Biogenic N-I-codoped TiO2 photocatalyst derived from kelp for efficient dye degradation†
Nan
Shi
a,
Xiaohui
Li
a,
Tongxiang
Fan
*a,
Han
Zhou
a,
Jian
Ding
a,
Di
Zhang
a and
Hanxing
Zhu
b
aState Key Lab of Metal Matrix Composites, Shanghai Jiaotong University, Shanghai, 200240, P. R. China. E-mail: txfan@sjtu.edu.cn; Fax: +86-21-34202079; Tel: +86-21-54747779
bSchool of Engineering, Cardiff University, Cardiff, CF24 3AA, UK
First published on 12th November 2010
Abstract
The design and fabrication of efficient, cost-effective photocatalysts are required in order to ease global energy and environmental issues. In this paper, we have obtained a biogenic-TiO2 photocatalyst through a simple one step infiltration process which can replicate well the hierarchical architecture of kelp from the macro- down to the nano-scale. In addition, the nitrogen and iodine, contained in the original plant corpus, are self-doped into the resulting samples. UV-Visible diffuse reflectance spectra of the biogenic-TiO2 indicate that it has efficient light-harvesting capacity, especially in the region from 400 nm to 550 nm, and compared with the common N-TiO2, its band gap absorption edge exhibits a clear red shift due to the self-doping. Moreover, the biogenic-TiO2 possesses excellent photocatalytic properties proved by the photocatalytic activity of methylene blue degradation under solar energy irradiation. This work may provide the inspiration for the synthesis of further high performance photocatalysts based on the kelp structure and a new methodology for application of nature's plants and utilization of solar energy with biogenic materials.
Broader contextThe global energy crisis and environmental pollution have become major concerns to the public in recent years. TiO2 is still the most commonly investigated photocatalyst which can use sunlight to destroy highly toxic molecules, remediate pollutants, produce clean energy as hydrogen and even convert solar energy to electrical power. Therefore, the development of efficient, cost-effective TiO2 photocatalysts may be an answer to ease the current environmental and energy problems. In this paper, biogenic-TiO2 with highly efficient light-harvesting and photocatalytic properties is fabricated by copying the elaborate architecture of kelp from the macro- and micro- down to the nanoscale and by self-doping of iodine and nitrogen, which are contained in the original plant corpus. Excellent photocatalytic properties are demonstrated due to the overall enhancement of light-harvesting by greater absorption and multiple scattering of light by the unique hierarchical architecture of kelp, as well as the narrowed band gap induced by doping. This approach demonstrates a new strategy for mimicking mother nature's elaborate creations in making materials for efficient photocatalysis and current artificial photosynthesis systems. |
Introduction
If only one thousandth of the solar energy that reaches the earth could be utilized, we could still have nine times the amount of energy we need.1 Nowadays, the global energy crisis and environmental pollution have become major threats to the lives of humans. Using solar energy more efficiently could help ease global energy and environmental issues. As photocatalysis can make use of sunlight to destroy highly toxic molecules, remediate pollutants, produce hydrogen and even electrical power,2 the design and fabrication of efficient, cost-effective photocatalysts are highly demanded. Titanium dioxide is one of the most viable materials for photocatalysis because of its high oxidative power, low cost, photostability and nontoxicity.3 Unfortunately, due to its wide band gap of 3.2 eV, it can only be activated under UV irradiation which is merely a small fraction (5%) of the solar spectrum.4 So far, tremendous efforts have been made to enhance its photocatalytic capacity by modifying both the structures and composition. One approach is to fabricate TiO2 with high specific surface area5 in the form of various nanostructures such as nanotubes, nanoplates, nanoparticles, etc. Another is to improve the composition of TiO2 by substituting metal ions6 (such as Mn4+, Cr3+, V3+) at Ti sites, or by doping TiO2 with non-metal elements (such as nitrogen,7sulfur,8carbon9) or noble metals.10 Although significant progress has been made, most research has only focused on one approach rather than both of them.Biogenic materials, which combine natural geometry with synthetic materials chemistry, are products (metals, oxides, carbides, nitrides, composites, etc.) synthesized through employing living things as templates with their structures resembling one another and elements being utilized.11Biogenic-TiO2 may realize the synergy of both structure and element-introduced improvements of TiO2 based on a single biotemplate, and be a major advance in photochemistry to develop novel and efficient photocatalysts. On the one hand, multidimensional, sophisticated and diverse biological structures give rise to specific functions, providing inspiration for the development of materials with promising photocatalytic, optical, and electronic properties.12 For instance, the hierarchical structures of leaves which are designed favorably for photosynthesis endow them with an extremely high light-harvesting efficiency. On the other hand, non-metallic elements derived from various kinds of organisms endowed by nature are expected to be self-doped into the prepared samples. For example, elements such as nitrogen present in plants in different forms have been successfully self-doped into TiO2.13
Here, we present a novel strategy to fabricate biogenic-TiO2 with highly efficient light-harvesting and photocatalytic properties by copying the elaborate architecture of kelp from the macro- and micro- down to the nanoscale and by the self-doping of iodine and nitrogen which are contained in the original plant corpus. In our experiments, we use algae rather than green leaves as a template due to the fact that the solar energy conversion efficiency is nearly 5% for water plants but less than 1% for land-based organisms.14 The structure of water plants, such as kelp with most of its chromatophores existing in the epidermis, can maximize the absorption of sunlight, which is beneficial to improving the light-harvesting efficiency. In addition, its porous structure increases the light scattering which enhances the solar energy conversion. Moreover, kelp is rich in natural iodine (including I−, IO3− and organic iodine, etc.15) which is expected to be self-doped into the resulting samples during synthesis, just as with nitrogen.13 Recently, many studies16 have reported that iodine-doped TiO2 shows outstanding visible light photocatalytic activity in the decomposition of dye and organic pollutants. UV-Visible diffuse reflectance spectra of this biogenic-TiO2 indicates that it has a highly efficient light-harvesting capacity, especially in the region from 400 nm to 550 nm. Compared with common N-TiO2 which is calcined under the same conditions without a template, its band gap absorption edge exhibits a clear red shift. Indeed, the superior photocatalytic activity of biogenic-TiO2 is proved by the degradation of methylene blue under solar energy irradiation. This work will probably provide a new methodology for the utilization of nature's plants and synthesis of light-harvesting biogenic materials.
Results and discussions
Structure of kelp
Kelp (Fig. 1a) is a type of large seaweed belonging to the brown algae (class Phaeophyceae) and is classified as the order Laminariales.17Scheme 1 illustrates the structures and functions of the natural kelp as well as the artificial kelp-TiO2 which is herein named biogenic-TiO2. Panel A shows the hierarchical structures of kelp at scales ranging from the macro- and micro- down to the nanoscale, with Fig. 1 showing images of the corresponding typical examples. As shown in the cross-section (Scheme 1(a)), it mainly contains epidermis, exodermis, endoderm and pith, endowing kelp with hierarchical porous and interconnected structures which are beneficial for efficient light-harvesting. Meanwhile, the corresponding confocal laser-scanning microscopy image (Fig. 1d) shows a cross-section of fresh kelp. The internal structure (Scheme 1(c)) of an individual epidermis cell reveals that it contains several chromatophores which make the cells look brown-green under the optical microscope (Fig. 1b), and which are observable with red fluorescence in a confocal laser-scanning microscope (Fig. 1c and e). Each chromatophore contains stacked nanolayered (about 50 nm) membranes (Scheme 1d, e), which are visible in the TEM images shown in Fig. 1f and g. A series of pigments18 of different colors including chlorophyll a and b and fucoxanthin, arranged in the membranes of the chromatophore, are responsible for light harvesting, transmission and conversion. In addition, such stacked nanolayered structures with high surface areas, allowing efficient interactions between the light-harvesting pigments and sunlight, are favorable for efficient light-harvesting and fast charge separation. In fact, the entire structure of kelp is greatly beneficial for light harvesting. For example, the less regularly arranged and porous architectures of the exodermis, endoderm and pith enhance the effective light path length and increase multiple light scattering to heighten light absorption.19 In addition, kelp becomes enriched in iodine as it grows, up to the maximum in its maturation period. Thus, besides replicating the hierarchical structures and self-doping nitrogen13 to achieve high energy conversion efficiencies, iodine is also expected to be self-doped into the resulting TiO2 samples during the synthesis process in our experiments. The main forms of iodine in kelp are I−, IO3− and organic iodine (Scheme 1C), and I− is the main species of iodine enriched by algae according to Küpper et al.20Iodine in kelp will be released rapidly after it is immersed into water. Fortunately, a number of insoluble organic iodine species can be preserved in kelp's remains.21 The whole process is based on structure inheritance of kelp and self-doping TiO2 with nitrogen and iodine originally existing in the kelp.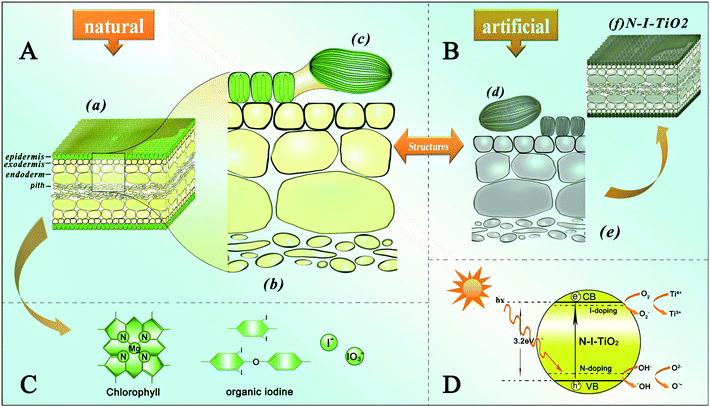 | ||
| Scheme 1 Schematic illustrations of A the hierarchical structures of kelp from the macro- to the nanoscale, B synthetic structures of the artificial oxide semiconductor photocatalyst, C the porphyrin ring of the chlorophyll and different forms of natural iodine and D a possible photocatalytic mechanism in the artificial semiconductor photocatalyst. | ||
 | ||
| Fig. 1 Characterization of natural kelp from the macro- to the nanoscale. a Digital picture of intact kelp. b The upper epidermis visualized by digital microscope (Keyence). c The upper epidermis visualized by fluorescence microscope. d Cross-section observed by confocal laser-scanning microscopy (CLSM). e Cross-section observed under confocal laser-scanning microscope. The epidermis cells are displayed by chlorophyll fluorescence (red) excited by 488 nm light, with insets of the magnified images of the lineate region (e1 by TEM, e2 by CLSM). f and gTEM images of a chromatophore. | ||
Synthetic procedure of biogenic-TiO2
Pieces of fresh kelp were first treated with a dilute HCl solution to remove Na, Ca, P ions etc., and to substitute Mg2+ ions in the porphyrins of the chlorophyll molecules with H+.22 Afterwards, the as-treated kelp samples were dipped into TiCl3 solution and left to stand under vacuum, with Ti3+ ions being introduced into the kelp by replacing the H+ ions in the pheophytins, and reacting with the abundant –OH functional groups on the cellulose and hemicellulose of the cells walls of the exodermis, endoderm, and pith. After calcination at 500 °C, 600 °C and 700 °C, respectively, the N–I-codoped biogenic-TiO2 with hierarchical porous structures was obtained by removing the biotemplates.Composition and structural characterization of biogenic-TiO2 derived from kelp
