Improved conductivity in dye-sensitised solar cells through block-copolymer confined TiO2 crystallisation†
Stefan
Guldin
a,
Sven
Hüttner
a,
Priti
Tiwana
b,
M. Christopher
Orilall
c,
Burak
Ülgüt
a,
Morgan
Stefik
c,
Pablo
Docampo
b,
Matthias
Kolle
ad,
Giorgio
Divitini
e,
Caterina
Ducati
e,
Simon A. T.
Redfern
f,
Henry J.
Snaith
b,
Ulrich
Wiesner
c,
Dominik
Eder
e and
Ullrich
Steiner‡
*a
aCavendish Laboratory, Department of Physics, University of Cambridge, J J Thomson Avenue, Cambridge, CB3 0HE, UK
bDepartment of Physics, Clarendon Laboratory, University of Oxford, OX1 3PU, UK
cDepartment of Materials Science & Engineering, Cornell University, Ithaca, New York 14853, USA
dNanoscience Centre, University of Cambridge, 11 J J Thomson Avenue, Cambridge, CB3 0FF, UK
eDeparment of Materials Science and Metallurgy, University of Cambridge, Pembroke Street, Cambridge, CB2 3QZ, UK
fDepartment of Earth Sciences, University of Cambridge, Downing Street, Cambridge, CB2 EQ, UK
First published on 20th October 2010
Abstract
Anatase TiO2 is typically a central component in high performance dye-sensitised solar cells (DSCs). This study demonstrates the benefits of high temperature synthesised mesoporous titania for the performance of solid-state DSCs. In contrast to earlier methods, the high temperature stability of mesoporous titania is enabled by the self-assembly of the amphiphilic block copolymer polyisoprene-block-polyethylene oxide (PI-b -PEO) which compartmentalises TiO2 crystallisation, preventing the collapse of porosity at temperatures up to 700 °C. The systematic study of the temperature dependence on DSC performance reveals a parameter trade-off: high temperature annealed anatase consisted of larger crystallites and had a higher conductivity, but this came at the expense of a reduced specific surface area. While the reduction in specific surface areas was found to be detrimental for liquid-electrolyte DSC performance, solid-state DSCs benefitted from the increased anatase conductivity and exhibited a performance increase by a factor of three.
Broader contextDye sensitised solar cells (DSCs) consist of an inorganic electron conducting skeleton, a light absorbing dye and a hole conductor. The inorganic component is typically assembled from randomly sintered 20 nm-sized TiO2 particles. Self-assembled block copolymers can be used to define structural motives on the nanometer scale. In combination with sol–gel chemistry, the macromolecules act as structure directing agent for inorganic material synthesis, offering a powerful tool for novel electrode architectures.This study presents a facile method for the synthesis of well-defined TiO2 morphologies and highlights its potential for the fabrication of highly conductive mesoporous films. The block-copolymer PI-b-PEO rapidly self-assembles to confine the TiO2 sol into an ordered spatial structure, replicating the organic matrix. This structural confinement allows extended annealing at high temperatures, which improves overall TiO2 crystallinity and promotes the growth of larger crystallites. The presented work spans from inorganic chemical synthesis over polymer self-assembly and the evolution of crystallinity to DSC device performance, which benefits from an improved TiO2 conductivity when annealed at high temperatures. |
Introduction
The interest in nanocrystalline, mesoporous TiO2 has emerged in recent years for a variety of “green” applications such as dye-sensitised solar cells (DSCs),1 photocatalysis,2 electrochemical capacitors,3 sensors,4 and electrochromic devices.5 Traditionally, mesoporous TiO2 is obtained by sintering a random network of 10–20 nm sized particles which are derived by sol–gel chemistry.6 The high surface area of these films is decisive for interface sensitive applications. Other important material properties include a large electronic band-gap, long-term stability against corrosion and a high surface affinity for dyes. The use of a network of randomly sintered particles has drawbacks, however. While the electron mobility in crystalline anatase TiO2 is usually on the order of μ ∼ 10 cm2/Vs, this value decreases by five to six orders of magnitude in nanoparticle films.7,8 The low electron mobility in nanoparticle TiO2 films appears to limit charge collection and thereby power conversion efficiency in solid-state DSCs.9 Furthermore, the lack of control over the pore size distribution may hinder pore infiltration by the viscous materials required to assemble this promising type of DSCs.10 Controlling the morphology and crystallinity on the 10 nm length scale in TiO2 films are therefore key factors for further development. A promising recipe for the synthesis of TiO2 with better controlled pore structures involves block copolymer directed self-assembly of the titania sol. Wiesner and coworkers11 as well as Stucky and coworkers12 pioneered this sol–gel approach for silica, which was subsequently extended to other ceramics.13Titania has attracted particular interest by a large number of groups.13–16 In these material systems, a sol selectively swells one part of an amphiphilic block-copolymer which typically microphase separates into ordered domains on the 5 to 50 nm length-scale. The incorporated inorganic material benefits therefore from the structure-directing property of the macromolecule even after the polymer is removed in a high temperature annealing step. Several groups have successfully used the structure directing agent Pluronic P123 (BASF) to derive mesoporous titania for dye sensitised photovoltaics,17,18 but several challenges remain, especially for solid-state devices. The synthesis is rather time-consuming due to the slow self-assembly process and can take several days.14 In order to prevent structural collapse, calcination is typically carried out at temperatures around 400 °C which leads to poor crystallinity (less than 50%19,20) in comparison to sintered nanoparticle films. Ozin and coworkers studied the evolution of P123-derived crystallinity in situ during the annealing process.19 Transformation into a fully crystalline material at high annealing temperatures leads to the collapse of the structure.20 This can be attributed to the fact that nanocrystals grow at typical crystallisation temperatures to diameters of 10–20 nm, which by far exceed the length scales of the self-assembled P123 structure and therefore causing the micro-morphology to break down during annealing. Alternative concepts include backfilling the mesopores with carbon or