IL-TEM investigations on the degradation mechanism of Pt/C electrocatalysts with different carbon supports†
Katrin
Hartl
ac,
Marianne
Hanzlik
b and
Matthias
Arenz
*c
aLehrstuhl Physikalische Chemie, Technische Universität München, Lichtenbergstr. 4, D-85748, Garching, Germany
bZentrum für Elektronenmikroskopie, Technische Universität München, Lichtenbergstr. 4, D-85748, Garching, Germany
cDepartment of Chemistry, CS06, University of Copenhagen, Universitetsparken 5, DK-2100, Copenhagen Ø, Denmark. E-mail: m.arenz@kemi.ku.dk
First published on 21st October 2010
Abstract
Utilizing our recently developed method of identical location transmission electron microscopy (IL-TEM) in combination with electrochemical surface area determination, the degradation behavior of different carbon supported Pt catalysts for polymer electrolyte membrane fuel cells (PEMFCs) is investigated. Two different Pt based catalysts supported on a low surface area (LSA) carbon are compared to a Pt catalyst with standard high surface area (HSA) carbon support. One of the LSA carbon supports is of conventional type, while the other is modified by a transition metal. Relative to the standard, both catalysts with LSA carbon support exhibit improved degradation behavior in terms of loss in active surface area upon accelerated stress tests. The catalyst with transition metal modified carbon support thereby exhibits by far superior improvements. The characterization of the bare carbon supports indicates that the observed differences between both catalysts with LSA carbon support are not related to the resistance of the support to complete oxidation to carbon dioxide. Instead, the IL-TEM results reveal that the improved properties of the catalyst with transition metal modified support are due to a stabilization of the Pt particles attached to the support. Particle detachment thus can be drastically reduced and the degradation is limited to a migration and coalescence or sintering mechanism.
Broader contextFuel cells are an efficient alternative to conventional power sources such as combustion engines. Instead of producing first heat, which has to be further transformed into mechanical energy, fuel cells convert chemical energy directly into useful electrical energy. An essential part of a fuel cell is the catalyst, where the chemical reactions take place. In general catalysts are composed of an active component of high surface to mass ratio, e.g.nanoparticles, and a support. In today's low temperature fuel cells, the active component of the catalyst must contain platinum, a rare and expensive metal. Thus there is a strong incentive to minimize the amount of platinum as well as to enhance the durability of the catalyst. Despite the significant progress in recent years, further breakthroughs are required. In order to optimize the durability of catalysts a detailed understanding of the degradation mechanism as a function of the applied conditions is necessary. The approach of identical location transmission electron microscopy (IL-TEM), recently developed in our group, combines accelerated stress tests with a detailed insight of the behaviour of single catalyst nanoparticles. Therefore this methodology offers a useful tool to screen the behaviour of fuel cell catalysts in an efficient way. |
1. Introduction
Due to their high efficiency converting chemical into electric energy polymer electrolyte membrane fuel cells (PEMFCs) play an important role in the widely discussed effort of substituting conventional vehicle motors by electric power-trains. Substantial progress has been made on many technical aspects concerning a commercial large-scale deployment of PEMFCs. The manufacturing of fuel cell stacks, their implementation in vehicles, and longtime field trials are nowadays standard practice. However, in order to render PEMFCs commercially viable for such demanding applications as electric vehicles their cathode electrocatalyst still needs improvements in terms of both activity1 and perhaps more importantly, stability.2,3The standard catalysts for PEM fuel cells are Pt-based nanoparticles on a high surface area (HSA) carbon support.4 In recent years the main research effort was to improve the power output per amount of Pt used in a fuel cell, i.e. the mass activity of the catalyst. It is now demonstrated by many experimental and theoretical investigations that the crucial cathode reaction—the oxygen reduction reaction—can be considerably increased by alloying platinum with a second transition metal such as Co, Ni, or Cu.5–7