The X-ray diffraction (XRD) patterns shown in Fig. 2 clearly verify that biogenic-TiO2 samples calcined at 500 °C, 600 °C and 700 °C are all pure anatase. All of the recorded peaks can be assigned to the tetragonal anatase TiO2 (I41/amd, No. 141, Joint Committee on Powder Diffraction Standards file number 71-1168), with lattice constants a = 3.797 Å, and c = 9.579 Å. It is commonly believed that the crystal phase is a crucial factor for the photocatalytic activity of TiO2,23 and that the anatase form is the active phase in photocatalytic reactions.24 Generally speaking, the rutile phase appears at a calcination temperature of 550 °C.25 According to Kröger et al.,26 the biogenic process may affect the crystal phase formation of TiO2. Different bio-processes have different influences. Here, our results suggest that using kelp as a biotemplate and the self-doping with N and I could suppress the phase transformation from anatase to rutile even at high temperatures. | ||
| Fig. 2 XRD patterns of biogenic-TiO2 and N-TiO2. | ||
Fig. 3a and b display the well retained structures of biogenic-TiO2 derived from kelp, which are similar to the original structures (Fig. 1d). From the FESEM images, we can see clearly the replicated hierarchical porous structures from the macro- to the nanoscale: a high surface area on the macroscale, a porous framework on the microscale, and a nanolayered structure on the nanoscale. The microstructure of the cross-section framework is highly porous with layers about 250 nm wide on the walls (Fig. 3b). The TEM image shows that the crystal sizes of biogenic-TiO2 calcined at 500 °C, all around 18 nm (Fig. 3d), are smaller on average than those of common N-TiO2 synthesized without a template of around 30 nm (Fig. 3c). The results coincide with the crystallite sizes of the samples calculated by employing the method proposed by Hall27 from the XRD pattern (Fig. 2). The interconnected nanolayered structure, which resembles the construction of membranes in the chromatophores of kelp, exists in the products as demonstrated also by TEM images (Fig. 3e). Each layer is around 30 nm thick, a little bit thinner than the original ones (∼50 nm), which can be attributed to the shrinkage resulting from calcination, and is confirmed to be the anatase crystalline phase only by the selected area electron diffraction (SAED) pattern in the inset of Fig. 3e. Such porous and nanolayered architectures with high surface areas are favorable for the absorption of light and the generation of photon-induced carriers, and thereby should enhance the photocatalytic properties.
 | ||
| Fig. 3 a FESEM image of a cross-section of biogenic-TiO2 derived from kelp calcined at 500 °C; the inset is an illustration of the cross-section of such a structure. b A magnification of the hole contained in the yellow rectangle in a. cTEM image of N-TiO2 synthesized without a template calcined under the same conditions as biogenic-TiO2, 500 °C. dTEM image of biogenic-TiO2 derived from kelp calcined at 500 °C. eTEM image of the layered nanostructure template from the chromatophore in biogenic-TiO2 calcined at 500 °C; the inset shows the SAED pattern. | ||
Fig. 4 shows the X-ray photoelectron spectroscopy (XPS) results for biogenic-TiO2 derived from kelp calcined at 500 °C. The high-resolution scanning spectrum of N 1s displayed in Fig. 4a shows two peaks, 398.1 eV and 399.8 eV. The 399.8 eV can be attributed to the N atoms located at the interstitial sites of the TiO2 lattice28 and the low binding energy component located at 398.1 eV could be due to substitutional N in the N–Ti–O structure, according to Sathish et al.29 The inset of Fig. 4b shows the contents of the main elements detected by XPS. Compared with carbonkelp (Fig. S1 of the supporting information†), elements like Na, P, Cl and Mg of the chlorophyll in the chromatophore were not detected in biogenic-TiO2 by XPS, proving that the dilute HCl has effectively removed them. In addition, the high-resolution XPS spectrum of the I 3d region (Fig. 4b) shows doublet peaks at 629.7 eV and 618.8 eV, respectively, suggesting that iodine exists in the biogenic-TiO2 sample. The binding energy at 629.7 eV and 618.8 eV should be ascribed to negatively charged iodine I− species.30 Compared with 630.5 eV and 619.2 eV in carbonkelp (Fig. S1 of the supporting information†) calcined under the same conditions as biogenic-TiO2, the binding energy shifts by 0.8 eV and 0.4 eV, respectively. Since the ionic radius of I− (0.216 nm) is much larger than that of O2− (0.124 nm) or Ti4+ (0.068 nm), the substitution of Ti4+ or O2− ions in the lattice by I− would obviously be difficult. So it is possible that the I− species is dispersed on the surface of anatase particles31 or at the interstitial sites of the TiO2 lattice. The atomic content of the I dopant is relatively low. As is well known, iodine in kelp can be released rapidly after it is immersed into water, of which 60–70% is in the form of I−. Fortunately, there is a dynamic process between the release and absorption of iodine; 10% of I− could be absorbed again into kelp by prolonging the soaking time appropriately.21 We will adjust the soaking time as well as the ratio of water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :kelp to improve the doping ratio in