silica21 or elaborate annealing and crystallisation protocols14 to enhance temperature stability or the addition of preformed TiO2 nanocrystals to decrease the crystallisation temperature.22 The simultaneous achievement of high crystallinity and structural integrity remains however a challenge. Furthermore phase impurities have repeatedly been reported with finite amounts of TiO2 (B)23 and traces of rutile.24 Both these phases are less favorable for solar cell operation.25 One of the main limiting factors is the relatively low molecular weight of the commercially available Pluronic compound. The resulting small pore sizes limit the infiltration of dye and hole-transporting phase, which poses a problem particularly for the assembly of solid-state devices.Recently, copolymer systems of higher molecular weights such as poly[(ethylene-co-butylene)-b-(ethylene oxide)] (PHB-b-PEO) were introduced, which offer significantly thicker pore walls compared to P123-derived materials, leading to more stable structures during annealing at up to 700 °C for short annealing times (10 min).16,20 The high molecular weight PI-b-PEO copolymer used in the present study exhibits several intrinsic characteristics which makes it well-suited as a structure-directing agent in polymer-derived inorganic nanostructures.11,26,27 Most important are the high Flory-Huggins interaction parameter of the PI-PEO couple of χ(T) = 65/T + 0.125 and the low glass transition temperatures of both blocks. The large degree of polymerisation of both blocks of the copolymer used in this study of NPEO = 190, and NPI = 400 exceed values of conventional structure-directing agents and enable robust and fast phase segregation which typically exhibits long-range order. In contrast to earlier studies using low molecular weight copolymers with long hydrophilic blocks, the large value of N and the high volume fraction (76.9%) of the hydrophobic PI block give rise to a porous network with large enough pores to facilitate the infiltration of the network with solid-state hole conducting materials28 as well as yielding a highly porous inorganic material which is less susceptible to shrinkage and structural collapse. Furthermore, the sp2 hybridized carbon atoms in PI can be converted to a carbonaceous scaffold under non-oxidising heating conditions, which helps to keep the mesostructure intact even at high temperatures.29,30 The insolubility of PI-b-PEO in aqueous solvents poses, however, a challenge when used with hydrolytic sol–gel chemistries, particularly in thin film processing.
Here, we report a material synthesis route using the amphiphilic PI-b-PEO block copolymer and sol–gel chemistry for the rapid and facile manufacture of well defined and temperature robust mesoporous TiO2 films. Three aspects were studied with the aim to improve mesoporous anatase scaffolds for DSCs. (1) A solvent exchange was introduced in the synthesis procedure to improve the film processing and the formation of the self-assembled mesostructure. (2) The time and temperature dependence of anatase formation was studied and correlated with the crystallite size, surface area, and conductivity of the mesoporous network. (3) Liquid and solid-state DSCs based on the anatase series from (2) were manufactured and their electronic properties are discussed in the context of crystallinity, porosity, and connectivity of the porous TiO2 scaffold.
Results and discussion
Structure formation and stability
In difference to Pluronic-based systems, PI-b -PEO is insoluble in water or ethanol. A typical approach for synthesising anatase TiO2 consists therefore of separately preparing a water-based sol containing a Ti salt or alkoxide and a solution of the block copolymer in tetrahydrofuran (THF). The two solutions are then mixed before allowing the solvents to evaporate, which results in a self-assembled organic-inorganic hybrid schematically shown in Fig. 1a. In thin film processing, the structure formation occurs on a much faster time scale. The differing boiling points of water and the organic solvents therefore often lead to quenched morphologies with poor structure control. For high inorganic loadings, the low solubility of polymer in the water rich solvent mixture imposes another challenge. We have addressed these limitations by adding a solvent exchange step to the sample preparation procedure. The initial mixture was slowly dried in a Petri dish and the resulting hybrid material was directly redissolved in a mixture of 1-butanol and toluene in a ratio that allowed the formation of an azeotrope31(a solvent mixture of equal boiling point). The azeotrope dissolves both the hydrophilic and hydrophobic phases of the hybrid material while exhibiting a common high boiling point. This results in a better solubility of the polymer and titania sol as well as a slower evaporation of the solvents, which gives rise to better ordered local structure and film formation of the dried hybrid material. Further experimental details about the redissolution step and its effect on the morphology of resulting films are given in the Supporting Information (SI).Fig. 1 demonstrates the structural control of this approach. The role of the amphiphilic block copolymer as structure-directing agent organising the inorganic titania sol in a well defined network is illustrated in Fig. 1a. Structural order after film deposition and high temperature annealing was probed by small angle X-ray scattering (SAXS), shown in Fig. 1b. The characteristic length-scale (d-spacing) of the self-assembled structure was 30 nm, shrinking to 27 nm after removal of the organic template for temperatures above 370 °C. The first-order peak at ∼0.23 nm−1 disappeared only at 740 °C, indicating a loss of structural order at this temperature. Electron microscopy confirmed these observations. Fig. 1c shows a TEM image of a hybrid film before calcination, exhibiting close-range order of the packed micelles. No straight lines were observed in any of the samples, eliminating interpretation as hexagonal cylinders. Note that this film was simply spin-coated from the 1-butanol/toluene solution without further time-consuming annealing procedures to promote phase separation. The cross-sectional SEM image of the heat-treated (500 °C) film in Fig. 1d shows that the self-assembled morphology remains intact after calcination of the organic component and crystallisation of inorganic matrix. The collapse of the pores observed at 740 °C is evidenced by the SEM cross-section in Fig. 1e.
