Polycyclic aromatic hydrocarbons and organochlorine pesticides in surface soils from the Qinghai-Tibetan plateau†
Shu
Tao
*,
Wentao
Wang
,
Wenxin
Liu
,
Qian
Zuo
,
Xilong
Wang
,
Rong
Wang
,
Bin
Wang
,
Guofeng
Shen
,
Yuanhe
Yang
and
Jin-shen
He
Laboratory for Earth Surface Processes, College of Urban and Environmental Sciences, Peking University, Beijing, 100871, China. E-mail: taos@urban.pku.edu.cn; Fax: +86 10-62751938; Tel: +86 10-62751938
First published on 16th November 2010
Abstract
Eighty eight surface soil samples were collected from the Qinghai-Tibetan Plateau (QTP) for determination of polycyclic aromatic hydrocarbons (PAHs) and organochlorine pesticides (OCPs), including dichlorodiphenyltrichloroethane and metabolites (DDXs) and hexachlorocyclohexane isomers (HCHs). The measured concentrations were 51.8 ± 38.5 ng g−1, 0.329 ± 0.818 ng g−1, and 0.467 ± 0.741 ng g−1 as means and standard deviations of PAHs, DDXs, and HCHs, respectively, which were 1–2 orders of magnitude lower than those reported for eastern China. Significant differences were also revealed among four sub-areas within QTP. PAHs detected in the samples from the remote sub-areas of T’ang-ku-la/Hoh Xil Mountains and along the Qinghai-Tibet highway in the west and northwest of QTP were 1 order of magnitude lower than those from Lhasa and east Qinghai. The differences in soil OCPs among the sub-areas were 2–7 times. Soil PAHs were significantly correlated with emission density and soil organic carbon content (SOC), while OCPs were correlated significantly with the population density and SOC. Based on the calculated backward air mass trajectories and geographical distributions of emission and population, it was revealed that PAHs and OCPs accumulated in the soils in the west and northwest QTP were primarily from long-range transport and may represent the background levels of East Asia. This part of QTP can also serve as an important receptor area for regional or even global long-range transport study. The elevated concentrations of PAHs and OCPs in Lhasa and east Qinghai were mainly from local sources, while PAHs from adjacent Lanzhou area also contributed considerably to the accumulation of PAHs in east Qinghai.
Environmental impactBecause of the unique geographical distribution pattern of population, production and application of OCPs occurred mostly in eastern China and so did energy consumption and PAH emission. Consequentially, the environment in western China is less contaminated than that in eastern China. This is particularly true for Qinghai-Tibetan Plateau (QTP), which covers a vast remote area of approximately 2 million km2 with a mean population density as low as 4.2 people per km2. QTP is often referred as a background area for persistent organic pollutants. In this study, PAHs and OCPs in surface soils from Qinghai-Tibetan Plateau were investigated based on 88 surface soil samples collected in the area. The key hypothesis tested was that this remote area can serve as an important background area as well as a receptor area for regional or even global long-range transport study. It was found that a part of Qinghai-Tibetan Plateau was contaminated by local sources and only the west and southwest part of the area, which received PAHs and OCPs primarily from long-range atmospheric transport, can serve as the background area. |
Introduction
Polycyclic aromatic hydrocarbons (PAHs) and organochlorine pesticides (OCPs), especially hexachlorocyclohexane isomers (HCHs) and dichlorodiphenyltrichloroethane (DDT), are among the most concerning toxic pollutants in China. The annual emission of 16 U.S. Environmental Protection Agency priority PAHs in China was over 110![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons.1 It was estimated that over 4 million tons of HCHs and approximately half a million tons of DDT were historically produced and applied in China.2 As semivolatile and persistent compounds, PAHs and OCPs can migrate to remote areas through long-range atmospheric transport. It has been reported that the ice sheet of Greenland serves as an important geochemical sink of PAHs from remote sources.3 Based on the results of field monitoring and air mass trajectory calculation, it was revealed that PAHs detected at Alert in the high Arctic atmosphere were originally from Eurasia.4
000 tons.1 It was estimated that over 4 million tons of HCHs and approximately half a million tons of DDT were historically produced and applied in China.2 As semivolatile and persistent compounds, PAHs and OCPs can migrate to remote areas through long-range atmospheric transport. It has been reported that the ice sheet of Greenland serves as an important geochemical sink of PAHs from remote sources.3 Based on the results of field monitoring and air mass trajectory calculation, it was revealed that PAHs detected at Alert in the high Arctic atmosphere were originally from Eurasia.4