our future experiments. Fig. 4c shows a Ti 2p XPS spectrum. Two peaks were observed at 464.25 eV and 458.45 eV, which were assigned to Ti 2p1/2 and Ti 2p3/2 respectively, agreeing well with Ti(IV) in pure anatase titanium.32 However, no trace of Ti3+ was observed by XPS, although it was observed by EPR spectroscopy (see Fig. 6). This may be attributed to either low amounts of Ti3+ which is hard to detect by the XPS technique or because the Ti3+ species exists in the subsurface region which is inaccessible by XPS.33 The O 1s XPS spectrum for biogenic-TiO2 shown in Fig. 4d can be fitted by three peaks at 532.37 eV, 531.35 eV and 529.72 eV, suggesting three independent environments for O within biogenic-TiO2. The peak component centred at 529.72 eV corresponds to oxygen in the oxide lattice while the 532.37 eV peak is due to surface OH groups.34 The component at intermediate binding energy (ca. 531.35 eV) can be assumed to be the result of Ti–N–O or N–Ti–O structures,35 as commented above in the case of the N 1s region, in agreement with Spadavecchia et al.36
:kelp to improve the doping ratio in our future experiments. Fig. 4c shows a Ti 2p XPS spectrum. Two peaks were observed at 464.25 eV and 458.45 eV, which were assigned to Ti 2p1/2 and Ti 2p3/2 respectively, agreeing well with Ti(IV) in pure anatase titanium.32 However, no trace of Ti3+ was observed by XPS, although it was observed by EPR spectroscopy (see Fig. 6). This may be attributed to either low amounts of Ti3+ which is hard to detect by the XPS technique or because the Ti3+ species exists in the subsurface region which is inaccessible by XPS.33 The O 1s XPS spectrum for biogenic-TiO2 shown in Fig. 4d can be fitted by three peaks at 532.37 eV, 531.35 eV and 529.72 eV, suggesting three independent environments for O within biogenic-TiO2. The peak component centred at 529.72 eV corresponds to oxygen in the oxide lattice while the 532.37 eV peak is due to surface OH groups.34 The component at intermediate binding energy (ca. 531.35 eV) can be assumed to be the result of Ti–N–O or N–Ti–O structures,35 as commented above in the case of the N 1s region, in agreement with Spadavecchia et al.36
 | ||
| Fig. 4 X-Ray photoelectron spectra of biogenic-TiO2 calcined at 500 °C. a N 1s XPS spectrum. b I 3d XPS spectrum, with the inset showing atomic contents of each element. cTi 2p XPS spectrum. d O 1s XPS spectrum. | ||
Fig. 5a shows the UV-vis diffuse reflectance spectra of biogenic-TiO2 derived from kelp and that of common N-TiO2. Compared with N-TiO2 nanoparticles, the absorption measurements of the biogenic-TiO2 samples show enhanced and stronger photoabsorption in the range of wavelengths from 400 nm to 550 nm. We attribute the light-harvesting enhancements to the hierarchical porous structures of biogenic-TiO2 from the macro- to the nanoscale: a high surface area on the macroscale, a porous framework on the microscale, and a nanolayered structure on the nanoscale. In addition, the band-gap absorption onsets, at the edge of UV and visible light, show red-shifts of more than 20 nm. This may be caused by the self-doping of N and I derived from kelp. The indirect relationship between the band gap Eg and the absorption coefficient α is also obtained from a plot of (αhν)nversus the photo energy (hν) using the following equation:37
| (αhν)n ∝ hν − Eg |
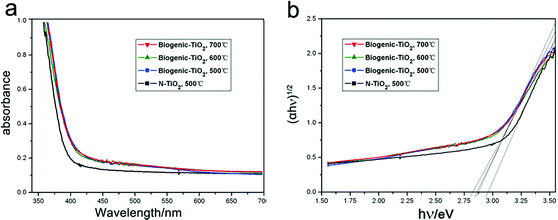 | ||
| Fig. 5 a UV-Vis diffuse reflectance spectra of biogenic-TiO2 and N-TiO2. b The corresponding Tauc plot of biogenic-TiO2 and N-TiO2. | ||
In order to examine the paramagnetic species in the biogenic-TiO2 samples, electron paramagnetic resonance (EPR) studies were also performed. Fig. 6 shows the EPR spectrum of biogenic-TiO2 (calcined at 500 °C) recorded at room temperature. The symmetric signal at g = 2.004 is clearly detected. It is the most prominent electron signal and is characteristic of paramagnetic materials containing “F-centers” or oxygen vacancies.39 Previous EPR studies suggest that signals with g values of less than 2.0 can be attributed to photogenerated electrons stabilized by Ti cations located at crystallization defects.40 It is well known that these trapped electrons could reduce Ti4+ to form Ti3+ paramagnetic species40 according to the following process: e− + TiO2(Ti4+) → TiO2(Ti3+), or alternatively, could reduce surface-absorbed O2 to form superoxide ions (O2−).33 The resonances at g values in the range of 2.0 to 2.08 are known to be due to photogenerated holes trapped by subsurface lattice oxygens.40 These holes, localized on the oxygen vacancies, react with the O2− and OH− to form O˙ and OH˙ radicals on the catalyst surface, both of which are reactive species and are responsible for the oxidative decomposition of organic pollutants. As shown in Fig. 6, the g values of trapped hole and electron are all observed, proving the presence of Ti3+ ions and the superoxide radical in the biogenic-TiO2 sample, which could facilitate photocatalysis.
