Ionic liquids in the biorefinery: a critical assessment of their potential
Annegret
Stark
*
Friedrich-Schiller-University Jena, Institute for Technical Chemistry and Environmental Chemistry, Lessingstr. 12, 07743, Jena, Germany. E-mail: annegret.stark@uni-jena.de; Fax: +49 3641 948402; Tel: +49 3641 948413
First published on 8th November 2010
Abstract
The combination of the concept of an integrated biorefinery with ionic liquid technology is critically assessed, and potentials for further research and development are identified.
Broader contextEver increasing resource requirements and concerns about dwindling fossil reserves have shifted biomass into the focus of many research endeavours. The concept of an integrated biorefinery combines the production of both fuels and chemicals, starting from the pretreatment of raw biomass, its separation in its main fractions and the conversion to platform chemicals required for various consumer products. Due to the unique solvent properties of ionic liquids, these solvents promise advantages with regard to conversion and selectivity, leading to improvements with respect to energy savings when compared to conventional solvents. However, this research field is still in its infancy, and important information such as recyclability of the often costly solvents, toxicology, environmental impact, waste disposal and long-term stability is scarce. This article critically assesses the state of the art on the example of four case studies, focussing on the carbohydrate fraction, and identifies a number of opportunities for further research and development. The next decades promise to be exiting times for biomass processing technology development in general. Time will show in which processes ionic liquids can excel. |
Introduction
This contribution aims at critically assessing the potential of ionic liquids in the manufacture of renewable resource-based products, i.e. chemicals and biofuels, from an industrial chemistry point of view. After discussing the principles of cellulose dissolution on the molecular level, the field of ionic liquid application in biomass processing is analysed on the examples of four case studies. Both potential pitfalls and research opportunities are identified.The biorefinery concept
Conventional fuel or petrochemical refineries can be subdivided schematically into various steps, during which the complexity of the feed crude oil, initially consisting of many thousand different organic structures, is progressively reduced, depending on the intended destiny: fuels or chemical products, respectively. The crude oil is first ‘cut’ into various fractions by distillation (i.e. physical separation), which differ in their boiling point ranges, before several chemical steps follow, in which(a) high boilers are thermally, catalytically or reductively cracked to yield lower boiling alkenes and alkanes. This improves the yield of the C5–C10 fraction for gasoline and the C12–C20 fraction for diesel,
(b) low(er) boilers, in particular heavy gasoline, are catalytically reformed, leading to the cyclisation of alkanes, aromatisation and isomerisation. This operation increases the research octane number and improves the performance of gasoline,
(c) defined feeds (propene and butenes) are transformed into higher isoalkenes (light-end processing), which are hydrogenated to isoalkanes for gasoline use, and
(d) raw oil-intrinsic catalyst poisons, such as sulfur- or nitrogen-containing organics and organometallic complexes, are removed by down-stream hydrotreating units, in which H2S, NH3 and metal particles are catalytically produced and subsequently removed from the feed.
Further separation units, especially distillation and extraction, are then applied to the various streams to obtain chemically pure bulk chemicals which are converted to various consumer goods.
According to the National Renewable Energy Laboratory, a biorefinery is defined as “a facility that integrates conversion processes and equipment to produce fuels, power and chemicals from biomass”.1 In analogy to fuel and petrochemical refineries, an integrated biorefinery will produce both product mixtures for fuel use and pure platform chemicals, which will be the basis of a family tree of compounds for the production of materials. In this sense, biorefineries are destined to complement petrol and chemical refineries. Currently in oil-based economies, about 90–95% of the raw oil is turned into fuels, and only 5–10% are converted for further chemical use.
A question which has to be answered in future is whether enough biomass is available (at reasonable cost and environmental impact) to substitute raw oil completely (2010: 150–185 × 109 GJ a−1).2 The yearly production of biomass is estimated at 170–200 × 109 t.3 If this biomass was wood only (i.e. high calorific value of approx. 15 GJ t−1), about 5–8% of the total biomass would be required to cover oil consumption alone (disregarding other fossil sources), in addition to the currently utilised biomass. It is unlikely that the exploitation of renewable resources in this magnitude can be conducted in a sustainable fashion. Therefore, it is paramount to reduce short-term the consumption of liquid fuels and make use of biorefinery products in the chemical sector.
Depending on the biomass used, the feed may or may not be as chemically diverse as crude oil. Overall, the 170–200 × 109 t annually produced consist of 75% carbohydrates (cellulose, starch, and saccharose), 20% lignin, and 5% other compounds, such as fats/oils, proteins, vitamins, dyes, essences, enzymes, hormones, etc.4,5 Presently, approximately 6 × 109 t a−1 of the total biomass are utilised for various applications by mankind, such as the generation of energy, food and feed, building materials, and clothing, of which 20 × 106 t are spent in non-food areas such as the chemical industry.4
In order to make use of the abundance of biomass, two general strategies are conceivable: the first postulates a thermochemical approach, applying syngas prescience to build up chemicals along the Fischer–Tropsch and other syngas pathways, leading to the same products established in oil- and coal-based economies. Ionic liquids have, with few exceptions only,6–10 not been applied in thermochemical conversion processes (gasification, pyrolysis) to yield either syngas or bio-oils. This is mainly due to their thermal instability under the harsh conditions used.11
The second strategy makes use of the synthetic accomplishment of nature, in which the predesigned chemical structures are preferentially retained (stereo- and regiochemically pure compounds). Especially if the high oxygen content can be maintained or selectively reduced, the need for capital-intensive oxidation processes required in petroleum-based chemistry can be mitigated. Hence, chemical operations will have to shift from oxidation (to incorporate functionality in petrochemicals) to reduction (to decrease the oxygen content).
In the context of the biorefinery, it remains still open which of the two strategies is economically and ecologically more beneficial. Possibly, future integrated biorefineries will combine both concepts. Much depends on diverse factors such as local climate and agricultural efficiency, politics, and, last but not least, consumer acceptance of biomass-derived products, which may possess somewhat different properties than already established products.