Because of the unique geographical distribution pattern of population, production and application of OCPs occurred mostly in eastern China and so did energy consumption and PAH emissions.1,2 Consequentially, the environment in western China is less contaminated than that in eastern China. This is particularly true for Qinghai-Tibetan Plateau (QTP), which covers a vast remote area of approximately 2 million km2 with a mean population density as low as 4.2 people per km2.5 The emission density of PAHs in QTP was orders of magnitude lower than that in eastern China and agricultural activities and OCP application in this area were very limited.1,6 Moreover, QTP is well beyond the influence of the East-Asia monsoon and the pollutants generated in east China can hardly migrate to there.7 Geographically, the extensive Himalayan range also forms a barrier that restricts the penetration of southwesterly monsoon air masses from tropical regions.8 Due to these reasons, it was suggested that QTP may serve as a background area for persistent organic pollutants.9
Even though the environment in QTP is less contaminated, PAHs and OCPs in air, soil, water, and sediment have been detected.6,9,10 It was found that mean concentrations of 16 parent PAHs were 50.8 ng m−3 in air in Lhasa and 82.5 ng g−1 in surface soil from Lharu wetland close to Lhasa and the latter was mainly from air-to-soil deposition.10 PAHs in soil samples from Mt Qomolangma area ranged from 168 ng g−1 to 595 ng g−1.9 Concentrations of HCHs, DDT and metabolites (DDXs) detected in the sediments from Lakes Yamzho Yumco (South Tibet) and Co Ngoin (North Tibet), particularly the latter, were significantly higher than those reported in Arctic lake sediment and long-range atmospheric transport from India was suggested to be the major source of the contamination.6
If the contaminants occurring in QTP primarily originated from long-range transport instead of local sources, this area could serve as another sink area, in addition to the Arctic, Greenland and other remote areas, for studying the global cycle of persistent organic pollutants. However, the origins of the observed OCPs and PAHs in QTP were not fully understood. Although the levels were very low in comparison with those observed in eastern China, the reported concentrations in various media varied greatly, implying the possibility of contributions from different sources. The objectives of this study were to investigate the levels, spatial distributions, and possible origins of PAHs, DDXs, and HCHs in surface soils from QTP based on an extensive survey and to test a hypothesis that the surface soils in QTP can serve as background media for East Asia.
Methodology
Soil sampling
41 and 47 surface soil samples (0–10 cm) were collected from QTP (ESI-1) in summers (July 20 – August 18) of 2005 and 2006, respectively. Detailed information on these sites is listed in ESI-2.† The samples were air-dried. After the samples reached dryness, they were ground to pass through a 70 mesh metal sieve and stored in glass bottles with ground glass stoppers immediately. Although the contamination of the soil in the laboratory could not be totally avoided, very low concentrations measured for the soils from several remote sites suggest that the soil samples were not contaminated notably in the laboratory. The 88 sampling sites presented in Fig. 1 were classified into four sub-areas: (1) an area between T’ang-ku-la and Hoh Xil Mountains in the middle of a vast no-man's land; (2) a narrow area along the Qinghai-Tibet highway from Golmud to Dangxiong with a very low population density; (3) Lhasa, the largest city in Tibet, and the surrounding area; and (4) East Qinghai, where over one third of population of QTP lives.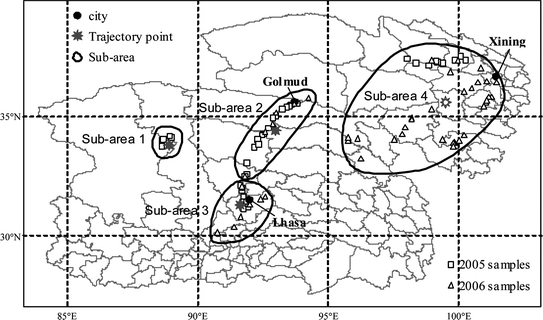 | ||
| Fig. 1 Soil sampling locations. The samples collected in 2005 and 2006 are labeled as squares (2005) and triangles (2006), respectively. The four sub-areas and the corresponding trajectory starting points are also shown. | ||
Reagents
Acetone, n-hexane and dichloromethane of analytical grade (Beijing Reagent, China) were purified by distillation. Granular anhydrous sodium sulfate (Beijing Reagent, China) was heated at 650 °C for 6 h. Silica gel (100–200 mesh, Qingdao Marine Chemical, China) was baked at 450 °C for 4 h and activated at 130 °C for 16 h prior to use. All glassware were cleaned in an ultrasonic cleaner and heated at 400 °C for 6 h.Sample extraction and cleanup
For the samples collected in 2005, a mixture of 10 g of soil sample and 10 g of anhydrous sodium sulfate was packed into a 34 mL extraction cell and extracted with 20 mL mixture of n-hexane and acetone (1:1 v/v) using an accelerated solvent extractor (Dionex ASE300, U.S.) to extract total extractable PAH and OCP residues. The extraction was carried out with a 7 min heating followed by a 5 min static extraction at 140 °C and 105 kg cm−2, and eluted with another 20 mL of the mixed solvent. For the samples collected in 2006, microwave-assisted extraction was applied using a procedure presented in a previous study.11 In brief, 5 g of soil sample was extracted with 25 mL mixture of n-hexane and acetone (1:1, v/v) in a 55 mL extraction vessel of a Microwave Accelerated Reaction System (CEM MARS Xpress, U.S.) at 110 °C and 1200 W for 20 min. The consistency of the two extraction procedures was checked and discussed in ‘Quality control’. The extract was evaporated to near dryness under reduced pressure at 35 °C with a rotary evaporator. The cleanup of the extract was conducted using a silica gel chromatography column (30 cm × 10 mm i.d.), which was eluted with 25 mL of n-hexane (discarded) and 50 mL mixture of dichloromethane and n-hexane (2:3 v/v) in sequence at a rate of 2 mL min−1. The eluent was concentrated with the rotary evaporator and the final volume was adjusted to approximately 1 mL under a nitrogen stream.
Sample analysis
An Agilent gas chromatograph 6890 coupled with a HP-5 column (30 m × 0.32 mm i.d. × 0.25 μm film thickness) was used. A 63Ni-ECD detector and a HP 5973 Mass Selective Detector (MSD) were used for analysis of OCPs and PAHs, respectively. The samples were injected in splitless mode with a venting time of 0.75 min. For PAHs, the temperature was programmed from 60 °C to 300 °C at a rate of 5 °C min−1, and then held isothermal for 20 min. The MSD was operated at 70 eV and the ion source temperature was 280 °C. Selected ion monitoring mode was performed to detect 15 parent PAHs including acenaphthylene (ACY), acenaphthene (ACE), fluorene (FLO), phenanthrene (PHE), anthracene (ANT), fluoranthene (FLA), pyrene (PYR), benz[a]anthracene (BaA), chrysene (CHR), benzo[b]fluoranthene (BbF), benzo[k]fluoranthene (BkF), benzo[a]pyrene (BaP), dibenz[a,h]anthracene (DahA), benzo[g,h,i]perylene (BghiP), and indeno[1,2,3-cd]pyrene (IcdP). For OCPs, the injector and detector temperatures were 220 °C and 280 °C, respectively. Oven temperature was held at 50 °C for 1 min, increased to 150 °C at 10 °C min−1, then to 240 °C at 3 °C min−1, and maintained for 15 min. The OCPs quantified were p,p′-DDT, p,p′-DDD, p,p′-DDE, o,p′-DDT, o,p′-DDD, o,p′-DDE, α-HCH, β-HCH, γ-HCH, and δ-HCH. Soil organic carbon content (SOC) was determined by a TOC analyzer (TOC-5000A, Shimadzu). The particle size distribution of the soil samples was not measured in this study since spatial distribution of hydrophobic organic contaminants in surface is predominantly controlled by organic matter content.12,13 All results are presented in dry-weight basis.Quality control
Reagent and procedure blanks were included in all test series. Quantification was conducted by internal standard method using 2-fluoro-1,1′-biphenyl and p-terphenyl-d14 for PAHs and 4,4′-dichlorobiphenyl for OCPs (J&K Chemical, U.S.). The detection limits (3 times noise) based on 10 g soil sample were 0.05–0.7 ng g−1 for PAHs, 0.01–0.06 ng g−1 for HCH isomers, and 0.04–0.4 ng g−1 for DDT and metabolites, respectively. The possible degradation of p,p′-DDT in the injector was routinely checked. Method recoveries were determined by spiking the sample media with mixed working standards of PAHs (Chem Service, U.S.) and OCPs (J&K Chemical, U.S.). The recoveries detected were from 63 to 103% for PAHs and 77 to 116% for OCPs. In addition, 20% of the samples were spiked with deuterated PAHs (ACY-d10, ACE-d10, ANT-d10, CHR-d12, and perelyne-d12) and 2,4,5,6-tetrachloro-m-xylene (TCMX) (J&K Chemical, U.S.) as surrogates for PAHs and OCPs, respectively to monitor the extraction and cleanup procedures. The recoveries of the surrogates were from 59 to 94% for the deuterated PAHs and 75% for TCMX. Two replicates were analyzed for each sample and the relative deviation between the replicates was below 30 and 25% for PAHs and OCPs, respectively. Since the samples were collected and analyzed in either 2005 or 2006, using two different methods, the consistency was checked by comparing the results of 8 pairs of the samples collected from the same sites in different years. No significant difference was found between the measurements of the two years (paired t-test, p < 0.01, ESI-3†).Air mass trajectory calculation and data analysis