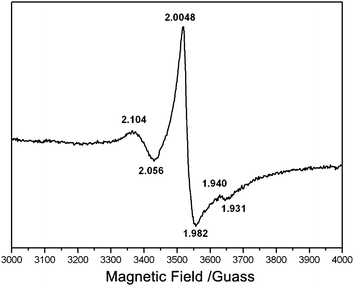 | ||
| Fig. 6 EPR spectrum of biogenic-TiO2 calcined at 500 °C. | ||
Photocatalytic activity for methylene blue degradation
Photocatalytic activities were examined for the degradation of methylene blue under solar irradiation as shown in Fig. 7. The degradation process obeys pseudo-first-order kinetics and the degradation rate can be extracted by plotting the natural logarithm of the absorbance against irradiation time.41 From Fig. 7, the mean decomposition rates of biogenic-TiO2 calcined at 500 °C, 600 °C and 700 °C under solar irradiation are (0.074 ± 0.003) min−1, (0.072 ± 0.004) min−1 and (0.077 ± 0.001) min−1, respectively, about 2.18, 2.12 and 2.26 times the rate of N-TiO2, which is (0.034 ± 0.002) min−1. Compared to N-TiO2, the biogenic-TiO2 is endowed with superior photocatalytic properties under solar irradiation. The enhanced catalytic activities of biogenic-TiO2 may result from the synergy of its structures and components. Hierarchical structures are favorable for enhancing the photocatalytic performance as highlighted above that the high surface area on the macroscale, the porous framework on the microscale, and the nanolayered structures on the nanoscale endow the samples with larger specific surface areas which could capture more photons and offer more absorption and reaction sites for the photocatalytic reaction. Meanwhile, the self-doping of nitrogen and iodine could enlarge the band gap absorption, and the presence of Ti3+ ions and the superoxide radical are both responsible for the degradation of methylene blue. Furthermore, the smaller mean diameter42 of biogenic-TiO2 and higher crystallinity of the sample were helpful. It is known that well crystallized anatase may facilitate the transfer of photoelectrons from the bulk to the surface and thus inhibit their recombination with photoholes, leading to enhanced quantum efficiency. | ||
| Fig. 7 Kinetic study of the degradation of methylene blue in the presence of biogenic-TiO2 and N-TiO2 under solar irradiation. The error bars in the figure represent the standard deviations of three independent measurements for each data point. | ||
Apart from its high activity, biogenic-TiO2 also shows good stability and durability during the photocatalytic degradation of methylene blue solution. As shown in Fig. 8, after recycling 3 times, the biogenic-TiO2 sample calcined at 500 °C can still completely degrade the methylene blue within 45 min, suggesting that biogenic-TiO2 has a good reusability.
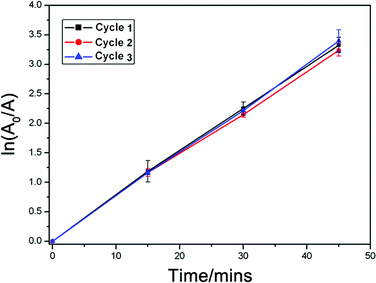 | ||
| Fig. 8 Recycling testing of the biogenic-TiO2 sample calcined at 500 °C for the photocatalytic degradation of methylene blue. The error bars in the figure represent the standard deviations of three independent measurements for each data point. | ||
Conclusions
Biogenic-TiO2, reproducing the structure of kelp and introducing nitrogen and iodine simultaneously in a self-doping manner, exhibits high light-harvesting and photocatalytic activity in methylene blue degradation under solar energy irradiation. Although the amount of doped iodine is not sufficient, which will be improved in future experiments, our results may represent an important first step towards the self-doping of metal oxides templated with natural plants besides N-doping. We believe that, as for TiO2, improvements in photocatalysis, luminescence or gas-sensing properties of oxides like ZnO, In2O3, CeO2, Cu2O and so forth could also be realized by copying the special structures or self-doping of non-metallic ions derived from various kinds of organisms endowed by nature.43 Besides using biogenic oxides in dye degradation, our strategy also provides inspiration for the further promise of applying them to produce hydrogen, solar cells, photoinduced sensors, photoelectrical devices and so forth.Mother nature is amazing; some special functions or structures we have been taking decades to work out may have existed on the earth for millions of years. For instance, no artificial water-splitting devices do better than chloroplasts in nature. Dye-sensitized solar cells, consisting of iodide/triiodide redox electrolytes between two electrodes, were recently noticed to have a similar sandwich structure to kelp, in which most of the chromatophores exist in the upper and lower epidermal layers and different forms of iodine (I−, IO3− and organic iodine, etc.15) are in the vacuoles of the interlayer. Therefore, inspired by the epidermal layers of kelp, a biogenic-TiO2 mesoporous film would be a novel, highly efficient photoanode structure for dye-sensitized solar cells.
Experimental
Materials
All chemical reagents were commercially available and chemically pure. Glutaraldehyde, sodium dihydrogen phosphate, disodium hydrogen phosphate, titanium trichloride and hydrochloric acid were all produced by Sinopharm Chemical Reagent Co., Ltd.Pretreatment
Fresh kelp samples were treated with 2% glutaraldehyde/phosphate buffered saline (PBS; pH = 7.2) solution at 4 °C for 6 h for the fixation of cells and tissues. The as-treated samples were then rinsed with 0.2% PBS and pure water and kept at 0 °C for further use.Synthesis procedure
The fixed kelp samples were treated with 5% HCl solution for 2 h. After rinsing with pure water, the as-treated samples were dipped into 5% TiCl3 solution and left to stand under vacuum for 24 h, with the color turning to purple brown. Afterwards, the samples were rinsed with pure water and were desiccated in an aerated oven at 40 °C, 60 °C and 80 °C successively for 2 h at each temperature. Finally, the samples were calcined in air at 280 °C for 2 h and then at 500 °C, 600 °C and 700 °C for 2 h, respectively, with a ramping rate of 1 °C min−1. Common N-TiO2 was prepared from a mixture of 25% ammonia solution and 15% TiCl3 solution with a 1:1 volume ratio, and calcined under the same conditions.