Much less is known about catalyst degradation. Despite the fact that several studies in PEM as well as phosphoric acid (PA) fuel cells showed that catalyst degradation is one of the major reasons for gradual performance losses, this topic only recently gained increased attention. In general the cause of catalyst degradation is related to a loss in accessible surface area of the active catalyst components.3 In addition, recent work also suggests that the de-alloying of Pt-alloy catalysts due to segregation and concomitant dissolution of non-noble components inflicts a continuous detrimental effect on the performance of low temperature fuel cells.8–10 For the loss in electrochemically active surface area (ECSA) of Pt in PEM fuel cells there are three fundamentally different mechanisms discussed.11 That is Pt dissolution, the migration and concomitant coalescence of Pt nanoparticles on the carbon support, and the detachment of Pt nanoparticles from the support. Pt dissolution can be further sub-divided into two processes, resulting either in Pt re-deposition onto larger particles (electrochemical Ostwald ripening) or in the precipitation of Pt crystallites close to the membrane electrolyte due to reduction by hydrogen gas diffusing through the membrane. In addition to these three processes concerning Pt also the complete oxidation of the carbon support to carbon dioxide can occur under certain conditions, which results in the loss of electrical contact of the Pt nanoparticles. In general it is assumed that a combination of these mechanisms occurs and that the degradation resistance of a catalyst can be improved by supports less prone towards total oxidation, i.e. that the particle detachment is linked to the oxidation of the support to carbon dioxide. Therefore, conventional high surface area supports are replaced by graphitized or low surface area carbons.
In the presented work the degradation behavior of two Pt based electrocatalysts, labeled as catalysts I and II, on low surface area (LSA) carbon supports is compared to a Pt based catalyst with standard high surface area (HSA) carbon, called standard. The two catalysts pursue different degradation mitigation strategies. For catalyst I the LSA carbon support was used in its original form, for catalyst II the LSA carbon support was modified by a transition metal prior to Pt impregnation. The bare carbon supports are characterized towards their corrosion resistance in membrane electrode assembly (MEA) tests, which indicate that the corrosion resistance of the carbon support of both catalysts is quite similar. The degradation of the synthesized Pt based catalysts is scrutinized by a combination of electrochemical half-cell measurements and identical location transmission electron microscopy (IL-TEM).
2. Experimental
2.1. Catalyst samples
The three investigated catalysts consisted of Pt nanoparticles about 3 nm in size supported on different carbon supports. As standard a catalyst with standard HSA carbon and 46 wt% Pt content was used, catalyst I has 27.8 wt% Pt content and is supported on a LSA carbon (CI), catalyst II has 29.8 wt% Pt content and is supported on a LSA carbon (CII) which was additionally modified by a transition metal. Catalysts I and II were synthesized, including the transition metal modification, by BASF, Germany. The BET surface areas of the LSA carbons were 30 and 53 m2 g−1, for catalysts I and II, respectively. After modification, the BET surface area of the LSA carbon support utilized for the synthesis of catalyst II was reduced to 28 m2 g−1.2.2. Stability test of carbon supports
The corrosion stability of the bare carbon supports was characterized in membrane electrode assembly (MEA) tests according to the procedure described in ref. 12. In short, carbon electrodes were prepared by casting a carbon/PTFE ink onto a highly hydrophobized carbon paper and subsequently hotpressed together with a standard Pt-containing GDE (gas diffusion electrode) on a Celtec® membrane. The corrosion tests were carried out in 50 cm2 single cell setups using specially coated graphite plates. The Pt-containing GDE acted as the reference and counter electrodes, whereas the carbon electrodes acted as working electrodes. The corrosion tests were performed at 1.2 V vs. RHE at 180 °C. The reference electrode was continuously purged by 10% H2 in N2 and the working electrode was purged by N2 containing 5% water partial pressure. The carbon loss was determined by detecting the CO2 release in the fuel cell exhaust.2.3. Electrochemical half-cell measurements
The electrochemical measurements were conducted in a three-compartment electrochemical Teflon-cell,13 using a rotating disc electrode (RDE) setup (Radiometer Analytical, France) and a home-built potentiostat. A saturated Calomel electrode (SCE), separated by an electrolytic bridge from the working electrode compartment, was used in each experiment. However, all potentials are given with respect to the reversible hydrogen potential (RHE), which was measured for every experiment. A graphite rod was used as a counter electrode. The electrolyte, 0.1 M HClO4, was prepared using Millipore Milli Q® water and concentrated HClO4 (Suprapure; Merck, Germany). In order to prepare a catalyst suspension the catalyst powders were dispersed ultrasonically for at least 10 minutes in ultrapure Millipore Milli Q® water to a concentration of 0.14 mgPt ml−1. Prior to preparation of a thin catalyst film onto the polished glassy carbon tip of a RDE, the catalyst suspension was sonicated for additional 3 minutes. Then 20 µl of the suspension were pipetted onto the RDE tip (5 mm diameter, 0.196 cm2 geometrical surface area), resulting in a Pt loading of 14 µgPt cm−2. The catalyst suspension on the glassy carbon electrode was dried in a nitrogen stream and the electrode tip was attached to a RDE. After transfer to the electrochemical cell, the tip was immersed under potential control at 0.05 V into argon-saturated 0.1 M HClO4 solution. The potential was then continuously cycled between 0.05 V and 1.0 V until a stable cyclovoltammogram was recorded. All measurements were performed at room temperature with iR-compensation of the solution resistance. The ECSA was determined by CO-stripping in Ar purged solution, after saturating the surface with CO at a potential of 0.05 V. As degradation treatment the potential of the electrodes was cycled between 0.4 and 1.4 VRHE with a scan speed of 1 V s−1. Each treatment procedure consisted of 3600 cycles, corresponding to 2 h. In some cases the treatment was repeated. The ECSA was determined before, at halftime, and at the end of the treatment procedures.2.4. Identical location transmission electron microscopy (IL-TEM)
For the IL-TEM14 investigations the catalyst suspension was diluted by a factor of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. A gold finder grid (400 mesh; Plano, Germany) was coated with an amorphous carbon film. The hydrophilicity of the carbon film, which is necessary for a uniform distribution of the catalyst particles, develops during a glow discharge exposure to an oxygen plasma. After this pre-treatment, 5 µl of catalyst suspension were pipetted onto the gold finder grid. In order to keep the catalyst loading as low as possible (to avoid overlapping of catalyst particles), the drop was delicately absorbed off the grid after approximately 10 seconds, using filter paper. The grid was dried, and then investigated using a JEM 2010 microscope (JEOL, Japan) with an accelerating voltage of 120 kV and a magnification of 150 k. TEM micrographs were recorded before and after the treatment procedures, i.e. 3600 potential cycles between 0.4 and 1.4 VRHE with a scan rate of 1 V s−1, on identical locations of the catalyst.
:10. A gold finder grid (400 mesh; Plano, Germany) was coated with an amorphous carbon film. The hydrophilicity of the carbon film, which is necessary for a uniform distribution of the catalyst particles, develops during a glow discharge exposure to an oxygen plasma. After this pre-treatment, 5 µl of catalyst suspension were pipetted onto the gold finder grid. In order to keep the catalyst loading as low as possible (to avoid overlapping of catalyst particles), the drop was delicately absorbed off the grid after approximately 10 seconds, using filter paper. The grid was dried, and then investigated using a JEM 2010 microscope (JEOL, Japan) with an accelerating voltage of 120 kV and a magnification of 150 k. TEM micrographs were recorded before and after the treatment procedures, i.e. 3600 potential cycles between 0.4 and 1.4 VRHE with a scan rate of 1 V s−1, on identical locations of the catalyst.
3. Results and discussion
3.1. Stability of bare carbon support materials
In order to investigate the intrinsic corrosion resistance of the carbon support materials MEA tests of the bare carbons were performed prior to the synthesis, i.e. supporting of Pt nanoparticles onto them. The carbon loss of the different samples determined by detecting the CO2 release in the fuel cell exhaust is plotted in Fig. 1. It is seen that under the applied conditions in the first five hours all LSA carbon samples exhibit higher corrosion resistance than the standard HSA carbon Vulcan XC72 which loses 50% of its mass. Furthermore, it is obvious that the transition metal modification of the initially poorly performing LSA carbon CII leads to a considerably improved corrosion resistance. The decrease in mass over time is almost identical for the LSA carbon CI and the modified LSA carbon CII. Thus the resistance towards complete oxidation of their support materials is similar for both catalysts I and II. | ||
| Fig. 1 Corrosion test of carbon supports before Pt impregnation. Dotted lines show exponential fits and are considered only as a guide for the eye. | ||
3.2. Electrochemical characterization of ECSA loss
In order to determine the ECSA loss of the synthesized catalysts I and II upon accelerated degradation tests, they were exposed to potential cycling treatments in an electrochemical half-cell. The treatments consisted of potential cycling between 0.4 and 1.4 VRHE with a scan rate of 1 V s−1. The cyclovoltammograms (CVs) of the particular catalysts in the initial state as well as after 2 and 4 hours of treatment are shown in Fig. 2. In its initial state the CV of catalyst I (Fig. 2a) exhibits the typical features of a Pt/C catalyst.15 The under potential deposition of hydrogen (Hupd) and the formation of OH and oxides on the Pt nanoparticles are well discernible and lie in the expected potential regions. Furthermore, it is evident from the reduced features in the CV that after potential cycling the ECSA of the catalyst is considerably decreased. By comparison the initial CV of catalyst II (Fig. 2b) clearly shows a modified signature and the typical Pt features are diminished due to the influence of the transition metal. Furthermore, the capacitance of the support is larger than for catalyst I. Based on the CVs the ECSA loss during degradation treatment is considerably inhibited compared to catalyst I. The inhibited Pt features, however, hamper a precise ECSA evaluation. In order to study the Pt surface area loss more accurately, CO stripping experiments were conducted (see ESI†). Thus determined ECSA values for the pristine catalysts are 52 m2gPt−1 and 67 m2gPt−1 for I and II, respectively. In Fig. 3 the ECSA loss due to the potential cycling treatment of the catalysts I and II is compared to the one of a standard high surface area Pt catalyst with similar particle size (∼3 nm). Both electrocatalysts supported on a LSA carbon exhibit an improved performance in the degradation test compared to the standard catalyst. Furthermore, it is evident that catalyst II exhibits a much less pronounced ECSA loss than catalyst I, especially after the first degradation treatment. The slowed-down ECSA loss in the second treatment indicates that catalyst II experiences an initial degradation process, but is afterwards stabilized. Catalyst I by comparison exhibits a continuous time-dependent degradation profile, similar as the standard catalyst, but inhibited.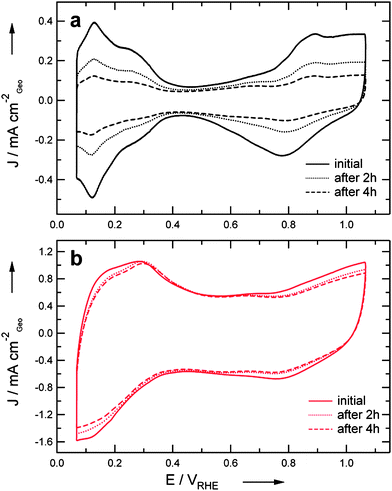 | ||
| Fig. 2 CVs of catalyst I (a) and catalyst II (b) in their initial state as well as after 2 h and 4 h accelerated degradation treatment. The treatment consisted of potential cycling between 0.4 and 1.4 VRHE at 1 V s−1 in argon saturated 0.1 M HClO4. | ||
 | ||
| Fig. 3 ECSA loss of catalysts I and II due to accelerated degradation treatment by potential cycling between 0.4 and 1.4 VRHE at 1 V s−1 determined by CO stripping measurements. Measurements of the standard catalyst are shown for comparison. | ||
3.3. IL-TEM analysis
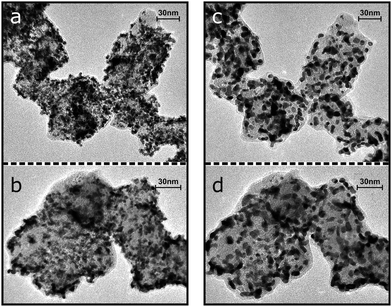