Backward air mass trajectories were calculated to probe the potential sources of PAHs and OCPs. Four sites (34.0°N, 88.7°E; 34.7°N, 92.9°E; 31.49°N, 91.9°E, and 35.6°N, 99.5°E, Fig. 1) at the centers of the four sub-areas were selected as the starting points. Ten-day backward trajectories starting at 200 m above the ground were calculated using HYSPLIT 4.8 with NCAR/NCEP re-analysis global meteorological data at 24 h intervals for 10 years from January 1, 1996 to December 31, 2005 to represent the air circulation in this area in general.14 The calculated trajectories for each sub-area were clustered into 3 or 4 categories with the total spatial variance below 25%. Statistica (v5.5, StatSoft) was applied for hypothesis tests with a significant level of 0.05. The measured results are generally presented in arithmetic means and standard deviations.Results and discussion
Concentrations of PAHs and OCPs in the surface soils
The means and standard deviations of PAHs, HCH isomers, DDT and metabolites are tabulated in the ESI-4.† In brief, the geometric and arithmetic means and standard deviations (n = 88) of PAHs were 41.0 ± 2.04 ng g−1 and 51.8 ± 38.5 ng g−1, respectively. Qi et al. measured PAHs in soils collected from Lharu wetland close to Lhasa and found similar results with arithmetic mean concentration of 82.5 ng g−1.10 These values were considerably lower than those reported for eastern China.15,16 Similarly, the observed geometric and arithmetic means and standard deviations (n = 88) were 0.182 ± 2.69 ng g−1 and 0.329 ± 0.818 ng g−1 for HCHs and 0.211 ± 3.75 ng g−1 and 0.467 ± 0.741 ng g−1 for DDXs, which were 1 order of magnitude lower than the levels found in eastern China.17,18,19Fig. 2 presents the levels of individual compounds of PAHs and OCPs as arithmetic means and standard deviations in log-scale. The recently reported data for North China Plain (NCP) are also shown in the figure for comparison (n = 303).20,21 The total concentrations of all PAHs in QTP were lower than those in NCP (552 ± 622 ng g−1), with the difference as high as 2 orders of magnitude for 4–6 ring compounds from BaA to BghiP and around 1 order of magnitude for 3-ring compounds. According to the emission inventory of PAHs in China, the composition profiles of PAHs emitted in QTP and NCP were similar to each other (ESI-5†), while the emission density was 0.51 kg m−2 y−1 in QTP compared to 39 kg m−2 y−1 in NCP.22 Low molecular weight PAHs are predominately in the gaseous phase while the high molecular weight compounds are mainly associated with particulates.23 The latter are more readily removed from the air through deposition.24 Therefore, the relatively high accumulation rates of high molecular weight PAHs in NCP can be explained by faster transport from air to soil, and a resulting lower potential for long-range atmospheric transport. Among the individual OCPs, the differences of p,p′-DDT and metabolites between QTP and NCP (51 ± 250 ng g−1 as p,p′-DDXs) were much larger than those of HCH isomers (1.3 ± 5.5 ng g−1 as HCHs in NCP), likely due to the difference in historical applications.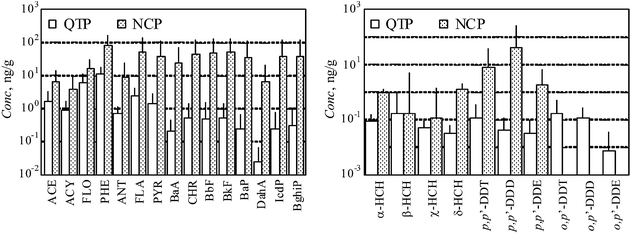 | ||
| Fig. 2 Comparison of individual PAHs, HCH isomers, and DDT metabolites in surface soils between the samples collected from QTP (n = 88) and North China Plain (n = 303).18 Data for o,p′-DDT and metabolites in NCP are not available. | ||
Geographical distributions of PAHs and OCPs
The total PAHs in the soils from the four sub-areas were 19 ± 8.6 ng g−1, 25 ± 11 ng g−1, 58 ± 31 ng g−1, and 66 ± 44 ng g−1, respectively. The levels of soil PAHs in the sub-areas 3 and 4 were significantly higher than those in the sub-areas 1 and 2 (log-transformed, ANOVA and multiple comparison, p < 0.01). In a global scale study on background soils, Nam et al. found the influences of population density and SOC (and black carbon) on soil PAHs and referred these as either source (population) or sink (SOC) related factors.13 A similar phenomenon was revealed in this study. Fig. 3 illustrates the similarity in spatial distribution patterns of SOC (left panel), soil PAHs (middle panel), and PAH emission density (right panel). Significant correlations (log-transformed, p < 0.01) between soil PAHs and SOC (r = 0.429) and between soil PAHs and emission density (r = 0.705) for the whole data set were revealed. Although PAHs may travel long distances in air, it was reported that soils close to emission sources were often heavily contaminated due to air-to-surface deposition.24,25 The correlation between PAHs and SOC can be explained by the following reason: high sorption affinity of the hydrophobic PAHs to SOC and sequestration of PAHs in fine structure of SOC can lead to accumulation of PAHs in soil with high SOC.13 Such a non-emission related interpretation was supported by the fact that within each of the four sub-areas, there were significant correlations (log-transformed, p < 0.01) between PAHs and SOC, but not between PAHs and emission density.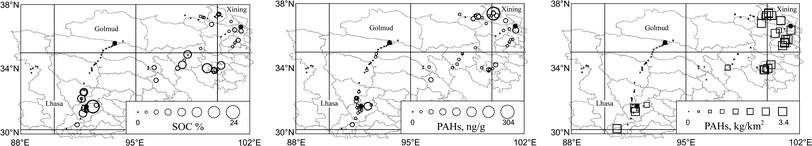 | ||
| Fig. 3 Geographical distributions of SOC, PAHs, and PAH emission density in QTP. The emission data were derived from a county-resolution emission inventory.20 | ||
Based on the data collected, a regression model was developed to predict PAHs using SOC based on the whole data set. Since SOC data are often readily available, this model can provide a rough estimation of soil PAHs in this area. Both models I (it was assumed that there was no variability in the independent variable) and II (variability in the independent variable was assumed) regressions were tested and it was found that the latter could provide a much better fitting to the data.26 After log-transformation, the regression model accounted for more than 47% of the total variation in soil PAHs (ESI-6†).
Significant difference in OCPs among the four sub-areas was also demonstrated (p < 0.01). HCHs were 0.091 ± 0.050 ng g−1, 0.11 ± 0.075 ng g−1, 0.22 ± 0.22 ng g−1, and 0.51 ± 1.1 ng g−1 and DDXs were 0.096 ± 0.097 ng g−1, 0.12 ± 0.15 ng g−1, 0.38 ± 0.76 ng g−1, and 0.72 ± 0.85 ng g−1 in the soils from the sub-areas 1 to 4, respectively. Similar to PAHs, the levels of OCPs in the sub-areas 3 and 4 were significantly higher than those in the other two sub-areas (log-transformed, ANOVA and multiple comparison, p < 0.01). The geographical distributions of HCHs (left panel) and DDXs (middle panel) are presented in Fig. 4. After log-transformation, both HCHs and DDXs were correlated significantly (p < 0.01) with SOC and population density. Without spatially resolved emission inventory, it has been anticipated that the emission of OCPs from their historical application was proportional to population. Positive correlations between OCPs and SOC were also significant within three out of the four classified sub-areas, within which population densities were relative uniform. The positive correlation between soil OCPs and SOC was reported in other areas as well,27 likely due to the same reason for PAHs. Linear regression models were also developed to provide a tool for a rough estimation of HCHs and DDXs in the surface soil of QTP based on the whole data set (ESI-7†) and the coefficients of determination were 0.397 and 0.267 for HCHs and DDXs, respectively.