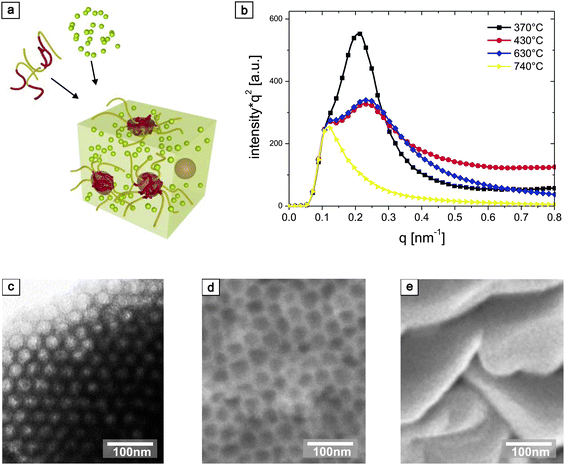 | ||
| Fig. 1 Film deposition and structural stability. (a) Hydrolytic TiO2 sol when mixed with a PI-b-PEO solution is structure-directed by copolymer self-assembly, giving rise to nanostructured TiO2 after high-temperature annealing. (b) Small-angle X-ray scattering (SAXS) spectra for copolymer-derived TiO2 annealed at 370 °C (black squares), 430 °C (red circles), 630 °C (blue diamonds) and 740 °C (yellow triangles). The spectra are scaled in intensity to overlap in the 0–0.1 nm−1q-range. (c) TEM image of the micellar hybrid film produced by spin-coating the redissolved material. (d) Cross-sectional SEM image of the nanostructured TiO2 film after calcination and crystallisation of the anatase matrix in air at 500 °C. (e) Cross-sectional SEM image of a film annealed at 740 °C which resulted in the transformation of the crystal phase from anatase to rutile causing the loss of the mesopores. | ||
An ordered micellar morphology which is well defined on the local scale is observed for a number of different copolymer architectures and sol-loadings. This suggests that the micro-morphology originates from the packing structures that probably already exist in solution. In contrast to structure formation in bulk samples produced by drop-casting (where an equilibrated microphase morphology is observed), spin-cast films are presumably kinetically hindered from forming a self-assembled morphology dictated by the copolymer architecture in the melt. Supported by our results discussed below, it is important to note that, despite the micellar assembly a 3D bicontinuous network of titania and pores is obtained with structure control (through the micellar size) on the 10-nm length scale. The robustness of the micellar morphology with respect to the various system parameters is an advantage, simplifying the comparison of differently produced samples.
Crystallisation kinetics and crystal sizes
The overall crystallinity and crystallite size strongly influence charge transport properties of polycrystalline wide band-gap semiconductors and therefore play important roles in device applications. Annealing times and temperatures during crystal formation are obviously important in controlling these parameters. Fig. 2a shows wide-angle X-ray scattering (WAXS) patterns of TiO2 films annealed for 400 min at temperatures between 400 °C and 700 °C. Up to annealing temperatures of 700 °C, all reflections can be indexed to anatase (PDF 71–1167), confirming phase purity for these annealing conditions. The smaller angular range of Fig. 2b shows the evolution of the width of the anatase (101) peak with temperature after an annealing time of 400 min. The Debye Scherrer analysis of the full-width at half maximum (FWHM) of the (101) peak was used to define the mean crystallite size of the polycrystalline assembly, shown in Fig. 2c. The final crystallite size was found to be independent of the heating rate in the range of 1–10 °C/min. In contrast to phase purity up to 700 °C, a significant rutile content can be observed after annealing at 740 °C, evidenced by the appearance of the (110) peak at 27.3° (PDF 76–1940). A comparison of the SAXS and XRD results of Fig. 1b and Fig. 2b suggests that the structural collapse at 740 °C is caused by the stresses involved in the reconstructional transformation from anatase to rutile. | ||
| Fig. 2 Crystallisation of TiO2 anatase crystals in block-copolymer derived confinement (a) Wide angle X-ray diffraction scans of mesoporous films annealed at 400 °C, 500 °C, 600 °C, and 700 °C (bottom to top). (b) Anatase (101) crystal footprint as a function of crystallisation temperature after annealing for 400 min. From bottom to top: 400 °C to 700 °C in 50 °C increments (structurally intact), and 740 °C (collapsed structure). (c) Debye-Scherrer mean crystallite sizes after annealing for 400 min as a function of temperature. (d) Evolution of crystallinity by monitoring the peak area as a function of time at 530 °C. (e) Crystallinity evolution with time as a function of temperature. Each stack represents an in situ experiment at a fixed temperature as shown in (d). (f) Dependence of anatase (101) peak on mass ratio of TiO2 to block copolymer. From bottom to top; black: 1to1 mass ratio (mean crystallite size: 9.5 nm); red: 2to1 mass ratio (14.7 nm); blue: sol only (23.6 nm). | ||
To investigate the time required for completion of the crystallisation process, we monitored the crystallite growth in situ during the calcination process by X-ray diffraction. The observed diffraction range for 2θ between 22° and 32° (Cu Kα radiation) contains the anatase (101) peak which was used for this analysis. A typical in situ scan of TiO2 during calcination at 530 °C is shown in Fig. 2d, indicating completion of the crystallisation process after about 100 min.
