMed.
- S. P. Choi, S. P. Kim, M. Y. Kang, S. H. Nam and M. Friedman, Protective effects of black rice bran against chemically-induced inflammation of mouse skin, J. Agric. Food Chem., 2010, 58(18), 10007–10015 Search PubMed.
- A. Ashkenazi and V. M. Dixit, Apoptosis control by death and decoy receptors, Curr. Opin. Cell Biol., 1999, 11(2), 255–260 Search PubMed.
- J.-C. Martinou, S. Desagher and B. Antonsson, Cytochrome c release from mitochondria: All or nothing, Nat. Cell Biol., 2000, 2(3), E41–E43 Search PubMed.
- J. Rodriguez and Y. Lazebnik, Caspase-9 and APAF-1 form an active holoenzyme, Genes Dev., 1999, 13(24), 3179–3184 Search PubMed.
- K. Arita, T. Utsumi, A. Kato, T. Kanno, H. Kobuchi, B. Inoue, J. Akiyama and K. Utsumi, Mechanism of dibucaine-induced apoptosis in promyelocytic leukemia cells (HL-60), Biochem. Pharmacol., 2000, 60(7), 905–915 Search PubMed.
- S. D. Ray, G. Balasubramanian, D. Bagchi and C. S. Reddy, Ca2+-calmodulin antagonist chlorpromazine and poly(ADP-ribose) polymerase modulators 4-aminobenzamide and nicotinamide influence hepatic expression of bcl-XL and p53 and protect against acetaminophen-induced programmed and unprogrammed cell death in mice, Free Radical Biol. Med., 2001, 31(3), 277–291 Search PubMed.
- S. Zinkel, A. Gross and E. Yang, BCL2 family in DNA damage and cell cycle control, Cell Death Differ., 2006, 13(8), 1351–1359 Search PubMed.
- Z.-Z. Su, I. V. Lebedeva, R. V. Gopalkrishnan, N. I. Goldstein, C. A. Stein, J. C. Reed, P. Dent and P. B. Fisher, A combinatorial approach for selectively inducing programmed cell death in human pancreatic cancer cells, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(18), 10332–10337 Search PubMed.
- B. Shashi, S. Jaswant, R. J. Madhusudana, S. A. Kumar and Q. G. Nabi, A novel lignan composition from Cedrus deodara induces apoptosis and early nitric oxide generation in human leukemia Molt-4 and HL-60 cells, Nitric Oxide, 2006, 14(1), 72–88 Search PubMed.
- S. Y. Olson and H. J. Garbán, Regulation of apoptosis-related genes by nitric oxide in cancer, Nitric Oxide, 2008, 19(2), 170–176 CrossRef CAS.
- X.-M. Yin, Signal transduction mediated by Bid, a pro-death Bcl-2 family proteins, connects the death receptor and mitochondria apoptosis pathways, Cell Res., 2000, 10(3), 161–167 Search PubMed.
- M. Friedman, B. E. Mackey, H.-J. Kim, I.-S. Lee, K.-R. Lee, S.-U. Lee, E. Kozukue and N. Kozukue, Structure–activity relationships of tea compounds against human cancer cells, J. Agric. Food Chem., 2007, 55(2), 243–253 Search PubMed.
- K.-R. Lee, N. Kozukue, J.-S. Han, J.-H. Park, E.-Y. Chang, E.-J. Baek, J.-S. Chang and M. Friedman, Glycoalkaloids and metabolites Inhibit the growth of human colon (HT29) and liver (HepG2) cancer Ccells, J. Agric. Food Chem., 2004, 52(10), 2832–2839 Search PubMed.
- M. Friedman, K.-R. Lee, H.-J. Kim, I.-S. Lee and N. Kozukue, Anticarcinogenic effects of glycoalkaloids from potatoes against human cervical, liver, lymphoma, and stomach cancer cells, J. Agric. Food Chem., 2005, 53(15), 6162–6169 Search PubMed.
- M. Friedman, T. McQuistan, J. D. Hendricks, C. Pereira and G. S. Bailey, Protective effect of dietary tomatine against dibenzo[a, l]pyrene (DBP)-induced liver and stomach tumors in rainbow trout, Mol. Nutr. Food Res., 2007, 51(12), 1485–1491 Search PubMed.
- M. Friedman, C. E. Levin, S.-U. Lee, H.-J. Kim, I.-S. Lee, J.-O. Byun and N. Kozukue, Tomatine-containing green tomato extracts inhibit growth of human breast, colon, liver, and stomach cancer cells, J. Agric. Food Chem., 2009, 57(13), 5727–5733 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |