Indoor and outdoor concentrations of fine particles, particle-bound PAHs and volatile organic compounds in Kaunas, Lithuania
Linas
Kliucininkas
*a,
Dainius
Martuzevicius
a,
Edvinas
Krugly
a,
Tadas
Prasauskas
a,
Violeta
Kauneliene
a,
Peter
Molnar
b and
Bo
Strandberg
b
aDepartment of Environmental Engineering, Kaunas University of Technology, Radvilenu pl. 19, LT50254, Kaunas, Lithuania. E-mail: linas.kliucininkas@ktu.lt
bOccupational and Environmental Medicine, Sahlgrenska Academy at the University of Gothenburg, Box 414, SE-40530, Goteborg, Sweden
First published on 16th November 2010
Abstract
This complex study presents indoor and outdoor levels of air-borne fine particles, particle-bound PAHs and VOCs at two urban locations in the city of Kaunas, Lithuania, and considers possible sources of pollution. Two sampling campaigns were performed in January–February and March–April 2009. The mean outdoor PM2.5 concentration at Location 1 in winter was 34.5 ± 15.2 µg m−3 while in spring it was 24.7 ± 12.2 µg m−3; at Location 2 the corresponding values were 36.7 ± 21.7 and 22.4 ± 19.4 µg m−3, respectively. In general there was little difference between the PM concentrations at Locations 1 and 2. PM2.5 concentrations were lower during the spring sampling campaign. These PM concentrations were similar to those in many other European cities; however, the levels of most PAHs analysed were notably higher. The mean sum PAH concentrations at Locations 1 and 2 in the winter campaign were 75.1 ± 32.7 and 32.7 ± 11.8 ng m−3, respectively. These differences are greater than expected from the difference in traffic intensity at the two sites, suggesting that there is another significant source of PAH emissions at Location 1 in addition to the traffic. The low observed indoor/outdoor (I/O) ratios indicate that PAH emissions at the locations studied arise primarily from outdoor sources. The buildings at both locations have old windows with wooden frames that are fairly permissive in terms of air circulation. VOC concentrations were mostly low and comparable to those reported from Sweden. The mean outdoor concentrations of VOC's were: 0.7 ± 0.2, 3.0 ± 0.8, 0.5 ± 0.2, 3.5 ± 0.3, and 0.2 ± 0.1 µg m−3, for benzene, toluene, ethylbenzene, sum of m-, p-, o-xylenes, and naphthalene, respectively. Higher concentrations of VOCs were observed during the winter campaign, possibly due to slower dispersion, slower chemical transformations and/or the lengthy “cold start” period required by vehicles in the wintertime. A trajectory analysis showed that air masses coming from Eastern Europe carried significantly higher levels of PM2.5 compared to masses from other regions, but the PAHs within the PM2.5 are of local origin. It has been suggested that street dust, widely used for winter sanding activities in Eastern and Central European countries, may act not only as a source of PM, but also as source of particle-bound PAHs. Other potential sources include vehicle exhaust, domestic heating and long-range transport.
Environmental impactThe paper addresses the issue of urban air quality, which becomes an increasingly important topic with tightening air quality requirements. Ambient air quality depends on number of factors and pollution sources, which often cannot be easily identified and managed. A complex urban air quality assessment study was conducted focusing on three key groups of urban air pollutants: particulate matter, PM-bound PAHs, and VOCs, estimating both local sources of pollution and contribution by a long distance transport. The paper reports information about the levels and distribution of the above compounds from the Baltic States, where such type of information is scarce. Our findings contribute to the sustainable management of the cities in the region as well as Europe and worldwide. |
Introduction
Particulate matter (PM) in the ambient air has been the subject of numerous studies and PM levels have been routinely monitored for many years. Recently, attention has focused on fine particles (PM2.5) due to their deep penetration into the respiratory system. The association of fine particles with mutagenic and carcinogenic compounds such as polycyclic aromatic hydrocarbons (PAHs) may contribute to acute health effects and potentially result in long-term health risks.1 PAHs are products of incomplete combustion and are often generated through open burning, incineration, industrial power generation, and vehicle emissions. Among other pollution sources surface depositions on roads/highways contain many toxic micropollutants such as heavy metals and PAHs.2,3 Typical outdoor concentrations of benzo(a)pyrene as reported in Duisburg, Amsterdam and Helsinki range from ca. 0.1 to 1 ng m−3.4 However, notably higher BaP outdoor concentration has been reported in Prague and Zagreb (3.0 and 3.2 ng m−3, respectively).4,5 Diverse PAHs, with varying properties and origins, may be adsorbed to particular matter, and increasing attention has been paid to the profiles in environmental samples.1,4,6–10 In fact, it has been well-documented that indoor PAH concentrations are influenced by both indoor and outdoor sources.11,12In recent decades, much concern about indoor and outdoor air quality has also focused on volatile organic compounds (VOCs), such as benzene, ethylbenzene, toluene, and xylenes (BETXs).13–17 This is because VOCs can readily enter the body via inhalation. Consequently, long-term exposure to VOCs presents a substantial health risk, even at low concentrations.17 In economically developed countries the main outdoor source of VOCs is road traffic. For instance, studies performed in Denmark in 1997/1998 indicated that benzene emissions from traffic strongly contribute not only to urban concentrations but also to exposure and domestic concentrations.18 This study reports outdoor urban concentrations for benzene, toluene, and the sum of xylenes of 9.6, 25.7, and 27.6 µg m−3, and indoor air concentrations of 3.5, 25.3, and 9.4 µg m−3, respectively. Similarly, a study on air pollution in Germany concluded that in street canyons, high-density traffic is the primary source of BETX compounds in indoor air.15,19
Despite the concern regarding VOCs, data on their levels and distribution in Lithuania are sparse, and to our knowledge limited to information collected in two outdoor measurement campaigns.20 Thus, the aim of this study was to investigate indoor and outdoor levels of PM, particle-bound PAHs and VOCs at two urban locations in Kaunas, Lithuania. In addition to presenting the results, possible sources of these pollutants, such as road traffic, aerosolized street dust and long-range transport are discussed.
Methods
Sampling locations and experimental setup
Two sampling locations in the city of Kaunas (pop. 361![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 930; total area 157 km2), Lithuania, were chosen. Both locations are urban and may be classified as “traffic” sampling sites, since they are located in close proximity to streets with dense traffic. The first location (henceforth referred to as Location 1) was beside a two-lane street (Vyduno al.) with an average daily traffic of over 12000 vehicles, with pronounced peaks during rush hours. This street connects the city centre to outer residential areas and carries a moderate level of bus traffic; approximately 300 diesel-fuelled public buses and 850 diesel-fuelled microbuses (important public transport vehicles in Kaunas) travel along it every day.
930; total area 157 km2), Lithuania, were chosen. Both locations are urban and may be classified as “traffic” sampling sites, since they are located in close proximity to streets with dense traffic. The first location (henceforth referred to as Location 1) was beside a two-lane street (Vyduno al.) with an average daily traffic of over 12000 vehicles, with pronounced peaks during rush hours. This street connects the city centre to outer residential areas and carries a moderate level of bus traffic; approximately 300 diesel-fuelled public buses and 850 diesel-fuelled microbuses (important public transport vehicles in Kaunas) travel along it every day.
The second location (Location 2) was on a one-way three-lane street (Kestucio g.). This street is in the city centre, with an average daily traffic of approx. 19000 vehicles, including 450 diesel public buses and 1820 microbuses. The traffic peaks were less pronounced in this location.