The evolution of the overall crystallinity with time was determined by comparing, at each temperature, the area of the anatase peak with the peak area for an annealing time of 400 min, which was defined as 100% crystallinity. The result of this analysis is shown in Fig. 2e, where the time required to reach values of the overall crystallinity of 50%–98% is plotted as a function of temperature. Each stack represents an in situ experiment at a fixed temperature as shown in Fig. 2d. While near-complete crystallinity (98%) was obtained after 100–150 min for temperatures between 530 °C and 690 °C, much longer times were needed for temperatures below 500 °C. The slow crystallisation for temperatures below 500 °C is noteworthy because mesoporous TiO2 employed in DSCs is often annealed at similarly low temperatures.
The careful investigation of all WAXS samples in reflection (Fig. 2a and b) and transmission (shown in the SI) showed no sign of an amorphous hump. Our results agree with a study by Fattakhova et al.20 reporting the rapid disappearance of the amorphous signal for annealing temperatures between 550 °C and 650 °C. Based on our measurements, we conclude that the amorphous content in all our samples was very low when annealed above 400 °C for several hours.
The role of the block-copolymer is further evidenced in Fig. 2f, which shows a strong dependence of the crystal size on the spatial confinement imposed by the organic template. Doubling the weight ratio of polymer to hydrolytic sol lead to a decrease in crystallite size by ≈30%. In the absence of the polymer compartmentalisation much larger crystals (>23nm) were observed at 500 °C. In addition, we have recently shown that the crystallization environment, tuned by the inorganic to organic ratio, is a decisive factor in determining the distribution of sub-bandgap electronic states and the associated electronic function in solid-state dye-sensitized solar cells.32
High resolution transmission electron microscopy (HRTEM) images of samples annealed at 400 °C and 650 °C are shown in Fig. 3a and b, respectively. The sample annealed for 400 min at 400 °C shows a heterogeneous assembly of small crystallites of 10 ± 5 nm. Increasing the temperature to 650 °C, lead to larger and more irregularly shaped crystals of around 20 nm, which probably derive from the coalescence of smaller grains. These results are consistent with the X-ray diffraction data analysis. The schematics in Fig. 3c and d illustrate crystal growth in copolymer derived confinement at low and high temperatures, respectively.
 | ||
| Fig. 3 Crystallite assembly within the mesoporous network. High resolution transmission electron microscopy (HRTEM) images of samples annealed at (a) 400 °C and (b) 650 °C. White dotted lines are superimposed on the images to visualize individual crystallites. (a) Sample annealed at 400 °C: small, randomly oriented crystals significantly smaller than the lengthscale of the porous matrix are observed. (b) Sample annealed at 650 °C: crystal size exceeds the length scale of the confinement. Anisotropic growth within the network is likely. (c,d) Schematic illustrating crystallite size and orientation in copolymer derived networks at (c) 400 °C and (d) 650 °C. | ||
Effect of high temperature annealing on conductivity and device performance
Dye sensitised solar cells consist of a bicontinuous random anatase network with porosity on the 10-nm scale. A molecular layer of dye is adsorbed onto the network surface and the pores are filled with either a liquid electrolyte or a molecular or polymeric hole-conducting material (solid-state DSC). The performance of DSCs hinges on the interplay of efficient charge generation and separation combined with a near-lossless charge transport through the n- and p-type materials to the respective electrodes. The morphology and electrical conductivity of the anatase network is therefore of central importance.Historically, copolymer-derived TiO2 films have been restricted to low annealing temperatures resulting in films which suffer from high resistivities. Since a low electron mobility has been identified as a limitation for the development of high-efficiency solid-state DSCs,9 this material synthesis route is particularly interesting for applications where poor charge carrier transport limits the device performance. Anatase resistivity is expected to depend sensitively on the degree of crystallinity, crystallite size, and topology of the porous network,33,34 all of which can be controlled by the present approach.
The annealing-temperature dependence of the resistivity of 900 nm thick mesoporous TiO2 films is shown in Fig. 4a. The film resistivity was determined by impedance spectroscopy measurements done on complete, liquid electrolyte based devices. The obtained data was fitted to a transmission line equivalent circuit model adapted from Ref. 35. Increasing the annealing temperature from 400 °C to 650 °C reduced the resistivity by over a factor of 3. We note that the sheet resistance of the bare FTO substrate increased slightly over this temperature range. When analysing these results in the light of the crystallization data shown in Fig. 2c, it is suggestive that film conductivity depends on the crystallite size, i.e. it is limited by the density of grain boundaries. This result illustrates the importance of annealing anatase at high temperatures for extended periods of time. Our results are in line with a recent study of Hartmann and coworkers who compared the photoelectrochemical properties of sol–gel derived and nanoparticle based electrodes. They associated enhanced electronic connectivity to the superior performance of sol–gel based materials.36
Fig. 4b shows the annealing temperature dependence of the relative amount of adsorbed dye (N719) per unit area, which is a measure for the accessible surface area in the film. The dye adsorption of the mesoporous film annealed at 400 °C was used as the baseline. At this temperature a surface area of 82 m2/g was measured by nitrogen physisorption (BET). The results suggest that the surface area accessible for the dye molecules varies only weakly in the 400–550 °C range, whereas it drops off for the highest annealing temperatures. This is in line with nitrogen physisorption measurements for bulk material. Fig. 4a and b illustrate a trade-off when choosing the optimal TiO2 sintering temperature for DSC production. High temperature annealing leads to a larger crystallite size and therefore a coarser crystallinity of the anatase network with a smaller number of grain boundaries. This benefits charge transport through the network at the expense of accessible surface area for dye adsorption. The coarser crystallinity comes at the expense of a reduced surface area leading to a reduction in dye adsorption and therefore to a reduction in charge generation per unit volume.