In order to examine the degradation process in detail, IL-TEM measurements were performed. In accordance with the electrochemical determination of the ECSA loss, the TEM micrographs of identical locations of catalyst I before and after potential cycling (see Fig. 4) demonstrate that the accelerated degradation treatment results in a considerable reduction in the amount of Pt nanoparticles on the carbon support. It is noteworthy in this respect that after the treatment individual Pt particles can be found on the amorphous carbon film of the TEM grid indicating a particle detachment process rather than the dissolution of Pt nanoparticles. At the same time no changes in the carbon support are discernible. In addition to particle detachment, the growth of larger Pt particles is evident as well. The formation of larger Pt nanoparticles seems to be increased in areas of high particle density. A preference for small particles to disappear,17 however, is not evident. The IL-TEM measurements thus indicate that under the applied accelerated degradation treatment the ECSA loss of catalyst I is a result of a particle detachment process superimposed by migration and coalescence and/or electrochemical Ostwald ripening. The particle detachment process on catalyst I is similar to the one observed in our previous study of a Pt catalysts with HSA carbon support.16 In the previous study, however, despite a larger ECSA loss the observed particle growth was considerably lower. | ||
| Fig. 4 IL-TEM of two representative areas of catalyst I before (a and b) and after (c and d) 2 h accelerated degradation treatment consisting of potential cycling between 0.4 and 1.4 VRHE at 1 V s−1. | ||
The IL-TEM micrographs of catalyst II are shown in Fig. 5. The micrographs of the identical locations of catalyst II clearly show that the loss in particles upon the accelerated degradation treatment is greatly reduced as compared to catalyst I. This finding is in line with the ECSA determination by CO stripping. While the loss in active material is considerably reduced and especially no significant particle detachment is apparent from the IL-TEM measurements, some changes in the catalyst are discernible. As an effect of the treatment, small Pt particles coalesce and form larger particles similar to a melting process. The large particles seem to be stabilized on the carbon support. As for catalyst I, for catalyst II no significant change in the carbon support is observed upon the treatment.
 | ||
| Fig. 5 IL-TEM of two representative areas of catalyst II before (a and b) and after (c and d) 2 h accelerated degradation treatment consisting of potential cycling between 0.4 and 1.4 VRHE at 1 V s−1. | ||
3.4. Comparison of the catalysts
Compared to the standard, catalysts I and II exhibit an improved performance in terms of ECSA loss. By comparison to the previous IL-TEM investigations16 the measurements indicate that the main degradation mechanism of particle detachment upon cycling treatment is still active for catalyst I with a conventional LSA carbon support. Most loss in ECSA is correlated to particle detachment, not Pt dissolution, in line with the observation of Pt particles on the carbon film of the TEM grids after treatment. In addition to particle detachment, there is a second, minor mechanism active, which is responsible for the Pt particle growth. Although it is difficult to distinguish between Pt dissolution and re-deposition, i.e. electrochemical Ostwald ripening, and a particle migration and coalescence mechanism, the fact that no preferential disappearance of small Pt particles is observed supports a migration and coalescence mechanism.18,19By comparison, the properties of catalyst II with a transition metal modified LSA carbon support are fundamentally changed. The combination of the ECSA loss determination together with the IL-TEM investigations unambiguously shows that the Pt nanoparticles are stabilized on the support. The particle detachment process is considerably inhibited and is not the main degradation mechanism anymore. Upon the accelerated degradation treatment the catalyst initially experiences a pronounced degradation (however, less than the other catalysts), but is then stabilized. The IL-TEM investigations indicate that the initial degradation is a result of the fusion of adjacent Pt particles to form larger stabilized particles. Hence, this process slows down with time—as would be expected—and thus leads to a considerable inhibition of the ECSA loss as observed in the second treatment of the electrochemical measurement. As for catalyst I the distinction between Ostwald ripening, and particle migration and coalescence is difficult, but no preferential disappearance of small Pt particles is observed.