Biorefinery processing—a general overview
In a fully integrated biorefinery, the feedstock (e.g. wood, grain, sugar beet/cane, soy, rape, etc.) is separated into its main, chemically well-defined fractions (cellulose, hemicellulose, lignin, extractives, etc.) in what could be considered a primary refinery. Fig. 1 shows a possible scenario of a future integrated biorefinery and examples of down-stream products derived from biomass.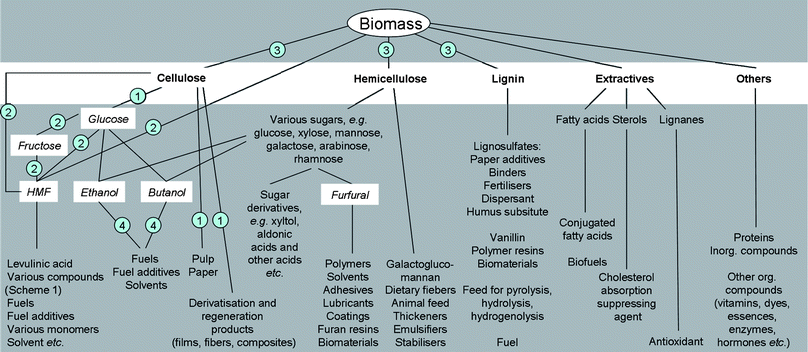 | ||
| Fig. 1 Schematic flow chart of a biorefinery (numbers indicate case studies discussed). | ||
Due to the low volatility of many bio-based fractions and products, separation units of oil-based refineries (mainly distillation) will have to be substituted by others, e.g. extraction. Simple adaptation of petrochemical separation processes to biomass will hence be relevant in rare cases only. The application of efficient extraction techniques implies that the biomass has to be homogenised, and most advantageously, liquefied. The liquefied biomass may then be treated by a series of selective extractants to remove valuable biogenic compounds (fragrances, pharmaceutically active compounds, alkaloids, fats and oils, proteins and lignin) from the carbohydrates (mono- and oligosaccharides, starch, cellulose, hemicellulose, and chitin).
After physical treatment by extraction, the fractions of the primary refinery are then distributed to various chemical or biochemical processes (secondary refineries), in which so-called platform chemicals are produced.
Due to the fact that dry biomass consists mainly of carbohydrates, this fraction is presently the most intriguing for potential biomass conversions12 in terms of proving the feasibility of the biorefinery concept, and will be considered in the following.
Fig. 2 shows examples of platform chemicals derived from the carbohydrate fraction. It also exemplifies that secondary refineries produce both innovative and established compounds. In the former instance, these chemicals and materials thereof have not yet entered (or are just about to enter) the market, and have to find acceptance there. In some cases, their properties, such as improved biodegradability, may allow for the substitution of the petrol-based competition products, as is the case for polylactic acid as substitute for PET.13 As examples for the latter instance, acrylic acid, 2,3-pentanedione and ethanol can be named. In September 2010, Braskem has inaugurated the operation of an ethene plant in Brazil, which produces polyethylene from bioethanol in pilot scale.
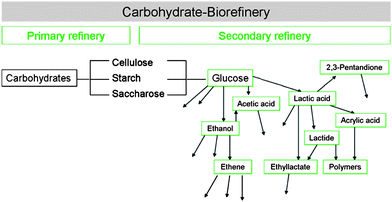 | ||
| Fig. 2 Examples of platform chemicals derived from the carbohydrate fraction. | ||
It needs to be stated at this point that an integrated biorefinery is at this moment a concept only, and presently running processes, such as the production of ethanol or lactic acid from glucose or starch, or glycerol and biodiesel from fats and oils, can be seen as dedicated product lines which may be incorporated into an integrated biorefinery in the future.
Challenges for the implementation of biorefineries
Several challenges result from the above discussion:(a) Liquefaction: most components in biomass, and particularly polysaccharides, are not soluble in water or other conventional solvents. What are the options for complete dissolution?
(b) Extraction: are solvents available to provide selective fractionation? How does the (hence large) solvent inventory affect the economical and ecological performance of the biorefinery?
(c) Chemical conversion: the break-down of polysaccharides into various products is structure-inherent, yet unselective. Is it feasible to control the pathways of decomposition to yield products, e.g. glucose from cellulose, selectively?
(d) Catalytic conversion: most industrially utilised catalysts have been designed to convert organic feed with little functionality (low degree of oxidation). Can established catalysts serve for the conversion of biomass? Are selective reduction methods available for bio-based feedstock? How can sufficient hydrogen be provided at lowest possible CO2-footprint?
Ionic liquids
Ionic liquids are salts which possess melting points below 100 °C, and can be designed to be liquid at room temperature.15–17 They are composed of large organic cations and inorganic or organic anions (Fig. 3), and the choice of these components determines the distinct physico-chemical properties (melting point, polarity, viscosity, chemical and thermal stability, solvation properties, and phase behaviour) of each ionic liquid.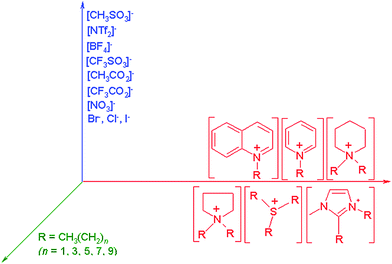 | ||
| Fig. 3 Some of the possible combinations of anions, cations and cation substituents to yield ionic liquids with distinct physico-chemical properties.14 | ||
Much research has been conducted to contribute to an understanding of how the complex interplay of van der Waals interactions, hydrogen bonding and Coulombic forces affect the various properties, so that the structure of an ionic liquid can be tuned to result in properties beneficial for the application in question. For example, densities and viscosities can be varied between 0.9 and 2.2 g L−1 and 14 and 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mPa s, respectively. The thermal stability can be designed to be as high as 400 °C, i.e. much higher than most organic solvents.
000 mPa s, respectively. The thermal stability can be designed to be as high as 400 °C, i.e. much higher than most organic solvents.
From a safety and environmental point of view, ionic liquids offer the potential of reducing both the risk of exposure of workers and the environment to gaseous emissions due to their low vapour pressure, and the risk of explosions due to their low flammability. Ionic liquids can feature low toxicities, and bring about facile product separation by tuning the solvation properties. An up-to-date state of the art on the application of ionic liquids in Green Chemistry has recently been published.18,19
In the past 20 years, ionic liquids have proven their potential as solvents for a wide variety of reactions and separation problems. While in most instances, petrochemical models were chosen and developed to reasonable maturity, the application of ionic liquid technology to biogenic feed is still in its infancy. However, especially due to the unique solvation properties, ionic liquids may contribute to tackle the challenges of biorefinery concepts pointed out above. Fig. 4 shows some of the tuneable ionic liquid properties which may be exploited in biomass processing.
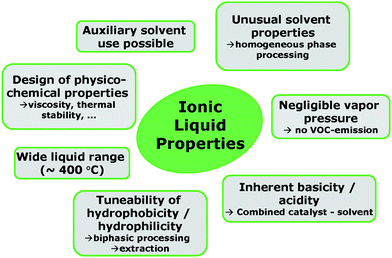 | ||
| Fig. 4 Some of the tuneable ionic liquid properties which may be exploited in biomass processing. | ||
The goal of this article is to provide a critical review of the state of the art and deduct a general understanding of ionic liquid interactions and potentials in applications rather than being a comprehensive19–26 compilation of the literature. Hence, after an analysis of the field, which gives examples of ionic liquid applications in biomass processing, the principles of cellulose dissolution are discussed on the molecular level. Then, several stages of the biorefinery where an ionic liquid could potentially be used are highlighted in the form of case studies:
1. cellulose processing,
2. 5-hydroxymethylfurfural production,
3. native biomass conversion and
4. down-stream product separation techniques (Fig. 1).
Finally, overall conclusions are drawn, pitfalls identified and potential areas of future research and development in this field highlighted.
Ionic liquids in the biorefinery
Analysis of the field
The non-derivatising dissolution† of cellulose in ionic liquids discovered in 200327–29 triggered an armada of publications23,30 and patents, highlighting the potentials of ionic liquid-based cellulose processes for the manufacture of fibres and other cellulosic materials. Building on this story of success, it was soon discovered that certain ionic liquids may even serve to dissolve complex biomaterials such as wood, straw, native chitin, wool keratin, suberin from cork, and silk.31–33 In the years following, ionic liquids have been applied as selective extractants. One impressive example is the removal of sulfur- and nitrogen-containing organics, which may be a technology to substitute hydrotreating units up-stream of catalytic processing facilities.34 Another example is the selective removal of lignin or butanol from fermentation broths by ionic liquid extraction.35,36With respect to chemical processes, ionic liquids can bring about the selective glycosidic cleavage of polysaccharides37,38 and high selectivities in the conversion of monosaccharides to platform chemicals, depending on the acidity of the medium38 and the water content.39
One very intensely investigated platform chemical based on saccharide fractions is 5-hydroxymethylfurfural (HMF). Ionic liquids improve the selectivity for HMF when starting from fructose,40–47 and act as bifunctional liquids with integrated catalytic activity for the combined isomerisation and condensation of glucose40,48,49 and cellulose50 to HMF. Furthermore, HMF conversion to possible biofuel additives has been demonstrated to work in ionic liquids.51–54
Another approach is the preparation of first generation biodiesel from the oil/fat fraction. The applicability of ionic liquids in both the lipase-catalysed transesterification55 and the acid-catalysed esterification of fatty acids56 has been demonstrated, where product isolation is achieved by simple decantation.
In the following, the solvation mechanism of cellulose and other bio-derived materials is discussed to lay a foundation on which ionic liquid application to biomass conversion is discussed on the example of several case studies.
Governing principles of cellulose in ionic liquids
Not many solvents57 are available which can be used on a technical scale to physically dissolve cellulose (and other biogenic compounds of high molecular weight). In these materials, the main structural motif yielding a high chemical stability and therefore insolubility is hydrogen bonding. Cellulose is the best-investigated compound in terms of understanding dominant solvent–solute interactions, which is a prerequisite for knowledge-based design of new ionic liquid candidates with improved performance. These interactions are summarised in the following.Depending on the ionic liquid's anion and the type of cellulose (degree of polymerisation) investigated, up to 20 wt%‡ solutions can be obtained. From a structural point of view, especially anions with high basicity, i.e. high hydrogen bond acceptor (HBA) strength (β-values in terms of Kamlet–Taft parameters),58 such as chloride, acetate, formate and diethylphosphate, give high concentrations of dissolved cellulose.28,25,59–61 Less basic anions, such as bromide, biscyanamide or thiocyanate, lead to swelling, but not to complete dissolution, while tetrafluoroborate, hexafluorophosphate and trifluoromethanesulfonate leave the hydrogen bonding network fully unaffected. The effect of the ionic liquid's cation is less well investigated, but solubility appears to decrease with increasing alkyl chain length, substitution of alkyl by alkenyl or alkynyl,62 and methylation in C2-position in 1-alkyl-3-methylimidazolium-based ionic liquids.63 Tetraalkylammonium-, N-alkyl-N-methylpyrrolidinium- and N-alkylpyridinium-based64 ionic liquids also yield lower solubility of cellulose than 1-alkyl-3-methylimidazolium-based ones. In certain cases, especially when high dissolution temperatures (>150 °C) are applied or ionic liquids containing traces of acids are used, dissolution is accompanied by polymer cleavage, leading to a reduced degree of polymerisation.25,28,32,59–65
It has been suggested that both the anion and the cation partake in the dissolution of cellulose by forming electron donor–electron acceptor complexes with the polymer's hydroxyl groups,66 hence separating the chains and affording dissolution. However, molecular dynamics studies of glucose as a model showed that a close contact of the chloride anion with the OH-groups occurs in 1,3-dimethylimidazolium chloride. When the ionic liquid is present in excess, three hydroxyl groups interact with three different chloride anions, while the remaining two OH-groups are involved in interactions with one chloride forming an OH–Cl–HO bridge. The ionic liquid's cation forms the outer shell, interacting only to a small percentage.6735/37Cl and 13C NMR T1 longitudinal relaxation measurements of the model cellobiose dissolved in either 1-butyl-3-methylimidazolium chloride ([C4mim]Cl) or 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc]) lead to the same conclusion.6813C NMR data of the β-1-4-linkage of cellulose and cellulose oligomers in 1-butyl-3-methylimidazolium chloride indicated a disordered structure, similar to that of other non-derivatising solvents such as DMSO/tetrabutylammonium fluoride.69
Our group studied the chemical shift of the hydroxyl group of ethanol by 1H NMR spectroscopy to measure the strength of the hydrogen bond interaction between hydroxyl groups and anions.14,70 These were found to be linearly correlated to the HBA strength of the anion58 (β-value vs. chemical shift of the hydroxyl group). The results were transposed to cellulose dissolution. The goal of this study was to understand how the solvation properties of an ionic liquid-cellulose solution may be affected by the addition of non-dissolving additives. The study, which was conducted at low cellulose concentrations (1–2 wt% Avicel), showed that in fact only those ionic liquids with a large downfield shift of the hydroxyl group of ≥4.3 ppm (i.e. β-values ≥ 0.85) gave clear solutions.71
Binary mixtures of ionic liquids (cation 1/anion 1 + cation 1/anion 2), composed of both a “dissolving” and a “non-dissolving” anion, were investigated with regard to cellulose dissolution ability.70 If the anions of the “non-dissolving” ionic liquid are of medium HBA strength (e.g. bromide), a lower amount of “dissolving” ionic liquid is necessary than when the “non-dissolving” ionic liquids show a reduced tendency to form hydrogen bonds (e.g. bis(trifluoromethanesulfonyl)amide, [NTf2]−). Hence, additive effects occur: in solutions of cellulose in ionic liquids, the anion interacts either with the cellulose–OH-groups or with the cation (or both). If the “non-dissolving” ionic liquid [C2mim][NTf2] is added to the dissolving [C2mim][OAc], the number of potential non-productive interactions of the acetate with the cation increases, especially since [NTf2]− does not interact much with its own cation. The HBA strength of the acetate is therefore decreased for interaction with cellulose. If the “non-dissolving” anion is a medium-strength hydrogen bond acceptor, such as bromide, it is involved in particular in cation–bromide hydrogen bonding, hence making the “dissolving” anion acetate available for stronger overall interactions with cellulose.
In fact, the 1H NMR investigations with ethanol showed differences between monoatomic and multiatomic anions in terms of their relative preference for interaction, where halides interacted stronger with their cations than multiatomic anions (Fig. 5).
![Correlation between 1H NMR chemical shift of the ethanol–OH and cationic C2–H proton in equimolar ethanol-[C2mim]-based ionic liquid mixtures.70](/image/article/2011/EE/c0ee00246a/c0ee00246a-f5.gif) | ||
| Fig. 5 Correlation between 1H NMR chemical shift of the ethanol–OH and cationic C2–H proton in equimolar ethanol-[C2mim]-based ionic liquid mixtures.70 | ||
Similar mechanisms must also be responsible for the fact that certain amounts of other, “non-dissolving” additives, such as water or DMSO, can be added to solutions of cellulose in ionic liquids without causing cellulose precipitation.28,71
Own results show70 that less water is tolerated, if higher concentrations of cellulose are dissolved. At low cellulose concentrations (2 wt% Avicel), approximately 2 and 0.5 molar equivalents of water can be added to [C2mim][OAc] and [C4mim]Cl, respectively, without leading to precipitation. This means that chloride is deactivated as cellulose solvent if a 2:1 complex has formed with water, while acetate is only deactivated at a much higher water content. Again, the higher HBA strength of the latter anion can be used as explanation of this phenomenon.
1H NMR studies on the influence of the cation (increase of the alkyl chain length, methylation in C2-position in 1-alkyl-3-methylimidazolium-based ionic liquids) on the downfield shift of the hydroxyl group of ethanol showed no alteration in the HBA strength of the interaction with the anion. This stands in contrast to experiments determining the solubility in cellulose, as discussed above. In this instance, accompanying parameters, such as increased viscosity, melting point and steric hindrance, may be responsible for reduced solubility at temperatures <100 °C. In conclusion, the solubility of cellulose can be described with the HBA strength of the anion of the ionic liquid. Anions which form strong hydrogen bonds are capable to interfere with the inter- and intramolecular hydrogen bond network of cellulose, leading in effect to dissolution.14,70
Case-study 1: cellulose processing
The discovery that ionic liquids can be used as non-derivatising cellulose solvents has lead to a huge body of work. Four general processing applications arise from this:1. regeneration,
2. chemical modification,
3. enzymatic hydrolysis, and
4. chemical depolymerisation.
| Feed | Entry | Conditions | Ionic liquid | Products | Ref. |
|---|---|---|---|---|---|
| a 1,8-Diazabicylo(5.4.0)undec-7-enium-cation. b Mixture of alkylbenzenesulfonate, consisting mainly of xylenesulfonate. | |||||
| Cellulose (dried) | 1 | 10 wt%, 100 °C, or, 25 wt%, microwave heating | [C4mim]Cl, [C2mim][OAc] | Cellulose regenerated | 27, 28 |
| 2 | 5 wt%, 130–150 °C, 10–180 min | [C4mim]Cl | Cellulose regenerated for successive cellulase hydrolysis | 82, 84 | |
| 3 | 2.5–5.0 wt%, 100 °C, 16 h (addition of water) | [C2mim][HSO4] | Lower oligomers, DP depending on processing conditions | 85, 86, 89, 90 | |
| 2.5–5.0 wt%, 100–120 °C, 4–16 h (addition of various acids) | [C2mim]Cl, [C4mim]Cl, [C4dmim]Cl | ||||
| 2.5–5.0 wt%, 80–120 °C, 24–96 h (addition of nucleophiles) | [C4mim]Cl | ||||
| 2.5–5.0 wt%, ultrasound | [C4mim]Cl | ||||
| 5 wt%, 100 °C, 5 h, Amberlyst 15DRY | [C4mim]Cl | • Cellulose oligomers | 86, 87, 88, 90 | ||
| • 30% total reducing sugars, of which 10% mono- and disaccharides | |||||
| • Precipitation of oligomers by addition of water | |||||
| Pine, poplar, eucalyptus, oak, straw, plywood, spruce, switchgrass | 4 | 5–10 wt%, 100–130 °C, 8–16 h,31 150 °C (micro-wave), 1 h105 | [C4mim]Cl and other chloride-based ionic liquids, [C2mim][OAc] | • Complete dissolution | 31, 32, 83, 105, 108 |
| • Cellulose separation from lignin and hemicellulose by | |||||
| - Precipitation of cellulose31 | |||||
| - Removal of lignin by extraction prior to precipitation of cellulose105 | |||||
| • Some depolymerisation | |||||
| • Direct hydrolysis with enzymes in ionic liquid not possible due to denaturation108 | |||||
| • Selective precipitation of cellulosic and hemicellulosic oligomers suitable for fast enzymatic digestion in aqu. medium83,108 | |||||
| • Direct acetylation32 | |||||
| Bagasse | 5 | 6–10 wt%, 190 °C, 1 h | [C4mim]Cl, +20 wt% aqu. NaOH/NaCl | • Complete dissolution | 106 |
| • Addition of aqu. NaOH-phase | |||||
| • Precipitation of cellulose | |||||
| • Lignin and hemicellulose dissolved in aqu. phase | |||||
| • Ionic liquid free of org. material | |||||
| • Precipitation of lignin by acidification of aqu. phase | |||||
| Poplar | 6 | 7 wt%, 100 °C, 24–72 h | [DBU-H]Xa, X− = Cl−, [CH3SO3]−, [HCO2]−, [CH3CO2]−, [(RO)2PO2]− | • Complete dissolution | 73 |
| Corn stalk, rice straw, pine, bagasse | 7 | 5 wt%, 100–120 °C, 5–60 min | [C4mim]Cl + aqu. HCl, [C4mim]Cl + aqu. CF3CO2H, [C4mim][HSO4] | • Complete dissolution of carbohydrates, not lignin | 37, 39, 91 |
| • Depolymerisation to lower oligomers | |||||
| • Monosaccharide selectivity is function of time, water and acid concentration (e.g. formation of HMF and furfural) | |||||
| Poplar, bagasse, maple | 8 | 5–10 wt%, 80–160 °C, 1.5–72 h (acetic acid)111 | [C1mim][CH3SO4], [C4mim][CF3SO3], [C2mim][ABS]b, [DBU-H]-Xa73 X−= [OTs]−, [CF3CO2]−, [HSO4]−, lactate, [SCN]− |
• Selective dissolution of lignin | 35, 73, 111 |
1. How can polar impurities which accumulate in the spinning bath be efficiently removed?
2. Is the price of ionic liquids prohibitive for cellulose processing?
Also, the application of ultra-sound85 or microwaves90 can help in the breakdown of the polymer chain. Examples of conditions investigated are shown in Table 1, entry 3. In general, acid hydrolysis of cellulose features higher rates of reaction, but lower selectivity than enzyme catalysis.
As with cellulase, in situ fermentation to convert the sugars to ethanol is currently not feasible. Furthermore, if depolymerisation is carried out with the goal of producing glucose, the problem of product separation arises, since solvent extraction was found to be inefficient.87,91,92 Higher oligomers may be precipitated by the addition of water.86 However, chemical conversion to 5-hydroxymethylfurfural can be achieved, as discussed in case study 2.
In summary, cellulose processing may benefit from the application of ionic liquids as solvents, although, as pointed out, several questions remain to be answered. In the area of homogeneous chemical modification, ionic liquids are one of the few available solvent classes which allow for the selective derivatisation of cellulose. For enzymatic hydrolysis in ionic liquids, specialised enzymes or whole cells may in future be used for the direct digestion, but even the faster rates obtained with ionic liquid-regenerated cellulose provide pronounced advantages. The chemical depolymerisation proceeds under moderate conditions, and the selectivity can be controlled by the choice of ionic liquid, reaction time and acid catalyst. However, the removal of lower glucose oligomers and glucose from the ionic liquid is yet unsolved, although first results show that it may be possible to use either an extraction technique with boronates93 or ion exclusion chromatography.39 Since enzymatic conversion appears to be hampered due to deactivating effects of the ionic liquids, in situ chemical conversion to down-stream products appears to be the only alternative, which has recently been exploited in the generation of 5-hydroxymethylfurfural. This is discussed in the following section.
Case study 2: 5-hydroxymethylfurfural production
5-Hydroxymethylfurfural (HMF)¶ is a platform chemical down-stream of monosaccharides such as glucose or fructose. It can in principle also be made directly from higher saccharides, a pathway which has recently found much attention in chemical research. As expected for a molecule bearing multiple functionalities (aldehyde, alcohol, ether, and diene), the number of reactions it undergoes is enormous (Scheme 1) and has been reviewed.47,94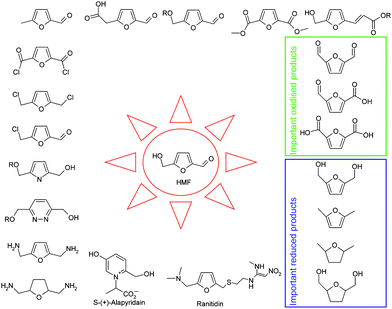 | ||
| Scheme 1 Examples of HMF-derivatives. | ||
Examples of possible fields of application include the monomer sector (e.g. as substitute for terephthalic acid), fuels and fuel additives, solvents, pharmaceuticals and fine chemicals, food additives, etc. Its oxidised form (2,5-dicarboxyfuran) has been selected by the Pacific Northwest National Laboratory as a Top 10 platform chemical in an evaluation of biorefinery products of the highest industrial relevance.95 Despite this broad applicability, it must be stated that relatively little is known about the specific properties of HMF-based products, in particular about the much-propagated polymers.
Additionally, it is unlikely that with today's state of the art, competition with petrochemical products in high volume markets is possible. Hence, especially the fine chemical and pharmaceutical markets appear lucrative, for which alapyridain and ranitidin may serve as examples.
HMF is in principle an intermediate in the condensation of saccharides to levulinic and formic acid. Hence, the chemical challenge is to choose reaction conditions by which side-product formation is avoided, hence keeping separation expenditure to a minimum. In fact, most publications deal with the optimisation of process parameters, disregarding product separation and purification. One example of the open literature underlines the complexity of efficient product separation: in order to obtain high yields of HMF, fructose is converted using HCl as catalyst in a reaction phase consisting of aqueous DMSO, which is modified with poly(1-vinyl-2-pyrrolidone) to improve selectivity. This process yields >80% HMF at 90% sugar conversion. The extractant is a solvent mixture consisting methyl-iso-butyl ketone and 2-butanol, both high-boiling solvents which have to be separated by distillation. This lowers the energy efficiency of the overall process dramatically.96–98
On a technical scale, HMF has been produced in a pressurised batch reactor in aqueous solution at pH = 1.8 (H2SO4) in 2 h at 150 °C. After precipitation of the catalyst by neutralisation with a base, filtration, removal of the solvent, followed by time- and energy-consuming ion exchange chromatography, a crystallisation step is required. At a conversion of 90%, yields between 40 and 50% are obtained.99 Using this method, a large batch has been produced and distributed by Südzucker.
Ionic liquids, in particular [C4mim]Cl,45 [C4mim][BF4] or [C4mim][PF6],43 and their mixtures with DMSO were used as solvents for the condensation of fructose to HMF, catalysed by acidic ion exchange resins, Brønsted acids or tungsten salts.46 Acido-basic media (nowadays a subgroup of ionic liquids), i.e. equimolar mixtures of organic bases (e.g. pyridine) with acids (e.g. HCl or p-toluenesulfonic acid) were developed to tune the acid strength and thus improve the selectivity of the system.40–44 Yields between 70 and 90% HMF can be obtained, fully avoiding the formation of levulinic acid or humines. Brønsted-acidic ionic liquids, in some cases chemically immobilised on silica, have also been described.100
In our laboratories,47 we have conducted systematic batch studies to elucidate the effect of temperature, reaction time, sugar and catalyst concentration, sugar source, as well as purity and water content of the ionic liquid in the condensation of fructose to HMF. Reactions were carried out with the ionic liquid [C4mim][CH3SO3] using methanesulfonic acid as catalyst. It was found that the reaction proceeded only slowly at temperatures below 80 °C, while at higher temperatures, the formation of side-products, indicated by a dark colour of the reaction mixture, occurred. Thermal activation can be achieved convectively, i.e. by using an oilbath or heating block, or by using a microwave apparatus. The type of activation did not affect selectivity or yield. Fructose concentrations between 4 and 30 wt% were investigated, showing that at lower concentrations, higher yields and selectivities were obtained (up to 90% within 60 min at 4.1 wt% fructose). Some side-product formation occurred via pathways which do not proceed from HMF as intermediate, as opposed to side-product formation in aqueous systems.96,99,101 Hence, for a future process, a balance between a low ionic liquid inventory and high selectivity must be found to achieve lowest possible process costs.47
Since water is liberated in this reaction, the effect of the water concentration was investigated. At molar ratios X(ionic liquid) < 0.5, i.e. in the water-rich region, the reaction was highly negatively affected (75–0% yield), while at X(ionic liquid) > 0.5, hardly any effect on the yield was found (75–90% yield). Hence, water must be removed regularly from the ionic liquid in a future process. However, the reaction outcome is relatively stable as long as the ionic liquid is present in excess.
In the pure ionic liquid, no conversion was observed. Small amounts of methanesulfonic acid (1.5–5.6 wt%) lead to a tremendous increase of activity, decreasing again at higher catalyst concentrations. In these latter cases, the selectivity decreased also, therefore, the acid concentration should not exceed 5 wt%.47
In order to investigate the effect of the type of ionic liquid chosen, various alternative ionic liquids were tested, giving similar yields and selectivities as [C4mim][CH3SO3]. This aspect allowed for the design of more cost effective ionic liquids, which could be obtained by a modified Radziszewski reaction yielding homosubstituted 1,3-dialkylimidazolium-based ionic liquids.102
Additionally, since the reaction is independent of the type of the ionic liquid, the anion and cation choice can be used as a tool to optimise the extraction efficiency in product recovery and recycling steps.
Several alternative saccharides were tested. Glycosidic cleavage occurred with any of the saccharides investigated, e.g. inulin or saccharose. However, only the fructose moiety was converted in this latter instance. On the other hand, high yields were obtained from inulin (90%, 3 h) and high fructose corn syrup (HFCS 50), containing about 42% fructose with respect to dry matter. Under optimum conditions, about 90% of the fructose content was converted to HMF, indicating that the system is flexible with regard to its fructose-containing feed source.47 The economic potential of HMF is closely connected to the type of bio-feedstock that is processable. Due to the lower availability of fructose, its cost and the fact that it is a food-stuff (food-versus-feed discussion), hexose alternatives need to be exploited.
As early as 1962, Mednick described the synthesis of HMF in 45% yield from glucose (or starch) in acido-basic media as catalysts, consisting e.g. of pyridine and phosphoric acid, in a mixture of water and dioxane as solvent at high temperatures (200–230 °C).40 The temperature can be significantly reduced by using metal-based catalysts. Hence, a groundbreaking improvement of HMF manufacture was achieved by Zhao et al. with a system that allows for the single-stage isomerisation of glucose to fructose prior to the condensation to HMF. Addition of catalytic amounts (0.6 mol% of CrCl2) to [C2mim]Cl gives a glucose conversion of 80–90% (yield: approx. 70%) at 100 °C. Although it is clear that from an environmental point of view, the use of chromium catalysts is problematic, this study demonstrates that glucose isomerisation is feasible using Lewis acid catalysts in ionic liquids.48 A recent example is the use of a paired CuCl2/CrCl2 catalyst in [C2mim]Cl, which yields 55% HMF within 8 h at 120 °C, starting from cellulose.103 Further improvements in yields of HMF from both fructose and glucose were achieved by ligand design. Yong et al. showed that 1,3-substituted imidazolidene ligands on Cr(II)chloride (10 mol% catalyst concentration) gave yields of 96 and 81% for fructose and glucose, respectively, using 10 wt% sugar in [C4mim]Cl.49
N,N-Dimethylacetamide (DMA)–LiCl mixtures are known cellulose solvents (solvation capacity up to 15 wt%). However, strong ion-pair formation seems to inhibit catalytic interaction with fructose in the conversion to HMF. On the other hand, softer halides (bromide) gave an excellent yield of 90%. Starting from glucose, DMA with CrCl2 or CrCl3 (modified with NaBr) resulted in yields of up to 80% HMF. From both purified cellulose and untreated corn stover, yields of up to 50% HMF were obtained in mixtures of DMA–LiCl, CrCl2, HCl and [C2mim]Cl at 4 wt% cellulose concentration.50
In summary, it has been demonstrated that ionic liquids can serve as solvents for the production of HMF from various saccharides, including glucose and cellulose. Before these processes can be included into an integrated biorefinery, focus must be placed in the near future on product separation and purification techniques to demonstrate economic viability.
Case study 3: native biomass conversion
After it became clear that ionic liquids can be used to dissolve cellulose, the problem of native biomass up-stream treatment was tackled. Although this area is still in its infancy, and—owing to the complexity of native biomass—little is yet known about behind-lying mechanisms, several conclusions can be drawn from the combination of the results. The major goal of biomass treatment is to allow for the separation of the main compounds, i.e. cellulose, hemicellulose and lignin. This separation must rely largely on the differences in chemical properties between the fractions: lignin is a heterogeneous group of phenolic macromolecules which are tightly linked. It is amorphous and of hydrophobic nature. Basic monomers include p-coumaryl alcohol, coniferyl alcohol and sinapyl alcohol. Hemicellulose is an amorphous, branched polymer structure of various monosaccharides. As opposed to hemicellulose, cellulose consists solely of anhydroglucose units, which are highly structured and possess areas of crystallinity. Its degree of polymerisation is higher than that of hemicellulose (7000–15000 and 500–3000 monomer units, respectively). Both hemicellulose and cellulose are hydrophilic in nature. Quite roughly it can be stated that solvents which dissolve lignin are less polar than those which are able to dissolve the celluloses. In the latter instance, the strong inter- and intramolecular hydrogen bonding interactions between the polymer chains have to be disrupted, which in most instances is achieved by using solvents which are able to engage in still stronger hydrogen bonds.
Particularly the removal of lignin from the (hemi)celluloses is important, as it inhibits the down-stream digestion of cellulose by enzymes. Conceptually, several strategies may be used:
1. Complete dissolution, and extraction of lignin.
2. Complete dissolution, and precipitation of lignin.
3. Selective dissolution of hemicellulose and cellulose, leaving lignin as solid behind.
4. Selective dissolution of lignin, leaving (hemi)cellulose as solid behind.
Especially interesting is the use of acido-basic media (Table 1, entry 6), such as [DBU-H]Cl (where DBU = 1,8-diazabicyclo[5.4.0]undec-7-ene), to achieve complete dissolution of the biomass. It can be expected that cellulose depolymerises under these conditions to some degree. Lignin can be precipitated by pH change (acidic to basic), leading to the liberation of DBU as free amine, which can be hence recovered.73
The ionic liquid-cellulose solution can be used for direct acylation to yield cellulose esters.32,76 Also, the regenerated cellulose was found to be suitable for enzymatic digestion in aqueous solutions due to its low crystallinity.83
The simultaneous dissolution and depolymerisation described here can be compared with two processes developed to overcome a shortage of sugar at the beginning of the 20th century:107 firstly, in the Scholler-Tornesch process,109 dried wood particles are subjected for 45 min to diluted sulfuric acid at 150–190 °C and 12–14 bar pressure in a percolator. Within 10–20 h, about 500 kg fermentable sugars are obtained per ton of wood. As disadvantages, the batchwise production mode, saline waste and a number of side-products due to unselective depolymerisation can be named.
Secondly, in the Bergius–Rheinau process,110 dried wood particles are exposed to a concentrated HCl (40 wt%) solution at 20 °C and ambient pressure, resulting in 600 kg total sugar, of which 280 kg are glucose hydrate per ton of wood, leaving lignin as insoluble residue. The formation of much saline waste (due to the neutralisation step), side-product formation, as well as corrosion are inherent problems of this process. The sugars were mainly used for the fermentation to yield ethanol.
Also, protonated DBU-salts based on anions such as tosylate, trifluoroacetate, lactate, hydrogensulfate, trifluoromethane-sulfonate or thiocyanate can be used to selectively dissolve lignin.73 As discussed above, structural analogues of these were claimed to bring about complete dissolution. How in the case of these protonated amines the choice of the anion influences dissolution selectivity is not yet understood. In general, however, the solubilisation of lignin occurs via strong ion–dipole and π–π-interactions.111
In summary, the handling of native biomass may profit from the unusual solvent properties of ionic liquids. As in the case of cellulose processing, little detailed data are momentarily available relating to product quality and properties, solvent regeneration and price, etc. However, the analysis of the literature shows clearly that ionic liquids can be tuned to selectively dissolve either lignin or (hemi)cellulose.
Presently, little attention is paid to the hemicellulose fraction, which remains dissolved in either the ionic liquid31,32,73,83,105,110 or is lost with the aqueous phase.106 Hemicellulose-based down-stream products are of economic interest (see Fig. 1). Hence, there is further R&D potential in the area of hemicellulose separation and processing using ionic liquids. Furthermore, it needs to be pointed out that dissolution in ionic liquids is only achieved if the biomass is predried, although a certain amount of water may even promote dissolution.71 Additionally, the dissolution process proceeds very slowly yielding highly viscous solutions.112
Both aspects may be connected to several problems in a large-scale process. It needs to be seen in the near future if using ionic liquids is a viable alternative for the Kraft-process, in particular with regard to the enormous solvent inventory that can be expected.
Case study 4: application in down-stream product separation
Ethanol and 1-butanol can be used as biofuels and/or fuel additives and are produced by fermentation. Alcohol recovery from ethanol or acetone–butanol–ethanol (ABE) fermentation broths is characterised by a high energy demand. For one reason, this is due to the low alcohol content achieved by yeasts (6 and 2 wt% for ethanol and ABE, respectively). Furthermore, in the case of ethanol separation, an azeotrope is formed, rendering the complete removal of water by distillation difficult.A recent patent reflects the attractiveness of the concept. Here, tricyanomethide-based ionic liquids are used to extract 1-butanol from the fermentation broth after separating and recycling the microorganisms. The 1-butanol-depleted aqueous phase is re-fed into the fermentation vessel, while 1-butanol is removed from the ionic liquid phase by distillation (Fig. 6).
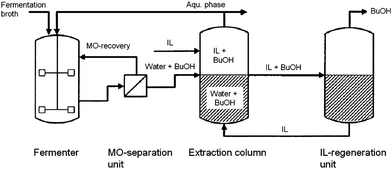 | ||
| Fig. 6 Process scheme detailing the extraction of 1-butanol from fermentation broth (adapted from ref. 113). | ||
The ionic liquids claimed for the process exhibit distribution coefficients DBuOH between 3.4 and 4.7 and selectivities α between 28 and 240, hence improving the performance when compared to both the hexafluorophosphate system discussed above and the often used oleyl alcohol (DBuOH = 3.5 and α = 230).113
![Separation factors as function of ethanol concentration in ethanol–water mixtures (IL = [C2mim][BF4]).114](/image/article/2011/EE/c0ee00246a/c0ee00246a-f7.gif) | ||
| Fig. 7 Separation factors as function of ethanol concentration in ethanol–water mixtures (IL = [C2mim][BF4]).114 | ||
Fig. 7 shows that entrainers enhance the separation factor, and that the ionic liquid is more efficient than ethanediol if similar concentrations are used. The separation factor increases further, if more ionic liquid is applied. The use of an ionic liquid entrainer reduces thus the number of plates and/or the recirculation ratio, leading to overall reduced separation costs.114
In conclusion, although the number of investigations is relatively small, ionic liquids have shown promise in the final separation and purification of biorefinery products. In most mature chemical processes, about 60–80% of the process costs are due to separation steps. As shown for the two examples of alcohol purification, ionic liquids may provide a higher energy efficiency than other methods.115
Conclusions
From the point of view of an ionic liquid chemist, the production and separation of platform chemicals and down-stream products deserves attention because of the large technical innovation potential, which might not be exploitable in already established and optimised petrochemical processes. The utilisation of ionic liquids, possibly in connection with other innovative tools such as supercritical fluid extraction, microwave-assisted synthesis or microreaction technology, may find entry into production in tandem with renewable resource utilisation.The analysis of the state of the art has demonstrated that ionic liquids containing anions of medium or high hydrogen bond acceptor strength can be used to dissolve biomass. The choice of the anion determines the selectivity of the dissolution process, hence leading to either biomass, cellulose or lignin solutions, or depolymerised oligo- and monosaccharide solutions, which can be sent to respective down-stream processing units. On the example of the production of 5-hydroxymethylfurfural, the selective preparation of a highly functional platform chemical has been demonstrated. Furthermore, ionic liquids display advantages in separation processes, examples being the enrichment of 1-butanol by extraction from fermentation broths, and the splitting of the ethanol–water azeotrope, where ionic liquids are exploited as entrainers.
However, especially ionic liquids of high HBA strength tend to possess high viscosities, low melting points, and may exhibit corrosive properties. Furthermore, they are characterised by lower thermal stabilities than their low HBA strength analogues. Hence, further research into ionic liquid design for biomass applications will certainly have to be in the focus of future research. Aspects relating to the recyclability, cost and large-scale availability, energy requirement, life-cycle analysis and toxicity must be considered (Fig. 8). However, the same applies to any other potential solvent candidate for biorefinery processing.
 | ||
| Fig. 8 General aspects to be addressed, relating to ionic liquids in biomass processing. | ||
Although ionic liquids feature unusual solvation properties, it is unlikely that they will be employed in the short-term in large-scale processes such as biorefineries, considering their current prices. However, the number of patents suggests strong industrial interest. On the other hand, it is interesting to note that the extraction of valuable products from biomass (i.e. vitamins, dyes, pharmaceuticals precursors, etc.)116 or the upgrading of chemicals and fuels by selective removal of nitrogen and sulfur compounds,117 which would immediately justify the usage of specialised solvents, is currently rarely investigated. These areas possess a large future research potential, since the recovery and commercial exploitation of high value products from biomass will improve the overall economics of a biorefinery.
Strategically, establishing innovative, ecologically sound technologies for the generation of upmarket products using ionic liquids may pave the way to later diffusion into large scale platform chemical production. More generally, this requires the adaptation of catalysts, solvent systems and processing techniques to enable the conversion of highly functionalised feedstock. In addition to chemistry and engineering challenges, the resulting products do often not find structural equivalence in already established products, offer other properties or target chemical product markets where the competition with cheap petrochemical-derived products is high (e.g. introduction of polylactic acid to the polymer market). Therefore, establishing novel products will be a long-term goal, and support by political and financial engagement is required.
Looking back at the development time required before other resources (coal, oil) were economically exploited may allow for an estimation of the time-scale. While over the past 100 years, fossil-based resource utilisation increased continuously, coal was the main source of most chemicals and fuels percentage-wise until about 1960 (peaking in the 1920s). It was complemented by oil and gas, peaking in 1980. An innovation cycle of about 50–60 years is followed, in which technology and infrastructure is established to make full use of a less-exploited resource. Hence, it is reasonable to expect that biomass conversion technology will be in place within 40–50 years. The next decades hence promise to be exciting times for biomass processing technology development in general. Time will show in which processes ionic liquids can excel.
Acknowledgements
The support granted by the Friedrich-Schiller University, the Institute for Technical Chemistry and Environmental Chemistry (B. Ondruschka), and the past and present members of my research group (especially C. Palik, J. Lifka, M. Sellin, M. Struempel, and J. Zimmermann) are gratefully acknowledged. Some aspects of this work were financially supported by the German Research Foundation (DFG) within the Priority Programme SPP 1191 Ionic Liquids (STA1027/2-2), BASF SE, and the German Federal Environmental Foundation (DBU, AZ 24762 and AZ 24621).References
- Homepage National Renewable Energy Laboratory, http:\www.nrel.gov/biomass/biorefinery.html, last accessed 28 June 2010.