In Location 1 air was sampled at an indoor site inside a room and an outdoor site on the balcony on the 3rd floor of a 5-storey university dormitory, located 12 m away from the street. The room was occupied by two non-smoking students who carried out their routine activities (no thermal aerosol generation was expected). Outdoor air was sampled at Location 2 from a storage facility on the first floor of a university faculty building, 3 m away from the street. The indoor site was an unoccupied room visited only occasionally by sampling crew and other university staff.
In order to estimate seasonal fluctuations in the atmospheric abundance of the two main PM size fractions (PM2.5 and PM10), and the particle-bound PAH content of these fractions, daily samples were taken at the outdoor sites over two-week periods in January and April of 2009 at Location 1, and in February and March of 2009 at Location 2. In addition, the levels of the PM4 fraction and VOCs at both indoor and outdoor sites were monitored in week-long daily sampling campaigns; at Location 1 samples were obtained in January and April of 2009, while at Location 2, samples were taken in February and March of 2009. In each case samples were collected (as described below) over eight hour periods during weekdays.
PM sampling and concentration measurement
Air-borne particulate matter was collected on glass microfiber filters (GF/A, Whatman International Ltd., Maidstone, UK; diameter 25 mm, pore size 1.6 µm) over a sampling period of 8 hours. Before use, the microfiber filters were heated in an oven for 4 hours at 500 °C and conditioned for 24 hours at 20 ± 1 °C before and after sampling. Finally, PM mass concentrations (µg m−3) were obtained from the gravimetric analysis of samples, using a MXA5 microbalance (Radwag, Poland).The PM10 and PM2.5 fractions were separated at an air sampling flow rate of 16.7 l min−1 (1 m3 h−1) by cyclones (URG Corp, Chapel Hill, NC, USA) with appropriate cut-off sizes, and separate rotary vane vacuum pumps equipped with flow-meters. To minimise indoor noise nuisance for residents, the PM4 fraction was sampled using a flow rate of 2.2 l min−1 (0.13 m3 h−1) by SKC Conductive Plastic Cyclones for respirable dust sampling with personal sampling pumps.
PAH analysis
After gravimetric analysis the filters were shipped to the Sahlgrenska Academy at the University of Gothenburg, Sweden, for PAH analysis. Deposited particles were extracted from the filters by means of a Sonica ultrasonic extractor (Soltec Srl, Italy) in 3 mL of dichloromethane for 10 min. Before extraction 40 µL of an internal standard mixture containing the 16 deuterated US EPA priority PAHs at a concentration of 1.0 ng µL−1 were added to the samples. The extract solutions were filtered through Pasteur pipettes filled with Na2SO4, then concentrated by purging with ultra-pure nitrogen gas to 200–300 µL. Prior to analysis a recovery estimation standard (octachloronaphthalene) was added, and the excess solvent was evaporated to the final volume of 20–30 µL. PAHs were then separated and detected by means of high-resolution gas chromatography/low resolution mass spectrometry (HRGC/LRMS). The MS instrument (a 5973 model connected to a 6890N GC, both made by Agilent Technologies, Inc., Santa Clara, CA, USA) was operated in electron impact (EI) ionization, selected ion monitoring (SIM) mode. The GC column was a non-polar capillary column (60 m × 0.32 mm id and 0.25 µm film thickness; J&W DB-5, Folsom, USA). The SIM descriptors included the most abundant ion of each native compound and the 2H-labeled PAH standards. In total, the concentrations of 32 PAHs were determined. It should be noted that although a relatively low flow rate was used in this study, the levels of the more volatile PAH (those with 2–4 rings) may have been somewhat underestimated due to losses from the particles during sampling.VOC sampling and analysis
VOCs were actively sampled using Perkin Elmer (PE) tube samplers containing 300 mg Tenax TA and a flow rate of 9.9 ± 0.4 mL min−1. After sampling, the time was noted and the samplers were retrieved, sealed and shipped to the laboratory at Sahlgrenska Academy at the University of Gothenburg, Sweden, for instrumental analysis.The samples were analyzed using a Unity Markes International Limited thermal desorber (Unity Ultra TD) connected to a gas chromatograph (6890, Agilent Technologies, Inc., Santa Clara, CA, USA), with a selective mass detector (5973, Agilent Technologies, Inc.). Each sample tube was thermally desorbed for 5 min at 320 °C, focused on a cold trap at −10 °C and desorbed at a maximum temperature of 320 °C for GC injection. The GC oven temperature was maintained at 50 °C, for 3 min, then raised at 10 °C min−1 to 180 °C, and finally at 10 °C min−1 to 300 °C, at which temperature it was held for 5 min. The VOCs (benzene, toluene, ethylbenzene, xylenes and naphthalene) were detected using SIM, including the two most abundant ions of each compound. Controls for the quantification and identification of target compounds were established by injecting standard solutions (5 µL) of all target compounds in methanol into Perkin Elmer tubes in a stream of helium (15 mL min−1).
Quality control
For the PAH analyses a certified reference material (SRM 1649a urban dust) was used as a quality control (QC) sample. The measured levels of the 12 PAHs rarely deviated more than 10% from the certified levels. For the VOC analyses, QC samples obtained from the National Physical Laboratory (NPL), Teddington, UK, were analyzed at the same time as the samples. The levels of the VOCs detected in the QC samples did not deviate more than 5% from the certified levels. The QC results were considered acceptable during the whole study. Blanks were processed in parallel with the samples in this study. Some PAH residues were found on blank filters and all VOC target compounds were found in the PE blanks, but in no case was the amount of any given compound in any blank greater than 10% of the amount found in any non-blank sample.Modelling long-distance transported pollution
Air mass back trajectories were computed using the NOAA ARL HYSPLIT Model to investigate the effect of long-distance transported (LDT) pollution.21 For each sampling day, 96-hour air mass back trajectories were computed at start-up time and stop time at three starting heights (100, 200, and 500 m above ground level). The trajectories were divided into four trajectory classes or sectors, representing different source areas—Eastern Europe, Western Europe, the Nordic region (Finland, Norway, Sweden and the Baltic countries) and an eastern sector named Russia (including Russia, Belarus and Ukraine)—or remained undetermined (for trajectories that shifted classes during the sampling day). These sectors are similar to those used in a recent publication from Estonia.22 The classification was based on the criterion that all trajectories during a sampling period must share a major path belonging to the same class.Statistical calculations were performed using Wilcoxon's rank sum test implemented in SAS System for Windows, version 9.2.23 Statistical significance refers to p < 0.05 in two-tailed tests.
Results and discussion
PM
The PM2.5 and PM10 outdoor concentrations measured in this study are compared with those reported in various other cities around the world in Table 1. The results from the winter campaign (January–February) revealed very minor differences in PM2.5 and PM10 concentrations (the average PM2.5/PM10 ratio was 0.95 at Location 1 and 0.90 at Location 2). This indicates that both fractions were generated by the same source. Most of the PM10 mass consisted of fine particles, which probably originated from street dust. The average concentrations of particulate matter in Kaunas were comparable to those in most other European sites. In general there was little difference between the PM concentrations at Locations 1 and 2. PM2.5 concentrations were lower during the spring sampling campaign, for PM10 no significant seasonal concentration variations were observed. The average measured PM2.5 and PM10 concentrations were compared to those obtained from the Lithuania Environmental Protection Agency's automated monitoring station at Kaunas. The average measured concentrations were comparable to EPA monitored concentrations (see Table 1). At Location 1, the winter PM2.5 concentration was 34.5 ± 15.2 µg m−3 while in spring it was 24.7 ± 12.2 µg m−3; at Location 2 the corresponding values were 36.7 ± 21.7 and 22.4 ± 19.4 µg m−3, respectively.| Kaunas, Lithuania | Oxford, Ohio | Madrid, Spain | Bern, Switzerland | Athens, Greece | Belgrade, Serbia | Beirut, Liban | Teheran, Iran | Istambul, Turkey | Barcelona, Spain | Berlin, Germany | San Paulo, Brazil | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Location 1 | Location 2 | Automated monitoring station, EPA | ||||||||||||
| a Winter sampling period. b Spring sampling period. | ||||||||||||||
| PM2.5a | 34.5 ± 15.2 | 36.7 ± 21.7 | 26.81 ± 12.43 | 15.7 | 34 | 24.6 | 40.2 | 61 | 40 | 24.3 | 27.7 | 27.7 | 30 | 20.9 |
| PM2.5b | 24.7 ± 12.2 | 22.4 ± 19.4 | 23.19 ± 11.75 | |||||||||||
| PM10a | 35.2 ± 14.6 | 39.7 ± 20.3 | 33.91 ± 20.03 | 16.3 | 48 | 40.2 | 75.5 | 72 | 76 | 122.1 | 40.4 | 40.6 | 38 | 10.7 |
| PM10b | 41.7 ± 11.8 | 34.2 ± 20.6 | 32.55 ± 22.88 | |||||||||||
| Current study | ref. 24 | ref. 25 | ref. 26 | ref. 27 | ref. 28 | ref. 29 | ref. 30 | ref. 31 | ref. 32 | ref. 33 | ref. 34 | |||
PAHs
Very little information on the abundance and distribution of PAHs in Lithuania is available. Previously published studies are limited to investigations of BaP concentrations at a background station located on the Baltic sea coast and levels of 16 PAHs in the air (using passive sampling on PUF type samplers) and soil at five sites in the country.35,36The outdoor PAH concentrations measured at the two sampling locations in this study are presented in Fig. 1. High molecular weight PAHs with five to six rings, such as benzo(a)anthracene, chrysene, benzo(a)pyrene, benzo(g,h,i)perylene, predominated, while low molecular weight compounds were present only in smaller quantities. High concentrations of the 4-ring PAHs, fluoranthene and pyrene, were also observed. These findings are as expected, since 90% of 5–6 ring PAHs are adsorbed on particles and only 10% partition into the vapour phase, 4-ring PAHs are found in similar amounts in both phases, while PAHs with 2–3 rings are found predominantly (>90%) in the vapour phase.37 Five PAH components, i.e.acenaphthylene, 2,3,5-trimethylnaphthalene, 1-methylfluoranthene, 1-methylfluorene, and 2-methylchrysene, were not detected in any samples.
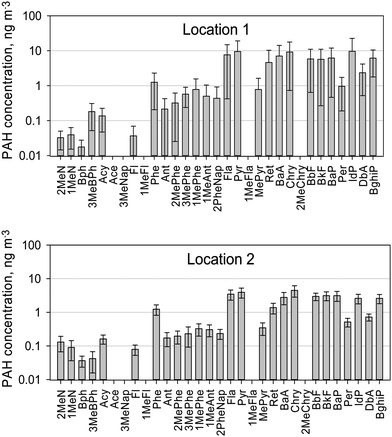 | ||
| Fig. 1 Mean concentrations of PAHs measured in the PM2.5 fraction in the winter sampling period. | ||
The mean sum PAH concentrations at Locations 1 and 2 in the winter campaign were 75.1 ± 32.7 and 32.7 ± 11.8 ng m−3, respectively. This difference is greater than expected from the difference in traffic intensity at the two sites, suggesting that there is another significant source of PAH emissions at Location 1 in addition to the traffic.
As shown in Table 2 the concentrations of most PAHs observed in this study are higher than those reported by other authors, particularly at Location 1. Comparison of benzo(a)pyrene levels in the PM2.5 fraction reported by Saarnio et al. (2008)4 showed that in Western European cities concentrations varied from 0.1 to 1.1 ng m−3 and in Eastern and Central European cities from 3.0 to 3.2 ng m−3. Benzo(a)pyrene levels at Location 2 were similar to those reported in Prague and Zagreb. It should be noted that the European limit value for benzo(a)pyrene is 1 ng m−3 in PM10.
| Compound | This study, Location 1 | This study, Location 2 | Kurkimäki, Finland, Hellen et al., 2008 | Bologna, Italy, Stracquandanio et al., 2007 | Duisburg, Germany | Prague, Czech R. | Amsterdam, Netherlands | Helsinki, Finland | Zagreb, Croatia, Sisovic et al., 2005 | Atlanta, USA, Liet al., 2009 |
|---|---|---|---|---|---|---|---|---|---|---|
| Saarnio et al., 2008 | ||||||||||
| Winter | Winter | Winter | Winter | Winter | Winter | Winter | Spring | Winter | Winter | |
| Phenanthrene | 1.3 ± 0.6 | 1.2 ± 0.4 | 2.5 | 0.9 | 0.7 | 4.8 | 0.8 | 0.3 | 0.15 | |
| Anthracene | 0.2 ± 0.1 | 0.2 ± 0.1 | 0.4 | 0.02 | ||||||
| Fluoranthene | 7.6 ± 3.4 | 3.5 ± 1.2 | 4.4 | 1.6 | 6.4 | 1.3 | 0.5 | 3.7 | 0.14 | |
| Pyrene | 9.5 ± 4.1 | 3.9 ± 1.3 | 4.0 | 1.4 | 1.3 | 5.6 | 0.9 | 0.4 | 4.7 | 0.17 |
| Retene | 4.6 ± 1.3 | 1.4 ± 0.5 | 0.14 | |||||||
| Benzo(a)anthracene | 7.0 ± 3.0 | 2.8 ± 1.1 | 1.3 | 1.1 | 0.19 | |||||
| Chrysene | 9.2 ± 4.1 | 4.5 ± 1.7 | 1.6 | 2.2 | 0.23 | |||||
| Benzo(b)fluoranthene | 5.8 ± 2.0 | 3.0 ± 0.7 | 2.5 | 3.5 | 0.61 | |||||
| Benzo(k)fluoranthene | 5.6 ± 1.9 | 3.1 ± 0.9 | 0.9 | 2.1 | 0.18 | |||||
| Benzo(a)pyrene | 6.2 ± 3.9 | 3.2 ± 1.0 | 1.3 | 1.7 | 1.1 | 3.0 | 0.3 | 0.1 | 3.2 | 0.42 |
| Indeno(1,2,3-c,d)pyrene | 9.6 ± 1.8 | 2.6 ± 0.8 | 2.2 | 0.59 | ||||||
| Dibenzo(a,h)anthracene | 2.3 ± 1.9 | 0.7 ± 0.2 | 0.4 | 0.02 | ||||||
| Benzo(g,h,i)perylene | 6.1 ± 2.9 | 2.6 ± 0.8 | 1.5 | 2.7 | 4.1 | 0.31 | ||||
| Sum PAHs | 75.1 ± 32.7 | 32.7 ± 11.8 | 3.17 | |||||||
The abundance of compounds such as benzo(a)anthracene, chrysene, benzo(a)pyrene, benzo(g,h,i)perylene that are characteristic of vehicular emissions suggests that traffic is probably one of the most important sources of PAH emissions at the investigated sites in Kaunas. However, although the traffic intensity is lower at Location 1 than at Location 2, levels of certain individual PAHs at Location 1 were found to be up to four times greater than at Location 2, possibly because most buildings are heated by the district supply system at Location 2, while at Location 1 most households have independent heating systems. To identify the sources of the various PAH emissions more clearly, the data were subjected to correlation analysis, factor analysis and a comparative analysis of indicative ratios and fractions. The intercorrelation matrix of the measured PAH concentrations is shown in Table 3.