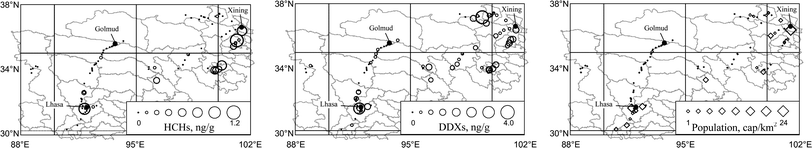 | ||
| Fig. 4 Geographical distributions of soil HCHs, soil DDXs, and population density in QTP. | ||
Origins of PAHs and OCPs
Both PAHs and OCPs are persistent and have potential for long-range transport.4,15,28,29 As the so-called “the third pole” on the earth, QTP is a remote area with limited human activities. As such, it is interesting to know if the observed PAHs and OCPs accumulated in the surface soils there originated from local emission or from remote sources via long-range atmospheric transport. If the latter is dominant, QTP can serve as a background area for regional soil pollution survey or for global cycling studies.As previously discussed, the observed PAH composition in the surface soil from QTP was characterized by a relatively low fraction of high molecular weight compounds, which was very different from that in eastern China (Fig. 2). When the four sub-areas were examined individually, the contributions of high molecular weight PAHs to the composition profile were even lower in the sub-areas 1 and 2 (ESI-8†). Such a pattern was evident in a plot (Fig. 5) of the factor score of the first principle component (F1) against the factor score of the second one (F2), derived from a principle component analysis using the observed PAHs in the surface soils from both QTP and NCP.21 With only a few exceptions, data points from QTP are well distinguished from those from NCP. Both F1 and F2 or at least one factor score of the soils from NCP were greater than 0, suggesting that the soils were relatively rich in either high or intermediate molecular weight PAHs according to the factor loadings as presented in the ESI-9.† In contrast, both F1 and F2 of most QTP soils were negative, indicating relatively high abundance of low molecular weight compounds. The composition profile of soil PAHs from NCP, characterized by relatively higher percentage of high molecular weight compounds, is similar to those observed in a number of other places in China.15,16 On the other hand, relatively low levels of high molecular weight PAHs were also reported for soils collected from Lharu wetland close to Lhasa and from east QTP.10,30
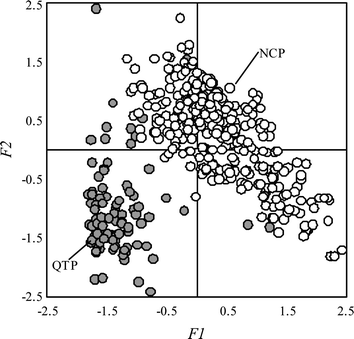 | ||
| Fig. 5 Plot of factor score 1 (F1) against factor score 2 (F2) based on a principal component analysis using the measured PAH concentrations in 88 soils from QTP (shaded) and 303 soils from NCP (open).19 | ||
Theoretically, two possible factors were responsible for the difference in PAH composition between QTP and NCP: (1) the emission sources were different; and (2) PAHs observed in QTP were largely from long-range transport and the composition altered on the way of the atmospheric migration. According to the PAH emission inventory at provincial level in China, emission compositions from the two areas were not significantly different (ESI-5†).22 Therefore, the relative accumulation of low molecular weight PAHs in the surface soils in QTP was likely due to the preferential removal of high molecular weight compounds during the atmospheric transport. Although the observed PAH composition in the environment depends on their sources originally, changes in relative abundance of various compounds on the way from sources to sinks were evident. For example, it was found that ANT degraded faster than PHE, leading to a reduced ANT/PHE isomer ratio in pine needles compared with that of emission sources.31 Schauer et al. proposed coefficients of fraction for individual PAHs, representing differential losses due to gravitational settling, chemical transformation and evaporation of PAHs in airborne particles.32 Based on multimedia modeling, significant differences in behaviors of paired PAH isomers and dramatic changes in PAH composition profiles from sources to sinks were quantitatively elaborated.33,34
To trace possible origins of the PAHs, backward air mass trajectories were calculated for each of the four sub-areas. The calculated daily trajectories for each starting point for 10 years were clustered into 3 to 4 classes and the results are presented as the mean trajectories of various classes in ESI-10.† Most air masses reaching QTP were from west and several exceptions were 42.5%, 34.8%, 30.0% of air masses from north, south and east to the sub-areas 2, 3, and 4, respectively. For the sub-areas 1 and 2, there were almost no local human activity and the air masses reached there passed a vast mountainous area without anthropogenic PAH input. The trace amount of PAHs in the surface soil there was most likely from remote sources through long-range atmospheric transport. In contrast, soil PAHs in the sub-area 3 was significantly higher than those from the sub-areas 1 and 2. Since two third of the air masses reached the sub-area 3 came from the west, the relatively higher PAH concentrations observed in the sub-area 3 should be mainly from local emission. Air masses from the south could bring in pollutants from India. However, considering the large distance, such influence was unlikely to be dominant. It should be noted that the sub-area 3 is the most populated area in Tibet and relatively high PAH levels in air and soil were previously reported.10 For the sub-area 4, relatively high population density, subsequently higher local emission, contributed partially to the accumulation of PAHs in the surface soil, which was significantly higher than those in the sub-areas 1 and 2. In addition, 30% of the air masses to the sub-area 4 were from Lanzhou and surrounding areas, where both population and PAH emission densities were much higher than those in QTP,35 providing additional input. This finding was further supported by the seasonality of air mass movement as shown on the right bottom corner of ESI-10† (the seasonal variations of air mass movement for the other three sub-areas are presented in the ESI-11†). In winter, when emissions of PAHs were significantly higher than those in the other seasons due to indoor heating,36 almost all air masses reached the four sub-areas were from west. Therefore, significant difference in the input from outside of the sub-areas was not expected.