Characterization
Pieces of fixed original kelp and Ti-substituted samples were embedded into a tissue freezing medium (Leica Instruments GmbH) at −25 °C. Cross-sections of 15 μm thickness were prepared using a cryo-microtome (Leica CM3050-Cryostat, Leica Instruments GmbH) at −24 °C, and collected on poly-L-lysine-coated slides. Fluorescence microscopy was carried out using a Laser-Scan-Microscope (LSM 510, Carl Zeiss) including an Axioveit 200M microscope. Chlorophyll fluorescence was excited by 488 nm light from a 30 mW argon ion laser. Excitation light was separated by a FT 488/543 dichroic mirror from emission light which was passed through a LP 560 band-pass filter. UV excitation light was split first by HFT 650 then NFT 545 and 490 dichroic mirrors from emission light which was passed through a BP 435–485 band-pass filter.The cross-sections of 15 μm samples were also collected on conductive glass slides, together with biogenic-TiO2 derived from kelp. They were sputtered with gold and observed using a field emission scanning electron microscope (FESEM; FEI SIRION 200) operated at 5 kV.
In order to observe the ultrastructure of chloroplasts and the layered nanostructure after converting the bio-precursor to biogenic-TiO2, the glutaraldehyde-fixed original kelp samples were further placed in 1% OsO4PBS solution at 4 °C for 2 h. The specimens, dehydrated in a graded EtOH series, were embedded in a low viscosity epoxy resin and cut on a Leica UCT ultra-microtome. Sections were collected on carbon-coated copper grids and stained with uranyl acetate and lead citrate. The biogenic-TiO2 was suspended in ethanol by ultrasonic treatment and then dropped onto copper grids. The samples were examined using a transmission electron microscope (TEM; JSM-2010, JEOL) equipped with energy dispersive X-ray spectroscopy capability (EDS, OXFORD INCA) at an applied voltage of 200 kV.
The images of the cuticle of the original leaves were obtained using a digital microscope (VHX-100, KEYENCE) and a fluorescence microscope (Lecia DM 2500).
X-Ray diffraction (XRD) measurements were performed to measure the crystal phase of the samples on a Bruker-AXS X-ray diffractometer system with Cu-Kα radiation at 40 kV and 20 mA. The spectra were recorded in the 2θ range from 20° to 90° with a scanning step of 0.02° s−1.
The X-ray photoelectron spectrum (XPS) of the artificial N-doped TiO2 photocatalyst was measured on a Thermo ESCALAB spectrometer with monochromatized Al Kα X-rays (hν = 1486.6 eV) at a pass energy of 20 eV. The C 1s peak of contaminant carbon was calibrated to 284.8 eV.
UV-Visible (UV-vis) diffuse reflectance spectra of all the samples were recorded using a Varian Cary UV-Vis-NIR spectrophotometer in the spectral range of 300–800 nm. 0.30 g of each sample was pressed between two pieces of quartz glass within the 3 × 3 cm2 area to cover the aperture through which excitation light passes. A BaSiO4 plate was used as a basis line for the spectra. All the samples were assumed to have the same quality during the measurement.
EPR measurements were carried out on a Bruker EMX-8/2.7 X-band EPR spectrometer operating in the X-band at 9.86 GHz and 2.005 mW. Portions of the solid samples (∼20 mg) were introduced into a spectroscopic quartz probe cell and the measurements were taken at room temperature.
The photocatalytic activity for methylene blue degradation was measured according to the following experiments. 0.1 g of the TiO2 sample was dispersed in 100 mL of 10−5 mol L−1methylene blue solution with pure water as the solvent. The TiO2 suspended solution was stirred in the dark for 1 h to reach the adsorption–desorption equilibrium and then placed under a 500 W Xe lamp. The degradation was monitored by taking aliquots every 5 min. These aliquots were centrifuged and then tested under a 25Lamda UV-vis spectrometer to obtain the absorption spectra. The rate of degradation was assumed to obey pseudo-first-order kinetics and the degradation rate constant, k, was obtained according to the following equation: ln(A0/A) = kt. A0 is the initial absorbance, namely, the characteristic absorbance peak of the original methylene blue solution. A is the characteristic absorbance peak after a degradation time t. In the recycling experiments, the degraded solution with biogenic-TiO2 photocatalyst suspended in it was centrifuged at 3000 r min−1 for 5 min. Then the supernatant was poured out and pure water was added to the remaining precipitate for washing. The performance was repeated three times to obtain the clean biogenic-TiO2 photocatalyst. The obtained wet biogenic-TiO2 was dried at 25 °C for the next degradation cycle.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 50972090), the National Basic Research Program of China (No. 2006CB601200), the Dawn program of the Shanghai Education Commission (08SG15) and the Shanghai Rising-Star Program (No. 10QH1401300).References
- A. E. Curtright, M. G. Morgan and D. W. Keith, Environ. Sci. Technol., 2008, 42, 9031 CrossRef CAS.
- M. Ni, M. K. H. Leung, D. Y. C. Leung and K. Sumathy, Renewable Sustainable Energy Rev., 2007, 11, 401 CrossRef CAS; T. L. Thompson and J. T. Yates Jr, Chem. Rev., 2006, 106, 4428 CrossRef CAS; J. L. Zhang, Y. M. Wu, M. Y. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715 Search PubMed.