| Nap | 2MeNap | 1MeNap | Bph | Acy | Fl | Phe | Ant | 2MePhe | 1MePhe | 1MeAnt | 2PhNap | Fla | Pyr | MePyr | Ret | BaAnt | Chry | BbF | BkF | BaP | Per | IdP | DbA | BghiP | ||
| Nap | 0.61 | 0.72 | 0.68 | 0.71 | 0.75 | 0.84 | 0.76 | 0.76 | 0.72 | 0.74 | 0.75 | 0.66 | 0.67 | 0.66 | 0.64 | 0.55 | 0.59 | 0.63 | 0.63 | 0.58 | 0.40 | 0.54 | −0.05 | 0.34 | Nap | |
| 2MeNap | 0.82 | 0.62 | 0.48 | 0.73 | 0.67 | 0.58 | 0.45 | 0.58 | 0.65 | 0.58 | 0.58 | 0.52 | 0.52 | 0.42 | 0.49 | 0.30 | 0.32 | 0.42 | 0.37 | 0.24 | 0.14 | 0.25 | 0.09 | −0.02 | 2MeNap | |
| 1MeNap | 0.83 | 0.97 | 0.79 | 0.34 | 0.59 | 0.46 | 0.13 | 0.30 | 0.25 | 0.27 | 0.28 | 0.14 | 0.14 | 0.11 | 0.12 | −0.04 | −0.03 | 0.08 | 0.06 | 0.00 | −0.22 | 0.20 | −0.41 | −0.21 | 1MeNap | |
| Bph | 0.53 | 0.77 | 0.82 | 0.60 | 0.78 | 0.59 | 0.46 | 0.49 | 0.45 | 0.49 | 0.51 | 0.38 | 0.37 | 0.35 | 0.35 | 0.22 | 0.27 | 0.34 | 0.31 | 0.22 | −0.07 | 0.26 | −0.35 | −0.16 | Bhp | |
| Acy | 0.77 | 0.90 | 0.93 | 0.72 | 0.92 | 0.92 | 0.88 | 0.85 | 0.82 | 0.80 | 0.77 | 0.71 | 0.70 | 0.66 | 0.69 | 0.60 | 0.64 | 0.65 | 0.63 | 0.57 | 0.45 | 0.37 | 0.21 | 0.38 | Acy | |
| Fl | 0.75 | 0.92 | 0.97 | 0.73 | 0.93 | 0.87 | 0.75 | 0.68 | 0.59 | 0.61 | 0.58 | 0.49 | 0.45 | 0.41 | 0.39 | 0.35 | 0.41 | 0.35 | 0.37 | 0.37 | 0.21 | 0.07 | 0.17 | 0.13 | Fl | |
| Phe | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 0.95 | 0.95 | 0.91 | 0.93 | 0.92 | 0.87 | 0.85 | 0.84 | 0.82 | 0.79 | 0.80 | 0.81 | 0.82 | 0.77 | 0.65 | 0.65 | 0.15 | 0.60 | Phe | |
| Ant | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 1.00 | 0.98 | 0.97 | 0.97 | 0.96 | 0.94 | 0.93 | 0.92 | 0.89 | 0.87 | 0.90 | 0.89 | 0.90 | 0.81 | 0.71 | 0.64 | 0.20 | 0.65 | Ant | |
| 2MePhe | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 1.00 | 1.00 | 0.98 | 0.99 | 0.98 | 0.96 | 0.95 | 0.93 | 0.91 | 0.91 | 0.91 | 0.92 | 0.93 | 0.87 | 0.78 | 0.70 | 0.21 | 0.70 | 2MePhe | |
| 1MePhe | 0.72 | 0.88 | 0.93 | 0.68 | 0.97 | 0.98 | 0.98 | 0.98 | 0.98 | 0.99 | 0.98 | 0.98 | 0.97 | 0.96 | 0.95 | 0.91 | 0.92 | 0.95 | 0.95 | 0.84 | 0.78 | 0.74 | 0.23 | 0.73 | 1MePhe | |
| 1MeAnt | 0.78 | 0.95 | 0.98 | 0.78 | 0.95 | 0.98 | 0.98 | 0.98 | 0.98 | 0.97 | 0.99 | 0.97 | 0.96 | 0.95 | 0.93 | 0.92 | 0.92 | 0.94 | 0.95 | 0.87 | 0.79 | 0.73 | 0.18 | 0.71 | 1MeAnt | |
| 2PhNap | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 1.00 | 1.00 | 1.00 | 0.98 | 0.98 | 0.98 | 0.97 | 0.96 | 0.93 | 0.91 | 0.92 | 0.93 | 0.95 | 0.86 | 0.76 | 0.77 | 0.13 | 0.70 | 2PhNap | |
| Fla | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 1.00 | 1.00 | 1.00 | 0.98 | 0.98 | 1.00 | 0.99 | 0.99 | 0.97 | 0.95 | 0.96 | 0.97 | 0.98 | 0.85 | 0.84 | 0.83 | 0.23 | 0.81 | Fla | |
| Pyr | 0.72 | 0.88 | 0.93 | 0.68 | 0.97 | 0.98 | 0.98 | 0.98 | 0.98 | 1.00 | 0.97 | 0.98 | 0.98 | 0.99 | 0.98 | 0.96 | 0.97 | 0.98 | 0.99 | 0.86 | 0.84 | 0.84 | 0.24 | 0.81 | Pyr | |
| MePyr | 0.77 | 0.88 | 0.93 | 0.67 | 0.90 | 0.98 | 0.95 | 0.95 | 0.95 | 0.97 | 0.97 | 0.95 | 0.95 | 0.97 | 0.98 | 0.97 | 0.97 | 0.99 | 0.99 | 0.87 | 0.86 | 0.85 | 0.23 | 0.83 | MePyr | |
| Ret | 0.75 | 0.92 | 0.95 | 0.73 | 0.98 | 0.97 | 1.00 | 1.00 | 1.00 | 0.98 | 0.98 | 1.00 | 1.00 | 0.98 | 0.95 | 0.93 | 0.93 | 0.97 | 0.97 | 0.84 | 0.82 | 0.83 | 0.20 | 0.78 | Ret | |
| BaAnt | 0.73 | 0.85 | 0.90 | 0.62 | 0.93 | 0.97 | 0.97 | 0.97 | 0.97 | 0.98 | 0.95 | 0.97 | 0.97 | 0.98 | 0.98 | 0.97 | 0.99 | 0.97 | 0.98 | 0.93 | 0.95 | 0.74 | 0.38 | 0.88 | BaAnt | |
| Chry | 0.73 | 0.53 | 0.58 | 0.33 | 0.62 | 0.63 | 0.63 | 0.63 | 0.63 | 0.65 | 0.62 | 0.63 | 0.63 | 0.65 | 0.73 | 0.63 | 0.75 | 0.97 | 0.98 | 0.92 | 0.93 | 0.75 | 0.38 | 0.87 | Chry | |
| BbF | 0.72 | 0.83 | 0.87 | 0.53 | 0.90 | 0.95 | 0.93 | 0.93 | 0.93 | 0.97 | 0.92 | 0.93 | 0.93 | 0.97 | 0.97 | 0.93 | 0.98 | 0.73 | 0.98 | 0.88 | 0.88 | 0.82 | 0.24 | 0.84 | BbF | |
| BkF | 0.73 | 0.85 | 0.90 | 0.62 | 0.93 | 0.97 | 0.97 | 0.97 | 0.97 | 0.98 | 0.95 | 0.97 | 0.97 | 0.98 | 0.98 | 0.97 | 1.00 | 0.75 | 0.98 | 0.90 | 0.89 | 0.83 | 0.27 | 0.85 | BkF | |
| BaP | 0.73 | 0.53 | 0.58 | 0.33 | 0.62 | 0.63 | 0.63 | 0.63 | 0.63 | 0.65 | 0.62 | 0.63 | 0.63 | 0.65 | 0.73 | 0.63 | 0.75 | 1.00 | 0.73 | 0.75 | 0.88 | 0.57 | 0.25 | 0.69 | BaP | |
| Per | 0.63 | 0.78 | 0.85 | 0.63 | 0.92 | 0.93 | 0.95 | 0.95 | 0.95 | 0.97 | 0.92 | 0.95 | 0.95 | 0.97 | 0.95 | 0.95 | 0.98 | 0.73 | 0.95 | 0.98 | 0.73 | 0.60 | 0.57 | 0.93 | Per | |
| IdP | 0.73 | 0.85 | 0.90 | 0.62 | 0.93 | 0.97 | 0.97 | 0.97 | 0.97 | 0.98 | 0.95 | 0.97 | 0.97 | 0.98 | 0.98 | 0.97 | 1.00 | 0.75 | 0.98 | 1.00 | 0.75 | 0.98 | −0.06 | 0.72 | IdP | |
| DbA | 0.53 | 0.45 | 0.47 | 0.10 | 0.63 | 0.57 | 0.58 | 0.58 | 0.58 | 0.65 | 0.50 | 0.58 | 0.58 | 0.65 | 0.58 | 0.58 | 0.67 | 0.67 | 0.75 | 0.67 | 0.67 | 0.63 | 0.67 | 0.59 | DbA | |
| BghiP | 0.73 | 0.85 | 0.90 | 0.62 | 0.93 | 0.97 | 0.97 | 0.97 | 0.97 | 0.98 | 0.95 | 0.97 | 0.97 | 0.98 | 0.98 | 0.97 | 1.00 | 0.75 | 0.98 | 1.00 | 0.75 | 0.98 | 1.00 | 0.67 | BghiP | |
| Nap | 2MeNap | 1MeNap | Bhp | Acy | Fl | Phe | Ant | 2MePhe | 1MePhe | 1MeAnt | 2PhNap | Fla | Pyr | MePyr | Ret | BaAnt | Chry | BbF | BkF | BaP | Per | IdP | DbA | BghiP |
As can be seen from the table, inter-correlation among PAH concentrations is generally very high for most of PAHs analysed. At Location 2 low molecular weight (LMW) compounds correlate with high molecular weight (HMW) compounds better than at Location 1, but the correlations of chrysene, benzo(a)pyrene and dibenzo(a,h)anthracene with other PAH concentrations are weaker. Diesel fuel reportedly contains high concentrations of LMW compounds, but because of pyrosynthesis during combustion in diesel engines the emission rate of HMW PAHs is greater than that of LMW PAHs.38,39 Therefore, the high correlations of LMW and HMW PAH concentrations may be due to emissions from diesel engines.
It should be noted that correlations of 1-methylphenanthrene with most of the other PAHs, including high molecular weight compounds, are very high. The high observed concentrations of alkylated PAHs relative to unsubstituted PAHs may indicate the presence of petrogenic sources.40–42 Although the concentrations of methylated compounds observed in this study were lower than those of the corresponding unsubstituted species, the above-mentioned correlations between the concentration of 1-methylphenanthrene and those of most of the other PAH analytes suggest that significant quantities of the observed PAHs were derived from unburned fuel.
Pyrene and chrysene are known to be emitted during industrial oil burning, fluoranthene and pyrene are typically emitted by both petrol and diesel vehicles, while chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene are characteristic of emissions from diesel vehicles.10 The high correlations between the concentrations of chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, fluoranthene and pyrene corroborate the importance of traffic as a source of PAH emissions at the investigated sites. However, it should be noted that the correlation of retene with HMW PAHs is also very high. Retene has been suggested as an indicator of biomass burning and has a unique formation mechanism or environmental pathway.1,43 Wood and peat are very commonly used fuels for heating individual houses in Kaunas.
To obtain further indications of the likely origins of the detected PAHs, sum concentrations of nine major combustion-derived PAHs (CPAHs: fluoranthene, pyrene, chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(a)pyrene, benzo[e]pyrene, indeno(1,2,3-c,d)pyrene, benzo(g,h,i)perylene) and CPAH/Sum PAH ratios were calculated. These ratios are reported to be 0.51, 0.41, and 0.30 for emissions from automobiles with catalytic converters, other automobiles, and heavy duty diesel trucks, respectively.44 The CPAH:Sum PAH ratio at Location 1 was as high as 0.78, suggesting that traffic is not the only important source of CPAHs in Kaunas.
Factor analysis of the measured PAH concentrations provided further insights into the influence of traffic and fuel combustion on the air quality in Kaunas (Table 4). The analysis revealed two factors influencing PAH concentrations at Location 2 and four factors at Location 1. It is apparent that the retene and dibenzo(a,h)anthracene observed at Location 1 arise from a different source from that of the other combustion PAHs. By contrast, at Location 2, it is likely that traffic is the major source of PAHs, with a small quantity of the most volatile PAHs arising from a different source.
| Location 1 | Location 2 | ||||||
|---|---|---|---|---|---|---|---|
| Factor 1 | Factor 2 | Factor 3 | Factor 4 | Factor 1 | Factor 2 | ||
| Nap | 0.65 | 0.50 | 0.54 | 0.07 | Nap | 0.31 | 0.85 |
| 2MeNap | 0.79 | −0.12 | 0.04 | 0.57 | 2MeNap | 0.35 | 0.92 |
| 1MeNap | 0.94 | −0.03 | 0.32 | −0.03 | 1MeNap | 0.62 | 0.76 |
| Bph | 0.81 | 0.18 | 0.43 | 0.27 | Bph | 0.65 | 0.63 |
| Acy | 0.99 | 0.12 | 0.04 | 0.03 | Acy | 0.86 | 0.49 |
| Fl | 0.99 | 0.07 | 0.04 | −0.09 | Fl | 0.85 | 0.52 |
| Phe | 0.98 | 0.16 | 0.11 | −0.04 | Phe | 0.90 | 0.41 |
| Ant | 0.94 | 0.30 | 0.16 | 0.05 | Ant | 0.90 | 0.43 |
| 2MePhe | 0.95 | 0.26 | 0.14 | 0.00 | 2MePhe | 0.90 | 0.42 |
| 1MePhe | 0.95 | 0.28 | 0.12 | 0.07 | 1MePhe | 0.91 | 0.40 |
| 1MeAnt | 0.90 | 0.33 | 0.24 | 0.11 | 1MeAnt | 0.92 | 0.39 |
| 2PheNap | 0.93 | 0.32 | 0.18 | 0.07 | 2PheNap | 0.92 | 0.38 |
| Fla | 0.82 | 0.53 | 0.13 | 0.18 | Fla | 0.91 | 0.41 |
| Pyr | 0.64 | 0.73 | 0.12 | 0.21 | Pyr | 0.90 | 0.43 |
| MePyr | 0.44 | 0.82 | 0.22 | 0.29 | MePyr | 0.88 | 0.47 |
| Ret | 0.17 | 0.58 | 0.32 | 0.73 | Re | 0.92 | 0.38 |
| BaA | 0.18 | 0.97 | 0.12 | 0.07 | BaA | 0.86 | 0.50 |
| Chry | 0.28 | 0.95 | −0.02 | 0.14 | Chry | 0.90 | 0.35 |
| BbF | 0.45 | 0.83 | 0.32 | 0.07 | BbF | 0.90 | 0.44 |
| BkF | 0.37 | 0.86 | 0.33 | 0.05 | BkF | 0.89 | 0.46 |
| BaP | 0.04 | 0.90 | 0.07 | 0.00 | BaP | 0.89 | 0.32 |
| Per | −0.17 | 0.96 | −0.18 | −0.07 | Per | 0.87 | 0.48 |
| IdP | 0.42 | 0.62 | 0.56 | 0.01 | IdP | 0.87 | 0.47 |
| DbaA | −0.35 | 0.04 | −0.91 | −0.09 | DbaA | 0.92 | 0.18 |
| BghiP | 0.05 | 0.88 | −0.24 | −0.09 | BghiP | 0.85 | 0.51 |
As discussed by Saarnio et al. (2008), neither the levels of individual PAHs nor the ratios of their concentrations can be regarded as highly specific indicators of emission sources for several reasons: PAHs originate from a large variety of combustion sources with only slightly different emission profiles; PAHs have widely differing vapour pressures and reactivities; and particulate PAH concentrations depend on the ambient temperature and solar radiation intensity as well as on the total particulate mass concentration.4 Furthermore, some compounds, especially those that are semi-volatile (2–4 rings), may be lost during long sampling campaigns. Nevertheless, their concentration ratios can give hints about the relative importance of possible sources (see Fig. 2).
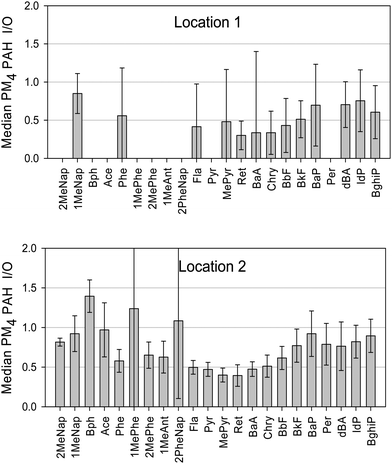 | ||
| Fig. 2 Ratios of indoor to outdoor (I/O) PAH concentrations in PM4 at Locations 1 and 2. | ||
In addition, indicative PAH concentration fractions and ratios were calculated, and the results were compared to data from a comprehensive study of PAHs in size-segregated particulate matter from European urban sites performed by Saarnio et al. (2008) and from a similar study by Tang et al. (2005) in China, Japan and Russia.4,7 Only data from cold seasons were included.
The observed indeno(1,2,3-c,d)pyrene:(indeno(1,2,3-c,d)pyrene + benzo(g,h,i)perylene) ratio is comparable to those reported in other studies, confirming that traffic is a significant source of PAH emissions at both locations examined. This conclusion is further supported by the benzo(a)anthracene:(benzo(a)anthracene + chrysene) and benzo(a)pyrene: (benzo(a)pyrene + chrysene) ratios. The influence of fossil fuel combustion on PAH levels in Kaunas is indicated by the benzo(a)pyrene:benzo(g,h,i)perylene and benzo(a)pyrene:chrysene ratios.
The ratio of air-borne PAH concentrations in indoor environments to those in outdoor environments (I/O ratios) can be used to determine whether the major PAH source(s) are located indoors (in which case I/O > 1) or outdoors (in which case I/O < 1). I/O ratios for PAHs bound to the PM4 fraction are presented in Fig. 2. The low observed I/O ratios may indicate that PAH emissions at the locations studied arise primarily from outdoor sources. It should be noted that the buildings at both locations have old windows with wooden frames that are fairly permissive in terms of air circulation. Thus, residents of older buildings that are close to roads may be significantly exposed to PAHs and PM in general.
VOCs
In developed and industrialized countries, traffic is the most important source of high levels of VOCs in towns and thus high exposure of the population in the ambient environment. The outdoor and indoor average concentrations, standard deviations, minimal and maximal values of BETX (benzene, ethylbenzene, toluene and xylene) and naphthalene observed in the present study are presented in Table 5. The observed outdoor VOC concentrations are lower than those reported in most previous studies. Monitoring of the outdoor air in front of the windows revealed that the concentration of benzene ranged from 0.3 to 1.7 µg m−3. The benzene concentration observed during January–February was somewhat higher (by 0.4 µg m−3) than that observed in March–April. Similar variations in benzene concentrations were observed in both locations. The results obtained for toluene exhibited significant variability, with the measured concentrations ranging from 0.4 to 9.5 µg m−3. The average outdoor concentration of toluene at Location 2 was 40% higher than that observed at Location 1. For comparison, Fernandez-Villarrenaga et al. (2004) carried out measurements in Coruna, a medium-sized town in Spain, and detected toluene, ethylbenzene, m + p-xylene and o-xylene at concentrations of 3.4, 23.6, 3.3, 5.1 and 2.7 µg m−3, respectively, while a study conducted in Sweden reported urban indoor and outdoor benzene levels to be in the range of 0.4–4 µg m−3.45,46 The average outdoor ethylbenzene concentrations ranged from 0.2 to 1.3 µg m−3, with the highest concentration being observed at Location 2 in April. The average outdoor concentration of the sum of all xylenes measured during January–February was 40% higher than that measured in March–April, and the concentrations of individual xylene fractions (m + p-xylene and o-xylene) exhibited similar trends to those of the sum of all xylenes. Average outdoor concentrations of naphthalene were higher during January–February than in March–April, and maximal (0.32 µg m−3) in mid-January.| Measurement week | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1a | 2b | 3c | 4d | ||||||
| Outdoor | Indoor | Outdoor | Indoor | Outdoor | Indoor | Outdoor | Indoor | ||
| a Location 1, winter. b Location 2, winter. c Location 1, spring. d Location, 2 spring. | |||||||||
| Benzene | Mean | 0.9 | 0.7 | 1.0 | 0.7 | 0.7 | 0.5 | 0.4 | 0.8 |
| Std. dev. | 0.5 | 0.4 | 0.5 | 0.3 | 0.3 | 0.2 | 0.2 | 0.4 | |
| Min | 0.4 | 0.3 | 0.5 | 0.4 | 0.4 | 0.3 | 0.3 | 0.3 | |
| Max | 1.7 | 1.3 | 1.6 | 1.3 | 1.0 | 0.7 | 0.6 | 1.4 | |
| Toluene | Mean | 2.7 | 5.3 | 2.0 | 1.8 | 1.2 | 0.9 | 3.5 | 7.0 |
| Std. dev. | 3.2 | 3.4 | 1.0 | 0.4 | 0.9 | 0.5 | 1.6 | 8.2 | |
| Min | 0.8 | 2.0 | 0.7 | 1.3 | 0.4 | 0.5 | 2.1 | 0.6 | |
| Max | 9.5 | 9.8 | 3.3 | 2.2 | 2.6 | 1.4 | 5.3 | 16.3 | |
| Ethylbenzene | Mean | 0.2 | 0.1 | 0.3 | 0.3 | 0.2 | 0.2 | 1.3 | 0.3 |
| Std. dev. | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 1.8 | 0.2 | |
| Min | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |
| Max | 0.6 | 0.2 | 0.4 | 0.4 | 0.4 | 0.3 | 2.0 | 0.5 | |
| Σ Xylenes | Mean | 0.9 | 0.6 | 1.3 | 1.1 | 0.7 | 0.7 | 0.6 | 1.7 |
| Std. dev. | 0.9 | 0.1 | 0.6 | 0.4 | 0.6 | 0.4 | 0.3 | 1.3 | |
| Min | 0.2 | 0.4 | 0.3 | 0.5 | 0.3 | 0.4 | 0.3 | 0.1 | |
| Max | 2.7 | 0.7 | 2.0 | 1.5 | 1.8 | 1.0 | 1.0 | 2.6 | |
| Naphthalene | Mean | 0.2 | 0.2 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 |
| Std. dev. | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |
| Min | 0.1 | 0.2 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | 0.1 | |
| Max | 0.3 | 0.3 | 0.3 | 0.2 | 0.2 | 0.2 | 0.2 | 0.2 | |
The indoor concentrations of benzene ranged from 0.3 µg m−3 to 1.4 µg m−3, and no significant differences in benzene concentration between samples taken in January–February and March–April were found. Like the outdoor concentrations of toluene (see above) there were wide variations in indoor concentrations of toluene, from 0.5 µg m−3 to 16.3 µg m−3. The indoor concentrations of ethylbenzene ranged from 0.1 µg m−3 to 0.5 µg m−3. For the sum of xylenes, the average of the values measured during January–February was 40% lower than the March–April average. The average indoor concentrations of naphthalene ranged from 0.1 µg m−3 to 0.3 µg m−3, and there were no show substantial differences between measurements obtained during the winter and spring periods.