Without detailed information on emission inventory, it is relatively difficult to identify the sources of OCPs. Still, the spatial distribution patterns suggest that the origins of OCPs in the surface soil were similar to those of PAHs. Without local input, the trace amounts of OCPs detected in the sub-areas 1 and 2 could only be from long-rang transport. For the sub-areas 3 and 4, local input should be one source, most likely the major source, of OCPs in the surface soil, leading to a significantly higher level of OCPs than those in the sub-areas 1 and 2.
Wang et al. reported that the total concentrations of 12 PAHs in surface soils from Mt. Qomolangma area of QTP ranged from 168 ng g−1 to 595 ng g−1 and they proposed that such levels can serve as the soil background values for mid-latitude northern hemisphere.9 According to our results, however, the background levels of PAHs in surface soils should be 13 ± 6.9 ng g−1 as represented by the mean and standard deviation derived from the sub-areas 1 and 2, which is lower than the reported background concentrations of 65 ng g−1 (also 15 PAHs) derived from 11 samples mostly collected from South and Southeast Asia.13 Similarly, the background concentrations of HCHs and DDXs are 0.10 ± 0.07 ng g−1 and 0.11 ± 0.14 ng g−1, respectively. It appears that the vast no-man's land in west QTP, rather than the entire QTP, can serve as a remote sink area for investigating long-range atmospheric transport of persistent organic pollutants in East Asia even Northern Hemisphere.
Conclusions
The measured concentrations of PAHs, DDXs, and HCHs were orders of magnitude lower than those reported for eastern China and there were significant differences among four sub-areas within QTP. Soil PAHs were correlated with emission density and SOC, while soil OCPs were population density and SOC dependent. PAHs and OCPs accumulated in the soils in the west and northwest QTP were primarily from long-range transport and may represent the background levels of East Asia.Acknowledgements
The funding of this study was provided by the National Basic Research Program (2007CB407301), the High Technology Research and Development Program of China (2007AA06Z408), and the National Science Foundation of China (Grant 40730737 and 140710019001). We thank Ms Julie Layshock for polishing the English of the manuscript.Notes and references
- Y. X. Zhang, H. Dou, B. Chang, Z. C. Wei, W. X. Qiu, S. Z. Liu, Y. Liu, W. X. Liu and S. Tao, Ann. N. Y. Acad. Sci., 2008, 1140, 218–227 CrossRef CAS.
- Y. F. Li and D. C. Li, Global Emission Inventories for Selected Organochlorine Pesticides. Internal Report, Meteorological Service of Canada, Environment Canada, Toronto, Canada, 2004 Search PubMed.
- P. Masclet, V. Hoyau, J. L. Jaffrezo and H. Cachier, Atmos. Environ., 2000, 34, 3195–3207 CrossRef CAS.
- C. J. Halsall, L. A. Barrie, P. Fellin, D. C. G. Muir, B. N. Billeck, L. Lockhart, F. Y. Rovinsky, E. Y. Kononov and B. Pastukhov, Environ. Sci. Technol., 1997, 31, 3593–3599 CrossRef CAS.
- National Bureau of Statistics, China Energy Statistical Yearbook (2004 editions), China Statistics Press, Beijing, 2004 Search PubMed.
- W. L. Zhang, G. Zhang, S. H. Qi and P. A. Peng, Geochimica, 2003, 32, 363–367 Search PubMed.
- W. D. Zhang, China Climate, China Meteorological Press, Beijing, 1991 Search PubMed.
- A. B. Shrestha, C. P. Wake, J. E. Dibb, P. A. Mayewski, S. I. Whitlow, G. R. Carmichael and M. Ferm, Atmos. Environ., 2000, 34, 3349–3363 CrossRef CAS.
- X. P. Wang, T. D. Yao, Z. Y. Gong, X. L. Yang, S. C. Kang and Y. Zhang, Chin. Sci. Bull., 2007, 52, 1405–1413 CrossRef CAS.
- S. H. Qi, G. Zhang, J. H. Liu and W. L. Zhang, China Environ. Sci., 2003, 23, 349–352 Search PubMed.
- W. T. Wang, B. J. Meng, X. X. Lu, Y. Liu and S. Tao, Anal. Chim. Acta, 2007, 602, 211–222 CrossRef CAS.
- Z. M. Gong, F. L. Xu, R. Dawson, J. Cao, W. X. Liu, B. G. Li, W. R. Shen, W. J. Zhang, B. P. Qin, R. Sun and S. Tao, Arch. Environ. Contam. Toxicol., 2004, 46, 432–437 CAS.
- J. J. Nam, A. J. Sweetman and K. C. Jones, J. Environ. Monit., 2009, 11, 45–48 RSC.
- R. R. Draxler and G. D. Rolph , HYSPLIT (HYbrid Single-Particle Lagrangian Integrated Trajectory) Model, access via NOAA ARL READY website (http://www.arl.noaa.gov/ready/hysplit4.html), 2003, NOAA Air Resources Laboratory, Silver Spring, MD Search PubMed.