Interestingly, the corrosion tests of the pure carbon supports demonstrate that the resistance of the CI carbon and the modified CII carbon towards complete oxidation to carbon dioxide is almost identical. This finding indicates that the complete oxidation of the carbon support to carbon dioxide does not play a decisive role in the particle detachment process, which is observed on catalyst I but not on catalyst II. This interpretation is consistent with the fact that in the IL-TEM measurements no change in the carbon support is apparent upon cycling treatment.
4. Conclusion
In conclusion, the behavior of two different electrocatalysts developed for degradation mitigation has been investigated by means of electrochemical and IL-TEM measurements. The carbon supports of both catalysts, a conventional and a transition metal modified LSA carbon, exhibit a similar corrosion resistance towards complete oxidation to carbon dioxide. The synthesized catalysts of similar Pt loadings and particle size, however, show a distinctively different degradation behavior. Upon exposure to accelerated degradation treatments the catalyst with an unmodified LSA carbon support exhibits a somewhat improved degradation resistance as compared to a standard. The mechanism responsible for the major loss in active surface area, i.e. a particle detachment process, however, is the same as that found in the previous studies. By comparison, the transition metal modified carbon support clearly leads not only to an inhibited ECSA loss, but furthermore results in a drastic change of the main degradation mechanism. The results indicate that after an initial degradation caused by the fusion of adjacent particles, the catalyst is stabilized against further treatment. The particle detachment process is successfully suppressed. Interestingly, the suppression of particle detachment is not correlated to the resistance of the support towards the complete oxidation to carbon dioxide.Acknowledgements
This work was supported by the German DFG through the Emmy-Noether project ARE852/1-1. Katrin Hartl acknowledges the German FCI for a chemistry funds scholarship. Dr. Ekkehard Schwab and Dr. Claudia Querner of BASF are acknowledged for the supply of the catalyst samples and the carbon corrosion tests.References
- T. R. Ralph and M. P. Hogarth, Platinum Met. Rev., 2002, 46, 3–14 CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- S. Mukerjee and S. Srinivasan, J. Electroanal. Chem., 1993, 357, 201–224 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS.
- P. Mani, R. Srivastava and P. Strasser, J. Phys. Chem. C, 2008, 112, 2770–2778 CrossRef CAS.
- H. R. Haas and M. T. Davis, ECS Trans., 2009, 25, 1623–1631 Search PubMed.
- K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef CAS.
- K. J. J. Mayrhofer, K. Hartl, V. Juhart and M. Arenz, J. Am. Chem. Soc., 2009, 131, 16348–16349 CrossRef CAS.
- P. J. Ferreira, G. J. la O', Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256–A2271 CrossRef.
- T. J. Schmidt, ECS Trans., 2006, 1, 19–31 Search PubMed.
- K. J. J. Mayrhofer, G. K. H. Wiberg and M. Arenz, J. Electrochem. Soc., 2008, 155, P1–P5 CrossRef CAS.
- K. J. J. Mayrhofer, S. J. Ashton, J. C. Meier, G. K. H. Wiberg, M. Hanzlik and M. Arenz, J. Power Sources, 2008, 185, 734–739 CrossRef CAS.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stab, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CAS.
- K. J. J. Mayrhofer, J. C. Meier, S. J. Ashton, G. K. H. Wiberg, F. Kraus, M. Hanzlik and M. Arenz, Electrochem. Commun., 2008, 10, 1144–1147 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- C. G. Granqvist and R. A. Buhrman, J. Catal., 1976, 42, 477–479 CrossRef CAS.
- M. S. Wilson, F. H. Garzon, K. E. Sickafus and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 2872–2877 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: CO stripping curves for the ECSA determination of the catalysts. See DOI: 10.1039/c0ee00248h |
| This journal is © The Royal Society of Chemistry 2011 |