The VOC I/O values at both locations are summarized in Table 6. For benzene, it is apparent that at Location 1 the most significant sources are indoors; the I/O ratios observed in January and April were 1.3 and 2.2, respectively. By contrast, the results from Location 2 indicate that the main sources of benzene are outdoors, with I/O ratios <1 throughout the campaign. For toluene, both the I/O values and the correlation coefficients exhibited a high degree of variability. For ethylbenzene and xylenes the concentrations in the indoor air were almost equal to those in front of the windows, except during the sampling week in April, when evidence of strong indoor sources of all the studied VOCs was detected. Thus, for ethylbenzene and xylenes, the outdoor air determines the indoor air, leading almost to equilibrium between indoor and outdoor air. This conclusion is supported by the high correlation coefficients between indoor and outdoor air concentrations of ethylbenzene and xylenes, as shown in Table 6. The results obtained for toluene, ethylbenzene and xylenes correspond with the findings of German authors (Ilgenet al., 2001).19 The I/O ratios and correlation coefficients for naphthalene exhibited a high degree of variability.
| Measurement week | ||||
|---|---|---|---|---|
| 1a | 2b | 3c | 4d | |
| a Location 1, winter. b Location 2, winter. c Location 1, spring. d Location, 2 spring. | ||||
| Concentration ratio, indoor to outdoor air (I/O) | ||||
| Benzene | 1.3 | 0.8 | 0.5 | 2.2 |
| Toluene | 3.6 | 1.1 | 0.9 | 1.7 |
| Ethylbenzene | 0.9 | 1.1 | 1.0 | 1.7 |
| Σ Xylenes | 1.0 | 1.0 | 1.0 | 1.8 |
| Naphthalene | 1.8 | 1.1 | 2.6 | 1.3 |
| Correlation coefficients between indoor and outdoor air | ||||
| Benzene | 0.10 | 0.20 | 0.89 | 0.50 |
| Toluene | −0.15 | 0.26 | −0.11 | 0.56 |
| Ethylbenzene | 0.87 | 0.57 | 0.68 | 0.95 |
| Σ Xylenes | 0.79 | 0.62 | 0.66 | 0.73 |
| Naphthalene | −0.88 | 0.41 | 0.52 | 0.66 |
Influence of long-distance transported pollution on PM2.5 and PAH levels
The trajectory analysis showed that air masses coming from Eastern Europe carried significantly higher levels of pollutants than those from other regions; air masses from other regions in Europe all had similar PM2.5 levels (see Table 7). Air masses from the Nordic countries usually carry lower pollutant loads, as the median levels suggest, but high concentrations in masses from the Nordic sector on some days probably explains the lack of significant differences in PM2.5 levels.22,47For a few of the PAHs (naphthalene, 2-methylnaphthalene, acenaphthylene, fluorene, phenanthrene, and anthracene; not presented in the table), significantly higher concentrations were observed on days when the air originated from Eastern Europe rather than from one or more of the other regions, but in most cases no such differences were observed.| PM2.5/µg m−3 | p-Value of the rank sum test | ||||
|---|---|---|---|---|---|
| Mean | Median | Russia | Western Europe | Nordic | |
| Eastern Europe (9) | 45.7 | 42.0 | 0.0318 | 0.0119 | 0.0464 |
| Russia (10) | 27.2 | 23.7 | 0.9575 | 0.4716 | |
| Western Europe (6) | 23.3 | 24.9 | 0.8388 | ||
| Nordic (13) | 25.5 | 15.4 | |||
| Unclassified (5) | 25.3 | 16.8 | |||
The higher concentrations of PM2.5 on days when the air originated from Eastern Europe were expected because anthropogenic emissions in Eastern Europe are higher than in the other regions. However, no consistent differences in levels of PAHs in air originating from different regions were observed, suggesting that local sources contribute most of the sampled concentrations of the PAHs.
Conclusions
The mean outdoor PM2.5 concentration at Location 1 in winter was 34.5 ± 15.2 µg m−3 while in spring it was 24.7 ± 12.2 µg m−3, at Location 2 the corresponding values were 36.7 ± 21.7 and 22.4 ± 19.4 µg m−3, respectively. The mean PM10 concentrations at Location 1 in winter and spring were 35.2 ± 14.6 and 41.7 ± 11.8 µg m−3, respectively, while at Location 2 the corresponding values were 39.7 ± 20.3 and 34.2 ± 20.6 µg m−3. PM2.5 concentrations were lower during the spring sampling campaign, but no significant seasonal variations in PM10 concentrations were observed. This study demonstrates that PM levels in Kaunas are comparable to those observed in many other European locations.The levels of most PAHs analyzed in this study, especially at Location 1, are notably higher than those reported elsewhere in Europe. The mean sum PAH concentrations at Locations 1 and 2 in the winter campaign were 75.1 ± 32.7 and 32.7 ± 11.8 ng m−3, respectively. The experimental results corroborated the hypotheses that 5–6 and 4-ring PAHs originate from automotive emissions, are bound to street dust and dispersed over the area. Thus, street dust may not only emit fugitive dust, but also be a substantial source of particle-bound PAHs. A trajectory analysis showed that long-range transport is also a source of PM2.5 in the region, but the PAHs within the PM2.5 are of local origin.
VOC concentrations were mostly low and comparable to those reported from Sweden. The mean outdoor concentrations of the VOCs, benzene, toluene, ethylbenzene, sum of m-, p-, o-xylenes, and naphthalene, were: 0.7 ± 0.2, 3.0 ± 0.8, 0.5 ± 0.2, 3.5 ± 0.3 and 0.2 ± 0.1 µg m−3, respectively. The outdoor concentrations of benzene and xylenes measured during January–February sampling campaigns were up to 40% higher than those observed during March–April sampling campaign. Indoor concentrations of benzene did not differ significantly between the January–February and March–April campaigns, but the concentration of xylenes was found to be 40% higher in January–February than in March–April.
Acknowledgements
This study was supported by the Lithuanian State Science and Studies Foundation (Grant No. T-103/09), and the Socrates/Erasmus student mobility programme. The authors are grateful to Ms Inga Vaskeviciute for the PAH laboratory analysis, to the Department of Solid Mechanics at Kaunas University of Technology (Head Assoc. Prof. Paulius Griskevicius), and to Ms Loreta Kregzdaite for providing access to measurement sites.References
- Z. Li, E. N. Porter, A. Sjodin, L. L. Needham, S. Lee, A. G. Russell and J. A. Mulholland, Atmos. Environ., 2009, 43, 4187–4193 CrossRef CAS.
- A. E. Barbosa and J. T. Hvitved, Sci. Total Environ., 1999, 235, 151–159 CrossRef CAS.
- R. Berbee, G. Rijs, R. de Brouwer and L. van Velzen, Water Environ. Res., 1999, 71, 183–190 CrossRef CAS.
- K. Saarnio, M. Sillanpaa, R. Hillamo, E. Sandell, A. S. Pennanen and R. O. Salonen, Atmos. Environ., 2008, 42, 9087–9097 CrossRef CAS.