- A. L. Linsebigler, G. Q. Lu and T. Yates, Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS.
- S. Yin, H. Yamaki, M. Komatsu, Q. Zhang, J. Wang, Q. Tang, F. Saito and T. Sato, J. Mater. Chem., 2003, 13, 2996 RSC.
- J. Zhang, F. Shi, J. Lin, D. Chen, J. Gao, Z. Huang, X. Ding and C. Tang, Chem. Mater., 2008, 20, 2937 CrossRef CAS; Z. G. Zhao and M. Miyauchi, Angew. Chem., Int. Ed., 2008, 47, 7051 CrossRef CAS; T. Kuo, C. Lin, C. Kuo and M. H. Huang, Chem. Mater., 2007, 19, 5143 CrossRef CAS; C. Zhang and Y. Zhu, Chem. Mater., 2005, 17, 3537 CrossRef CAS.
- M. Takeuchi, H. Yamashita, M. Matsuoka, M. Anpo, T. Hirao, N. Itoh and N. Iwamoto, Catal. Lett., 2000, 67, 135 CrossRef CAS; H. Yamashita, M. Harada, J. Misaka, M. Takeuchi, B. Neppolian and M. Anpo, Catal. Today, 2003, 84, 191 CrossRef CAS; T. Ikeda, T. Nomoto, K. Eda, Y. Mizutani, H. Kato, A. Kudo and H. Onishi, J. Phys. Chem. C, 2008, 112, 1167 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS; H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS; M. Miyauchi, A. Ikezawa, H. Tobimatsu, H. Irie and K. Hashimoto, Phys. Chem. Chem. Phys., 2004, 6, 865 RSC.
- T. Ohno, T. Mitsui and M. Matsumura, Chem. Lett., 2003, 32, 364 CrossRef CAS; T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. Ingler, Science, 2002, 297, 2243 CrossRef CAS; T. Ohno, T. Tsubota, K. Nishijima and Z. Miyamoto, Chem. Lett., 2004, 33, 750 CrossRef CAS.
- M. Valden, X. Lai and D. W. Goodman, Science, 1998, 281, 1647 CrossRef CAS; D. R. Rolison, Science, 2003, 299, 1698 CrossRef CAS.
- T. X. Fan, S. K. Chow and D. Zhang, Prog. Mater. Sci., 2009, 54, 542 CrossRef CAS.
- A. R. Parker, R. C. Mcphedran, D. R. Mckenzie, L. C. Botten and N. P. Nicorovici, Nature, 2001, 409, 36 CrossRef CAS; K. H. Jeong, J. Kim and L. P. Lee, Science, 2006, 312, 557 CrossRef CAS; C. Sanchez, H. Arribart and M. M. G. Guille, Nat. Mater., 2005, 4, 277 CrossRef CAS; M. Sarikaya, C. Tamerler, A. K.-Y. Jen, K. Schulten and F. Baneyx, Nat. Mater., 2003, 2, 577 CrossRef CAS.
- X. F. Li, T. X. Fan, H. Zhou, S. K. Chow, W. Zhang, D. Zhang, Q. X. Guo and H. Ogawa, Adv. Funct. Mater., 2009, 19, 45 CrossRef CAS.
- D. Gust, D. Kramer, A. Moore, T. A. Moore and W. Vermaas, MRS Bull., 2008, 33, 383 CAS.
- X. L. Hou, C. F. Chai, Q. F. Qian, X. J. Yan and X. Fan, Sci. Total Environ., 1997, 204, 215 CrossRef.
- X. T. Hong, Z. P. Wang, W. M. Cai, F. Lu, J. Zhang, Y. Z. Yang, N. Ma and Y. J. Liu, Chem. Mater., 2005, 17, 1548 CrossRef CAS.
- C. K. Tseng and C. F. Chang, Acta Bot. Sin., 1961, 9, 316 Search PubMed.
- X. F. Wang, C. H. Zhan, T. Maoka, Y. J. Wada and Y. Koyama, Chem. Phys. Lett., 2007, 447, 79 CrossRef CAS.
- E. Shimoni, O. Rav-Hon, I. Ohad, V. Brumfeld and Z. Reich, Plant Cell, 2005, 17, 2580 CrossRef CAS; E. H. Delucia, K. Nelson, T. C. Vogelmann and W. K. Smith, Plant, Cell Environ., 1996, 19, 159 CrossRef; M. E. Poulson and T. C. Vogelmann, Plant, Cell Environ., 1990, 13, 803 CrossRef.
- F. C. Küpper, N. Schweigert, E. A. Gall, J. M. Legendre, H. Vilter and B. Kloareg, Planta, 1998, 207, 163 CrossRef.
- S. X. Wu, W. H. Xin and Q. Y. Chao, J. Environ. Sci., 2005, 17, 241 Search PubMed.
- H. Kupper, F. Kupper and M. Spiller, Photosynth. Res., 1998, 58, 123 CrossRef CAS; P. Nyitrai, K. Boka, L. Gaspar, E. Sarvari, K. Lenti and A. Keresztes, J. Plant Physiol., 2003, 160, 1175 CrossRef CAS; A. D. Matuszek, A. Skalan, A. Karocki, G. Stochel and L. Fiedor, J. Biol. Inorg. Chem., 2005, 10, 453 CrossRef.
- B. Ohtani, Y. Ogawa and S. Nishimoto, J. Phys. Chem., 1997, 101, 3746 Search PubMed.
- Z. Ding, G. Q. Lu and P. F. Greenfield, J. Phys. Chem. B, 2000, 104, 4815 CrossRef CAS.