- J. Z. Ni, Y. M. Luo, R. Wei and X. H. Li, Eur. J. Soil Sci., 2008, 59, 1020–1026 CrossRef CAS.
- K. Y. Wang, Y. T. Shen, S. C. Zhang, Y. B. Ye, Q. Shen, J. D. Hu and X. J. Wang, Environ. Geol., 2008, 56, 1041–1050.
- F. Wang, X. Jiang, Y. R. Bian, F. X. Yao, H. J. Gao, G. F. Yu, J. C. Munch and R. Schroll, J. Environ. Sci. China, 2007, 19, 584–590 Search PubMed.
- X. X. Ma, Y. Ran, J. Gong and M. Y. Zou, Environ. Monit. Assess., 2007, 145, 453–464.
- Z. Wang, J. W. Chen, P. Yang, F. L. Tian, X. L. Qiao, H. T. Bian and L. K. Ge, Environ. Sci. Technol., 2009, 43, 1336–1341 CrossRef CAS.
- Y. Li, Residue of Organochlorine Pesticides in Soils from Hebei, China, MS Thesis, Peking University, Beijing, 2006 Search PubMed.
- Q. Zuo, PAHs in Surface Soils from the Western Watershed of Bohai Sea, China, Ph.D. Dissertation, Peking University, Beijing, 2007 Search PubMed.
- Y. X. Zhang, S. Tao, J. Cao and R. M. Coveney, Environ. Sci. Technol., 2007, 41, 683–687 CrossRef CAS.
- T. Harner, T. F. Bidleman and J. Chem, J. Chem. Eng. Data, 1998, 43, 40–46 CrossRef CAS.
- S. C. Zhang, W. Zhang, Y. T. Shen, K. Y. Wang, L. W. Hu and X. J. Wang, Atmos. Res., 2008, 89, 138–148 CrossRef CAS.
- M. A. Naeth and S. R. Wilkinson, Can. J. Environ. Qual., 2008, 37, 1675–1684 Search PubMed.
- R. R. Sokal and F. J. Rohlf, Biometry, W.H. Freeman and Co, San Francisco, 1981 Search PubMed.
- P. B. Kurt-Karakus, T. F. Bidleman and K. C. Jones, Environ. Sci. Technol., 2005, 39, 8671–8677 CrossRef CAS.
- J. Y. Lee, Y. P. Kim, C. H. Kang, Y. S. Ghim, N. Kaneyasu and J. Geophys, J. Geophys. Res., 2006, 111, D11303 CrossRef.
- T. Primbs, S. L. Simonich, D. Schmedding, G. Wilson, D. Jaffe, A. Takami, S. Kato, S. Hatakeyama and Y. Kajii, Environ. Sci. Technol., 2007, 41, 3551–3558 CrossRef CAS.
- N. Sun, C. G. Lu, X. Gao, C. L. Li and L. M. Chen, Environ. Sci., 2007, 28, 565–569 Search PubMed.
- H. M. Hwang, T. L. Wade and J. L. Sericano, Atmos. Environ., 2003, 37, 2259–2267 CrossRef CAS.
- J. J. Schauer, W. F. Rogge, L. M. Hildemann, M. A. Mazurek and G. R. Cass, Atmos. Environ., 1996, 30, 3837–3855 CrossRef CAS.
- X. L. Zhang, S. Tao, W. X. Liu, Y. Yang, Q. Zuo and S. Z. Liu, Environ. Sci. Technol, 2005, 32, 9190–9114.
- C. Lang, S. Tao, X. J. Wang, G. Zhang and J. M. Fu, Environ. Toxicol. Chem., 2008, 27, 1–9 CrossRef.
- W. Y. Zhao, M. Han, S. G. Dai, J. Xu and P. Wang, Chemosphere, 2006, 62, 1623–1629 CrossRef CAS.
- D. G. Wang, M. Yang, H. L. Jia, L. Zhou and Y. F. Li, J. Environ. Monit., 2008, 10, 1076–1083 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1. Study area; 2. Sampling site; 3. Comparison between the paired samples collected in 2005 and 2006; 4. The measured concentrations of PAHs and OCPs; 5. Composition profiles of emission inventories of PAHs in QTP and NCP; 6. Relationship between PAHs and SOC; 7. Relationship between OCPs and SOC; 8. Composition profiles of PAHs in the soils from the four sub-areas; 9. Factor loadings of the principle component analysis; 10. Air mass trajectories; 11. Seasonal variations of air mass trajectories. See DOI: 10.1039/c0em00298d |
| This journal is © The Royal Society of Chemistry 2011 |