 | ||
| Fig. 4 Effects of crystallinity on device performance. (a) Resistivity determined by impedance spectroscopy of 900 nm thick films annealed at 400 °C, 500 °C, and 650 °C. (b) Relative amount of dye adsorbed per unit area. The dye adsorption of the mesoporous film annealed at 400 °C was used as the baseline. (c) Current–voltage characteristics for liquid electrolyte DSC devices. (d) solid-state DSC performance of samples annealed at 350 °C to 700 °C. Higher temperatures led to a continuous increase in efficiency from 0.8% (450 °C) to 2.5% (700 °C) for 400 nm thick films sensitised with D102. Liquid and solid-state devices were characterised under standardised AM 1.5 illumination of 100 W/cm2. See the supporting information for a summary of all photovoltaic device performance parameters. | ||
This trade-off was further explored in liquid electrolyte and solid-state DSCs. For liquid electrolyte DSCs, TiO2 films were annealed at temperatures ranging from 400 °C to 650 °C. In this study, 900 nm thick anatase films were used to avoid the formation of cracks which typically occurs for thicker films. While devices annealed at 400 °C and 500 °C had very similar photon-to-electron conversion efficiencies (EQEs) with maxima of 22.6%, annealing at 650 °C resulted in a reduction of the EQEmax to 18.6%. This reduction in EQE most likely correlates with the reduced dye loading in Fig. 4b. The corresponding current–voltage (J–V) curves are shown in Fig. 4c. While all devices had comparable open-circuit voltages Voc, their short-circuit current Jsc decreased with increasing anatase annealing temperature. The related power conversion efficiencies η were 2.3%, 2.0% and 1.5% for 400 °C, 500 °C, and 650 °C, respectively. The decrease in Jsc and η follows the trend in dye-adsorption (Fig. 4b) and EQE. While the values for the anatase annealing temperatures of 400 °C and 500 °C are comparable, reflecting the plateau in Fig. 4b, the reduced values for the 650 °C data-set correlates with the drop-off in specific surface area in Fig. 4b for this temperature. This result is not surprising. In liquid cells, charge recombination is believed to be not a limiting factor as it occurs via a slow two-electron process.37 The increased TiO2 conductivity does therefore not outweigh the loss in specific surface area for this cell type.
The situation is very different for solid-state DSCs. To this end, Fig. 4d shows the J–V curves of solid-state DSCs employing the D102 dye and spiro-OMeTAD as hole-conducting medium (see Experimental Section for details on device assembly). 400 nm-thick TiO2 films were used. An increase in the sintering temperature in the range from 350 °C to 650 °C resulted in a continuous increase in all relevant device performance parameters (Voc, Jsc, FF, and η). The decrease in Voc for samples annealed at 700 °C might be partially due to the fact that the sheet resistance of the FTO doubled to 40 Ω/□ at these high temperatures. Nevertheless, the power conversion efficiency increased from 0.78% for 450 °C to 2.48% for samples annealed at 700 °C. This is in contrast to earlier Pluronic-based approaches, where annealing is typically limited to 450 °C.17 Since the specific surface area and dye loading are reduced for the highest anatase annealing temperatures (Fig. 4b), these results imply that the solid-state DSCs strongly benefit from the increased TiO2 conductivity as evidenced in Fig. 4a. This result underlines the importance of anatase conductivity for solid-state DSCs, the performance of which is limited by charge recombination. This effect is expected to be even more pronounced in thicker devices.
We are currently working on techniques to increase the thickness of crack free films which should enable us to increase the light absorption while maintaining the excellent conductivity afforded by our anatase synthesis route. We note that the TiO2 films were not post treated with TiCl4 further underlining the quality of the anatase produced by this approach. The for their thickness unusual high efficiencies of the solid-state DSCs also prove that the titania network, though micellar, is well interconnected38 and allows sufficient pore infiltration with a solid-state hole conductor.
Conclusions
We present a facile but powerful method for the synthesis of highly conductive mesoporous TiO2via sol–gel chemistry. In contrast to to earlier work, our method maintains structural integrity of the porous titania scaffold up to 700 °C, enabling to study the effect of high annealing temperatures on device performance of DSCs. The self-assembling block-copolymer PI-b-PEO is highly effective in controlling anatase crystallisation in mesoporous networks. It rapidly assembles the TiO2 sol into an ordered spatial confinement which promotes crystallite growth replicating the organic matrix. The resulting anatase network consists of relatively large crystallites, while maintaining the structural integrity dictated by the polymer scaffold.When used in DSCs, the control over anatase crystal parameters afforded by our protocol presents an interesting dilemma: while anatase conductivity is markedly improved by high temperature annealing, this comes at the expense of specific surface area of the mesoporous scaffold. For liquid DSCs in which charge carrier recombination is not a limitation the use of high temperature annealed anatase films therefore has a detrimental effect on device performance. In solid-state DSCs, on the other hand, the reduced dye loading of the less grainy TiO2 network is outweighed by the improved anatase conductivity, leading to an increase in power conversion efficiency of the cell employing anatase annealed at 700 °C by a factor of 3 compared to materials made by the previously established route.
In summary, we have demonstrated a DSC device manufacture protocol that combines the detailed control over anatase morphology on the 20-nm length scale with simultaneous control over anatase crystallinity. This protocol is highly promising for the improvement of device efficiency of solid-state DSCs. Future work will now concentrate on the manufacture of thicker, crack free films, to fully exploit the potential of this methodology.
Experimental section
Synthesis
Titania containing sol was synthesised by quickly adding 0.69 ml of HCl (37%) into 1 ml of titanium ethoxide (purum) under vigorous stirring at ambient conditions.39 0.873 ml sol was subsequently mixed with 0.1g PI-b -PEO copolymer (Mn = 35.7g/mol, 23 wt% PEO40) in 7 ml THF (99.97%) and slowly evaporated in a Petri dish at 50 °C before the hybrid material was redissolved in an azeotrope solvent mixture of toluene (apolar, 99.8%) and 1-butanol (polar, 99%). This step is undertaken since the hydrolytic sol contains a large amount of water and ethanol (over 70 wt%), which is unfavourable for the simultaneous continuous evaporation of the hydrophobic and hydrophilic solution components (see SI). A 20 wt% solution was then spin-coated (5 s, 1000 rpm) on a (100) silicon substrate and the sample was dried for several hours at room temperature, resulting in 1 μm thick films. For assembly in DSCs, films were similarly fabricated on transparent conductive substrates. All residual solvent was subsequently evaporated by heating the samples in a tube furnace at 130 °C followed by calcination in a tube furnace or a X-ray diffractometer with an integrated oven (Bruker D8 Advance) employing a heating rate of 10 °C/min unless otherwise stated.Wide-angle X-ray scattering
Wide angle X-ray scattering experiments during in situ heating were carried out using a Bruker D8 Advance diffractometer in an angular range of 22°–32° with a step size of 0.0143° and a total scan time of 5 min. A heating rate of 10 °C/min was used unless otherwise stated. Samples were measured during annealing in air for up to 7 h, representing 84 consecutive X-ray scans. Because of its crystal orientation, the single crystalline silicon substrate is not expected to contribute to a signal in the observed angular range. The measurements covered a temperature range between 380 °C and 740 °C in intervals of around 50 °C. The obtained raw data was corrected for the CuKα2 radiation by the Bruker software EVA. To obtain the peak broadening caused by the size of the anatase crystallites, a Voigt function was fitted to the peaks. The peak width (full width at half maximum) of the Lorentzian and the Gaussian parts of the Voigt function were determined by the approximation of Olivero and Longbothum.41 The peak width was then corrected for machine broadening by subtracting (by deconvolution) the broadening of a monocrystalline silicon reference sample. The Debye-Scherrer equation was subsequently employed to obtain a mean crystallite diameter of the peak broadening42 Like in similar studies, the dimensionless number K = 0.89 was used as Debye-Scherrer constant.19,43 Ex-situ X-ray diffraction of separately crystallised samples was carried out on a Bruker D8 diffractometer with a position sensitive detector (LynxEye).Small-angle X-ray scattering (SAXS)
Small-angle X-ray scattering of CuKα radiation was collected in transmission geometry at a sample-detector distance of 1.05 m (Bruker AXS Nanostar, Karlsruhe, Germany). Acquisition times of 7500 s were used. The scattered intensity was collected using a 2-dimensional Hi-Star gas-filled wire detector with 1024 × 1024 channels and the incident beam-path directed towards its centre. A lead beam-stop was suspended in front of the detector in the beam path, to prevent damage due to unattenuated X-rays; this obscured any scattering below q = 0.08 nm−1. ‘Background’ scans were performed with an empty camera; this was used to correct the scattering measured for specimens, after allowing for transmission. In each case, the scattering patterns were found to be isotropic (i.e. no preferred orientation). Radial scattering intensity profiles were obtained by averaging over the azimuthal angle (from 0 to 360°) with 0.005° increments in scattering angle.Transmission electron microscopy (TEM)
Samples annealed at different temperatures were dispersed on holey carbon coated TEM copper grids and imaged using a FEI Tecnai F20 microscope (200 kV). For high resolution transmission electron microscopy, a Jeol JEM 4000EX (400 kV) was used.Impedance spectroscopy
A Gamry Reference 600 potentiostat with the EIS300 software package was employed to measure impedance spectra. Spectra were collected in the dark under forward bias that is adjusted to match the open circuit potential under AM 1.5. The AC amplitude used was 10 mV and the spectra were collected from 10 mHz to 10 kHz with 10 frequencies per decade. The analysis of the spectra were performed using ZView v2.8 using a circuit model adapted from Ref. 35.Dye absorption measurements
In order to quantify the accessible surface area for dye sensitization, dye desorption studies were carried out similar to Neale et al.44 Mesoporous TiO2 films were immersed in a 0.5 mM solution of cis-di(thiocyanato)-bis(2,2′-bipyridyl-4-carboxylate-4′-carboxylic acid)-ruthenium(II) (termed N719) in a mixture of acetonitrile:tert-butyl alcohol (volume ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and kept in the dark for 16 h at room temperature. The samples were then thoroughly rinsed with acetonitrile and subsequently submerged into a 1.0 mM KOH solution (3 ml) for 3 h to fully desorb the dye. The absorbance of the resulting light pink solutions was measured with an UV-vis spectrophotometer (HP 8453 UV/vis spectrophotometer, wavelength range 190–1100 nm, 1 nm slit width, 0.03% stray light). The relative amount of dye adsorbed was determined by comparing the peak height of the respective solution with the reference obtained from desorption of the film annealed at 400 °C.
:1) and kept in the dark for 16 h at room temperature. The samples were then thoroughly rinsed with acetonitrile and subsequently submerged into a 1.0 mM KOH solution (3 ml) for 3 h to fully desorb the dye. The absorbance of the resulting light pink solutions was measured with an UV-vis spectrophotometer (HP 8453 UV/vis spectrophotometer, wavelength range 190–1100 nm, 1 nm slit width, 0.03% stray light). The relative amount of dye adsorbed was determined by comparing the peak height of the respective solution with the reference obtained from desorption of the film annealed at 400 °C.
Liquid electrolyte DSC assembly
Fluorine doped SnO2 (FTO) coated glass sheets (Nipon Sheet Glass, TEC15) were manually cleaned with Hellmanex soap, acetone, and isopropanol followed by 10 min in an O2 plasma etcher. 900 nm thick anatase films were then spin-cast onto the substrates and annealed as described above. The samples were sensitised with N719 as described above.The counter electrodes used for these devices were sheets of pre-drilled platinized FTO glass. The platinization was carried out by coating the FTO with hexachloroplatinic (IV) acid from an anhydrous isopropanol solution (50 mM) followed by heating to 450 °C for 15 min. The dyed TiO2 electrodes were washed in acetonitrile, dried and assembled in a sandwich type cell configuration with the platinized counter electrodes, using a ‘hot-melt’ 50 μ m thick surlyn spacer (Dupont) to seal the working and counter electrode together. A drop of ‘Robust’ liquid electrolyte (0.8 M PMII, 0.15 M iodine, 0.1 M GuNCS, and 0.5 M NMBI in 3-methoxypropionitrile)45 was dispensed into a hole in the counter electrode and the cell was filled via vacuum backfilling and sealed with polyimide tape (Dupont).
Solid-state DSC assembly
Solid-state DSCs were fabricated using the method outlined in Ref. 46. The TiO2 films were dye-sensitized with D102 (indolin-based organic dye) for 1 h. Spiro-OMeTAD (2,2′,7,7′-tetrakis(N,N-di-methoxyphenyamine)-9,9′-spirobifluorene) dissolved in chlorobenzene was used as the hole-transporting material (HTM), with tert-butyl pyridine (tBP) and bis(trifluoromethyl-sulfonyl)amine lithium salt (Li-TFSI) (dissolved separately in acetonitrile) used as additives for improved device performance.46 Finally, 150 nm thick silver electrodes were evaporated over the HTM layer under high vacuum (10−6 mbar) to complete the devices.DSC characterisation
The solar cells were measured under AM 1.5 simulated sun light generated by a 300 W Oriel solar simulator. The power of the simulated light was measured using a calibrated Si photo diode (Fraunhofer Institute for Solar Energy Systems). The mismatch factor to the solar spectrum was calculated over the entire spectral response region of both the test solar cells and calibration diode following the method of Seaman.47 The active area of the DSCs was 0.13 cm2. The current–voltage measurements were recorded by a Keithley 237 sourcemeter connected to a computer.Acknowledgements
This work was funded in part by the EPSRC Nanotechnology Grand Challenges Energy grant (EP/F056702/1), and EP/F065884/1, the Department of Energy (DE-FG02 87ER45298) through the Cornell Fuel Cell Institute (CFCI), the National Science Foundation (DMR-0605856), and the Cornell Universiy KAUST Center for Research and Education. SG acknowledges support by the Studienstiftung des deutschen Volkes and CD thanks the Royal Society for funding. We thank T. Abraham for help with the XRD measurements, P. Laity for help with SAXS measurements, and P. Müller-Buschbaum for useful discussions.References
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- A. Linsebigler, G. Lu and J. Yates, Chem. Rev., 1995, 95, 735–758 CrossRef CAS.
- T. Brezesinski, J. Wang, J. Polleux, B. Dunn and S. H. Tolbert, J. Am. Chem. Soc., 2009, 131, 1802–1809 CrossRef CAS.
- O. Varghese, D. Gong, M. Paulose, K. Ong and C. Grimes, Sens. Actuators, B, 2003, 93, 338–344 CrossRef; L. D. Bonifacio, D. P. Puzzo, S. Breslav, B. M. Willey, A. McGeer and G. A. Ozin, Towards the Photonic Nose: A Novel Platform for Molecule and Bacteria Identification, Adv. Mater., 2010, 22, 1351–1354 CrossRef CAS.
- P. Bonhote, E. Gogniat, F. Campus, L. Walder and M. Gratzel, Displays, 1999, 20, 137–144 CrossRef CAS.
- S. Burnside, V. Shklover, C. Barbe, P. Comte, F. Arendse, K. Brooks and M. Gratzel, Chem. Mater., 1998, 10, 2419–2425 CrossRef CAS.
- T. Dittrich, E. Lebedev and J. Weidmann, Phys. Status Solidi A, 1998, 165, R5–R6 CrossRef CAS.
- P. Tiwana, P. Parkinson, M. B. Johnston, H. J. Snaith and L. M. Herz, J. Phys. Chem. C, 2010, 114, 1365–1371 CrossRef CAS.
- H. J. Snaith and M. Grätzel, Adv. Mater., 2007, 19, 3643 CrossRef CAS.
- L. Schmidt-Mende and M. Grätzel, Thin Solid Films, 2006, 500, 296–301 CrossRef CAS.
- M. Templin, A. Franck, A. DuChesne, H. Leist, Y. Zhang, R. Ulrich, V. Schadler and U. Wiesner, Science, 1997, 278, 1795–1798 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. Fredrickson, B. Chmelka and G. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- P. Yang, D. Zhao, D. Margolese, B. Chmelka and G. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- E. Crepaldi, G. Soler-Illia, D. Grosso, F. Cagnol, F. Ribot and C. Sanchez, J. Am. Chem. Soc., 2003, 125, 9770–9786 CrossRef CAS.
- S. Choi, M. Mamak, N. Coombs, N. Chopra and G. Ozin, Adv. Funct. Mater., 2004, 14, 335–344 CrossRef CAS.
- B. Smarsly, D. Grosso, T. Brezesinski, N. Pinna, C. Boissiere, M. Antonietti and C. Sanchez, Chem. Mater., 2004, 16, 2948–2952 CrossRef CAS.
- M. Zukalova, A. Zukal, L. Kavan, M. Nazeeruddin, P. Liska and M. Grätzel, Nano Lett., 2005, 5, 1789–1792 CrossRef CAS.
- M. Wei, Y. Konishi, H. Zhou, M. Yanagida, H. Sugihara and H. Arakawa, J. Mater. Chem., 2006, 16, 1287–1293 RSC.
- S. Choi, M. Mamak, S. Speakman, N. Chopra and G. Ozin, Small, 2005, 1, 226–232 CrossRef CAS.
- D. Fattakhova-Rohlfing, M. Wark, T. Brezesinski, B. M. Smarsly and J. Rathousky, Adv. Funct. Mater., 2007, 17, 123–132 CrossRef CAS.
- J. N. Kondo and K. Domen, Chem. Mater., 2008, 20, 835–847 CrossRef CAS.
- J. M. Szeifert, D. Fattakhova-Rohlfing, D. Georgiadou, V. Kalousek, J. Rathouský, D. Kuang, S. Wenger, S. M. Zakeeruddin, M. Grätzel and T. Bein, Chem. Mater., 2009, 21, 1260–1265 CrossRef CAS.
- M. Zukalova, M. Kalbac, L. Kavan, I. Exnar and M Grätzel, Chem. Mater., 2005, 17, 1248 CrossRef CAS.
- K. Frindell, M. Bartl, M. Robinson, G. Bazan, A. Popitsch and G. Stucky, J. Solid State Chem., 2003, 172, 81–88 CrossRef CAS.
- I. Kartini, D. Menzies, D. Blake, J. da Costa, P. Meredith, J. Riches and G. Lu, J. Mater. Chem., 2004, 14, 2917–2921 RSC.
- G. Floudas, R. Ulrich and U. Wiesner, J. Chem. Phys., 1999, 110, 652–663 CrossRef CAS.
- S. Renker, S. Mahajan, D. Babski, I. Schnell, A. Jain, J. Gutmann, Y. Zhang, S. Gruner, H. Spiess and U. Wiesner, Macromol. Chem. Phys., 2004, 205, 1021–1030 CrossRef CAS.
- M. Nedelcu, J. Lee, E. J. W. Crossland, S. C. Warren, M. C. Orilall, S. Guldin, S. Hüttner, C. Ducati, D. Eder, U. Wiesner, U. Steiner and H. J. Snaith, Soft Matter, 2009, 5, 134–139 RSC.
- J. Lee, M. C. Orilall, S. C. Warren, M. Kamperman, F. J. Disalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS.
- M. Nedelcu, S. Guldin, M. C. Orilall, J. Lee, S. Hüttner, E. J. W. Crossland, S. C. Warren, C. Ducati, P. R. Laity, D. Eder, U. Wiesner, U. Steiner and H. J. Snaith, J. Mater. Chem., 2010, 20, 1261–1268 RSC.
- J. Ponton. Azeotrope Databank, http://www.chemeng.ed.ac.uk/people/jack/azeotrope/ Search PubMed.
- P. Docampo, S. Guldin, M. Stefik, P. Tiwana, M. C. Orilall, S. Hüttner, H. Sai, U. Wiesner, U. Steiner and H. J. Snaith, Adv. Funct. Mater., 2010, 20, 1787–1796 CAS.
- T. Dittrich, J. Weidmann, F. Koch, I. Uhlendorf and I. Lauermann, Appl. Phys. Lett., 1999, 75, 3980–3982 CrossRef CAS.
- K. Benkstein, N. Kopidakis, J. van de Lagemaat and A. Frank, J. Phys. Chem. B, 2003, 107, 7759–7767 CrossRef CAS.
- Q. Wang, S. Ito, M. Graetzel, F. Fabregat-Santiago, I. Mora-Sero, J. Bisquert, T. Bessho and H. Imai, J. Phys. Chem. B, 2006, 110, 25210–25221 CrossRef CAS.
- P. Hartmann, D. K. Lee, B. M. Smarsly and J. Janek, ACS Nano, 2010, 4, 3147–3154 CrossRef CAS.
- N. Duffy, L. Peter, R. Rajapakse and K. Wijayantha, J. Phys. Chem. B, 2000, 104, 8916–8919 CrossRef CAS.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu, X. Wu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS.
- P. Alberius, K. Frindell, R. Hayward, E. Kramer, G. Stucky and B. Chmelka, Chem. Mater., 2002, 14, 3284–3294 CrossRef.
- J. Allgaier, A. Poppe, L. Willner and D. Richter, Macromolecules, 1997, 30, 1582–1586 CrossRef CAS.
- J. Olivero and R. Longbothum, J. Quant. Spectrosc. Radiat. Transfer, 1977, 17, 233–236 CrossRef.
- P. Scherrer, Nachrichten von der Gesellschaft der Wissenschaften zu Goettingen, mathematisch-physikalische Klasse, 1918, 98–100 Search PubMed.
- D. Eder and A. H. Windle, Adv. Mater., 2008, 20, 1787 CrossRef CAS.
- N. Neale, N. Kopidakis, J. van de Lagemaat, M. Gratzel and A. Frank, J. Phys. Chem. B, 2005, 109, 23183–23189 CrossRef CAS.
- D. Kuang, S. Ito, B. Wenger, C. Klein, J. Moser, R. Humphry-Baker, S. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 4146–4154 CrossRef CAS.
- H. J. Snaith, L. Schmidt-Mende, M. Gratzel and M. Chiesa, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 045306 CrossRef.
- C. Seaman, Sol. Energy, 1982, 29, 291–298 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Further information. See DOI: 10.1039/c0ee00362j |
| ‡ Also at: Freiburg Institute for Advanced Studies (FRIAS), Universität Freiburg, Albertstraße 19, D-79104 Freiburg, Germany. |
| This journal is © The Royal Society of Chemistry 2011 |