- L. E. Depero, P. Bonzi, M. Zocchi, C. Casale and G. D. Michele, J. Mater. Res., 1993, 8, 2709 CAS.
- N. Kröger, M. B. Dickerson, G. Ahmad, Y. Cai, M. S. Haluska, K. H. Sandhage, N. Poulsen and V. C. Sheppard, Angew. Chem., Int. Ed., 2006, 45, 7239 CrossRef CAS.
- W. H. Hall, J. Inst. Met., 1949, 75, 1127 Search PubMed; G. K. Williamson and W. H. Hal, Acta Metall., 1953, 1, 22 CrossRef CAS.
- C. D. Valentin, G. Pacchioni, A. Selloni, S. Livraghi and E. Giamello, J. Phys. Chem. B, 2005, 109, 11414 CrossRef CAS; S. Livraghi, M. C. Paganini, E. Giamello, A. Selloni, C. D. Valentin and G. Pacchioni, J. Am. Chem. Soc., 2006, 128, 15666 CrossRef CAS.
- M. Sathish, B. Viswanathan, R. P. Viswanath and C. S. Gopinath, Chem. Mater., 2005, 17, 6349 CrossRef CAS.
- T. Sachiko, T. Takashi, F. Mamoru and M. Tetsuro, J. Phys. Chem. C, 2008, 112, 14948 CrossRef CAS.
- W. Y. Su, Y. F. Zhang, Z. H. Li, L. Wu, X. X. Wang, J. Q. Li and X. Z. Fu, Langmuir, 2008, 24, 3422 CrossRef CAS.
- J. F. Zhu, F. Chen, J. L. Zhang, H. J. Chen and M. Anpo, J. Mol. Catal. A: Chem., 2004, 216, 35 CAS.
- G. D. Tang, Z. Jiang, H. H. Shi, T. C. Xiao and Z. F. Yan, J. Mater. Chem., 2010, 20, 5301 RSC.
- C. L. Bianchi, G. Cappelletti, S. Ardizzone, S. Gialanella, A. Naldoni, C. Oliva and C. Pirola, Catal. Today, 2009, 144, 31 CrossRef CAS; T. L. Thompson and J. T. Yates, Top. Catal., 2005, 35, 197 CrossRef CAS.
- F. Spadavecchia, G. Cappelletti, S. Ardizzone, C. L. Bianchi, S. Cappelli, C. Oliva, P. Scardi, M. Leoni and P. Fermo, Appl. Catal., B, 2010, 96, 314 CrossRef CAS; E. György, A. Pérezdel Pino, P. Serra and J. L. Morenza, Surf. Coat. Technol., 2003, 173, 265 CrossRef CAS.
- F. Spadavecchia, G. Cappelletti, S. Ardizzone, C. L. Bianchi, S. Cappelli, C. Oliva, P. Scardi, M. Leoni and P. Fermo, Appl. Catal., B, 2010, 96, 314 CrossRef CAS.
- Y. M. Wu, M. Y. Xing, J. L. Zhang and F. Chen, Appl. Catal., B, 2010, 97, 182 CrossRef CAS; J. Tauc, Mater. Res. Bull., 1970, 5, 721 CrossRef CAS.
- J. A. Rengifo-Herrera, K. Pierzchala, A. Sienkiewicz, L. Forro, J. Kiwi and C. Pulgarin, Appl. Catal., B, 2009, 88, 398 CrossRef CAS; H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS.
- I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205 CrossRef CAS; E. A. Reyes-Garcia, Y. Sun, K. R. Reyes-Gil and D. Raftery, Solid State Nucl. Magn. Reson., 2009, 35, 74 CrossRef CAS; E. Serwicka, Colloids Surf., 1985, 13, 287 CrossRef CAS.
- S. K. Joung, T. Amemiya, M. Murabayashi and K. Itoh, Chem.–Eur. J., 2006, 12, 5526 CrossRef CAS; J. M. Coronado, A. J. Maira, J. C. Conesa, K. L. Yeung, V. Augugliaro and J. Soria, Langmuir, 2001, 17, 5368 CrossRef CAS; N. M. Dimitrijevic, Z. V. Sapomjic, B. M. Rabatic, O. G. Poluektov and T. Rajh, J. Phys. Chem. C, 2007, 111, 14597 CrossRef CAS.
- S. C. Pillai, P. Periyat, R. George, D. E. McCormack, M. K. Seery, H. Hayden, J. Colreavy, D. Corr and S. J. Hinder, J. Phys. Chem. C, 2007, 111, 1605 CrossRef CAS.
- X. T. Hong, Z. P. Wang, W. M. Cai, F. Lu, J. Zhang, Y. Z. Yang, N. Ma and Y. J. Liu, Chem. Mater., 2005, 17, 1548 CrossRef CAS.
- D. H. Lee, T. Gutu, C. Jeffryes, G. L. Rorrer, J. Jiao and C. H. Chang, Electrochem. Solid-State Lett., 2007, 10, K13 CrossRef CAS; C. Jeffryes, T. Gutu, J. Jiao and G. L. Rorrer, J. Mater. Res., 2008, 23, 3255 CrossRef CAS; J. W. Galusha, M. R. Jorgensen and M. H. Bartl, Adv. Mater., 2010, 22, 107 CrossRef CAS; J. W. Galusha, L. R. Richey, M. R. Jorgensen, J. S. Gardner and M. H. Bartl, J. Mater. Chem., 2010, 20, 1277 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: XPS pattern in all elements binding energy region of kelp. See DOI: 10.1039/c0ee00363h |
| This journal is © The Royal Society of Chemistry 2011 |