- A. Sisovic, Z. Vadjic, K. Sega, I. Beslic and V. Vadjic, Bull. Environ. Contam. Toxicol., 2005, 75, 121–126 CrossRef CAS.
- H. Guo, S. C. Lee, K. F. Ho, X. M. Wang and S. C. Zou, Atmos. Environ., 2003, 37, 5307–5317 CrossRef CAS.
- N. Tang, T. Hattori, R. Taga, K. Igarashi, X. Y. Yang, K. Tamura, H. Kakimoto, V. F. Mishukov, A. Toriba, R. Kizu and K. Hayakawa, Atmos. Environ., 2005, 39, 5817–5826 CrossRef CAS.
- M. Stracquadanio, G. Apollo and C. Trombini, Water, Air, Soil Pollut., 2007, 179, 227–237 CrossRef CAS.
- H. Helleén, H. Hakola, S. Haaparanta., H. Pietarila and M. Kauhaniemi, Sci. Total Environ., 2008, 393, 283–290 CrossRef.
- N. Rajput and A. Lakhani, Environ. Monit. Assess., 2009, 150, 273–284 CrossRef CAS.
- J. C. Chuang, G. A. Mack, M. R. Kuhlman and N. K. Wilson, Atmos. Environ., Part B, 1991, 25, 369–380 CrossRef.
- W. Ott, N. Wilson, N. Klepeis and P. Switzer, Air and Waste Management Association, Pittsburgh, PA, 1994 Search PubMed.
- A. B. Hansen and F. Palmgren, Sci. Total Environ., 1996, 189–190, 451–457 CrossRef.
- V. Cocheo, P. Sacco, C. Boaretto, E. De Saeger, P. Ballesta, H. Skov, E. Goelen, N. Gonzalez and A. B. Caracena, Nature, 2000, 404, 141–142 CrossRef CAS.
- E. Ilgen, N. Karfich, K. Levsen, J. Angerer, P. Schneider, J. Heinrich, H. E. Wichmann, L. Dunemann and J. Begerow, Atmos. Environ., 2001, 35, 1235–1252 CrossRef CAS.
- G. Bertoni, C. Ciuchini, A. Pasini and R. Tappa, J. Environ. Monit., 2002, 4, 903–909 RSC.
- A. J. Buczynska, A. Krata, M. Stranger, A. F. L. Godoi, V. Kontozova-Deutsch, L. Bencs, I. Naveau, E. Roekens and R. Van Grieken, Atmos. Environ., 2009, 43, 311–318 CrossRef CAS.
- H. Skov, A. B. Hansen, G. Lorenzen, H. V. Andersen, P. Lofstrom and C. S. Christensen, Atmos. Environ., 2001, 35, 2463–2471 CrossRef CAS.
- E. Ilgen, K. Levsen, J. Angerer, P. Schneider, J. Heinrich and E. Wichmann, Atmos. Environ., 2001, 35, 1265–1279 CrossRef CAS.
- L. Kliucininkas, unpublished work.
- R. R. Draxler and G. D. Rolph, HYSPLIT (Hybrid Single-Particle Lagrangian Integrated Trajectory) Model, NOAA Air Resources Laboratory, Silver Spring, MD, 2003 Search PubMed.
- H. Orru, V. Kimmel, U. Kikas, A. Soon, N. Kunzli, R. P. F. Schins, P. J. A. Borm and B. Forsberg, Sci. Total Environ., 2010, 408, 1515–1522 CrossRef CAS.
- SAS, SAS Statistical Software Version 9.2, SAS Institute Inc., Cary, NC, 2008 Search PubMed.
- B. Wojas and C. Almquist, Atmos. Environ., 2007, 41, 9064–9078 CrossRef CAS.
- B. Artínano, P. Salvador, D. G. Alonso, W. Querol and A. Alastuey, Environ. Pollut., 2003, 125, 453–465 CrossRef CAS.
- C. Hueglin, R. Gehrig, U. Baltensperger, M. Gysel, C. Monn and H. Vonmont, Atmos. Environ., 2005, 39, 637–651 CrossRef CAS.
- A. Chaloulakou, P. Kassomenos, N. Spyrellis, P. Demokritou and P. Koutrakis, Atmos. Environ., 2003, 37, 649–660 CrossRef CAS.
- M. D. Tasic, S. F. Rajsic, V. T. Novakovic, Z. R. Mijic and M. N. Tomasevic, Phys. Scr., T, 2005, 118, 29–30 Search PubMed.
- H. Shaka and N. A. Saliba, Atmos. Environ., 2004, 38, 523–531 CrossRef CAS.
- H. Kakooei and A. A. Kakooei, J. Appl. Sci., 2007, 7, 3081–3085 Search PubMed.
- F. Karaca, O. Alagha and F. Erturk, Chemosphere, 2005, 59, 1183–1190 CrossRef CAS.
- X. Querol, A. Alastuey, S. Rodriguez, F. Plana, C. R. Ruiz, N. Cots, G. Massague and O. Puig, Atmos. Environ., 2001, 35, 6407–6419 CrossRef CAS.
- P. Lenschow, H. J. Abraham, K. Kutzner, M. Lutz, J. D. Preuss and W. Reichenbacher, Atmos. Environ., 2001, 35, S23–S33 CAS.
- R. Miranda and E. Tornaz, Atmos. Res., 2008, 87, 147–157 CrossRef CAS.
- A. Milukaite, Atmos. Environ., 2006, 40, 2046–2057 CrossRef CAS.
- A. Milukaite, J. Klanova, I. Holoubek, I. Rimselyte and K. Kvietkus, Lith. J. Phys., 2008, 48, 357–366 Search PubMed.
- M. Odabasi, N. Vardar, A. Sofuoglu, Y. Tasdemir and T. M. Holsen, Sci. Total Environ., 1999, 227, 57–67 CrossRef CAS.
- C. H. Lim, G. A. Ayoko, L. Morawska, Z. D. Ristovski and E. R. Jayaratne, Atmos. Environ., 2005, 39, 7836–7848.
- L. C. Marr, T. W. Kirchstetter, R. A. Hartey, A. H. Miguel, S. V. Hering and S. K. Hammond, Environ. Sci. Technol., 1999, 33, 3091–3099 CrossRef CAS.
- R. Simo, J. O. Grimalt and J. Albaiges, Environ. Sci. Technol., 1997, 31, 2697–2700 CrossRef CAS.
- K. E. Gustafson and R. M. Dickhut, Environ. Sci. Technol., 1997, 31, 140–147 CAS.
- H. S. Soderstrom and P. A. Bergqvist, Environ. Sci. Technol., 2003, 37, 47–52 CrossRef.
- J. D. McDonald, B. Zielinska, E. M. Fujita, J. C. Sagebiel, J. C. Chow and J. G. Watson, Environ. Sci. Technol., 2000, 34, 2080–2091 CrossRef.
- W. F. Rogge, L. M. Hildemann, M. A. Mazurek, G. R. Cass and B. R. T. Simoneit, Environ. Sci. Technol., 1993, 27, 636–651 CAS.
- V. Fernandez-Villarrenaga, P. Lopez-Mahia, S. Muniategui-Lorenzo, D. Prada-Rodriguez, E. Fernandez-Fernandez and X. Tomas, Sci. Total Environ., 2004, 334–335, 167–176 CrossRef CAS.
- B. Strandberg, A. L. Sunesson, M. Sundgren, J. O. Levin, G. Sallsten and L. Barregard, Atmos. Environ., 2006, 40, 7686–7695 CrossRef CAS.
- S. Johanesson, P. Gustafson, P. Molnar, L. Barregard and G. Sallsten, J. Exposure Sci. Environ. Epidemiol., 2007, 17, 613–624 Search PubMed.
| This journal is © The Royal Society of Chemistry 2011 |