- Estimation on 9th March 2010, Department of Energy.
- Biorefineries—Industrial Processes and Products, Status Quo and Future Directions, ed. B. Kamm, P. R. Gruber and M. Kamm, Wiley-VCH, 2006, vol. 1 Search PubMed.
- H. Röper, Perspektiven der industriellen Nutzung nachwachender Rohstoffe, insbesondere von Stärke und Zucker, Mitteilung der Fachgruppe Umweltchemie und Ökotoxikologie der Gesellschaft Deutscher Chemiker, 2001, vol. 7(2), p. 6 Search PubMed.
- M. Baerns, A. Behr, A. Brehm and J. Gmehling, Technische Chemie: Lehrbuch, Wiley-VCH, 2006 Search PubMed.
- G. N. Sheldrake and D. Schleck, Green Chem., 2007, 9, 1044 RSC.
- B. Argyropoulos, US Pat., 20080185112, 2008, to North Carolina State University.
- X. Guo, Y. Zheng and B. Zhou, CN101235312, 2008, to Shanghai University.
- M. H. Gurin, US Pat., 20070161095, 2007.
- J. Ji, F. Yu, N. Ai, D. Ji, Z. Xu and Y. Yu, CN101085924, 2007, to Zhejiang University of Technology.
- P. J. Scammells, J. L. Scott and R. D. Singer, Aust. J. Chem., 2005, 58, 155 CrossRef CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411 CrossRef CAS.
- R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835 CrossRef CAS.
- A. Stark, Top. Curr. Chem., 2009, 209, 41.
- A. Stark and K. R. Seddon, in Kirk-Othmer Encyclopedia of Chemical Technology, ed. A. Seidel, John Wiley and Sons, Inc., Hoboken, New Jersey, 5th edn, 2007, vol. 26, p. 836 Search PubMed.
- J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS.
- Ionic Liquids in Synthesis, ed. P. Wasserscheid and Th. Welton, Wiley-VCH, 2nd edn, 2007 Search PubMed.
- Handbook of Green Chemistry: Green Solvents, Vol. 6—Ionic Liquids, ed. P. Wasserscheid and A. Stark, Wiley-VCH, 2010 Search PubMed.
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1 CrossRef CAS.
- P. Mäki-Arvela, I. Anugwom, P. Virtanen, R. Sjöholm and J.-P. Mikkola, Ind. Crops Prod., 2010, 32, 175 CrossRef.
- A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712 CrossRef CAS.
- S. S. Y. Tran and D. R. MacFarlane, Top. Curr. Chem., 2009, 209, 311.
- S. Zhu, Y. Wu, Q. Chen, G. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325 RSC.
- Y. Zhang and J. Y. G. Chan, Energy Environ. Sci., 2010, 3, 408 RSC.
- H. Ohno and Y. Fukaya, Chem. Lett., 2009, 2 CrossRef CAS.
- S. Murugesan and R. J. Linhardt, Curr. Org. Synth., 2005, 2, 437 Search PubMed.
- R. P. Swatloski, R. D. Rogers and J. D. Holbrey, WO03029329, 2003, to The University of Alabama.
- R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS.
- U. Vagt, in Handbook of Green Chemistry: Green Solvents, Vol. 6—Ionic Liquids, ed. P. Wasserscheid and A. Stark, Wiley-VCH, 2010, p. 115 Search PubMed.
- H. Zhang, J. Wu, J. Zhang and J. He, Macromolecules, 2005, 38, 827.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63 RSC.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142 CrossRef.
- (a) Y. Wu, T. Sasaki, S. Irie and K. Sakurai, Polymer, 2008, 49, 2321 CrossRef CAS; (b) S. E. Hecht, R. L. Niehoff, K. Narasimhan, C. W. Neal, P. A. Forshey, D. V. Phan, A. D. M. Brooker and K. H. Combs, WO2006116126, 2006, to Proctor & Gamble; (c) H. B. Xie, S. H. Li and S. B. Zhang, Green Chem., 2005, 7(8), 606 RSC; (d) H. Garcia, R. Ferreira, M. Petkovic, J. L. Ferguson, M. C. P. N. Rebelo and C. S. Pereira, Green Chem., 2010, 12, 357 Search PubMed; (e) D. M. Phillips, L. F. Drummy, D. G. Conrady, D. M. Fox, R. R. Naik, M. O. Stone, P. C. Trulove, H. C. De Long and R. A. Mantz, J. Am. Chem. Soc., 2004, 126, 14350 CrossRef CAS; (f) D. M. Phillips, L. F. Drummy, R. R. Naik, H. C. De Long, D. M. Fox, P. C. Trulove and R. A. Mantz, J. Mater. Chem., 2005, 15, 4206 RSC.
- L.-L. Xie, A. Favre-Reguillon, S. Pellet-Rostaing, X.-X. Wang, X. Fu, J. Estager, M. Vrinat and M. Lemaire, Ind. Eng. Chem. Res., 2008, 47, 8801 CrossRef CAS.
- S. H. Lee, Th. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368 CrossRef CAS.
- A. G. Fadeev and M. M. Meagher, Chem. Commun., 2001, 295 RSC.
- Ch. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177 RSC.
- L. Vanoye, M. Fanselow, J. D. Holbrey, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390 RSC.
- J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516 CrossRef CAS.
- M. L. Mednick, J. Org. Chem., 1962, 27, 398 CrossRef CAS.
- N. H. Smith, US Pat., 3118912, 1964, to Rayonier Inc.
- C. Fayet and J. Gelas, Carbohydr. Res., 1983, 122, 59 CrossRef.
- C. Lansalot-Matras and C. Moreau, Catal. Commun., 2003, 4, 517 CrossRef CAS.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165 CrossRef CAS.
- X. Qi, M. Watanabe, T. M. Aida and R. L. Smith, Green Chem., 2009, 11, 1327 RSC.
- I. Y. G. Chan and Y. Zhang, ChemSusChem, 2009, 2, 731 CrossRef.
- A. Stark and B. Ondruschka, in Handbook of Green Chemistry: Green Solvents, Vol. 6—Ionic Liquids, ed. P. Wasserscheid and A. Stark, Wiley-VCH, 2010, p. 85 Search PubMed.
- (a) H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS; (b) H. Zhao, J. E. Holladay and Z. C. Zhang, WO2008019219, 2008, to Battelle Memorial Institute.
- G. Yong, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2008, 47, 9345 CrossRef CAS.
- I. B. Binder and R. T. Raines, J. Am. Chem. Soc., 2009, 131, 1979 CrossRef CAS.
- G. J. M. Gruter and F. Dauzenberg, WO2007104514, 207, to Avantium Int. BV.
- A. J. Sanborn and P. D. Bloom, WO2006063287, 2006, to Archer Daniels Midland Co.
- G. J. M. Gruter, L. E. Manzer, D. A. S. V. De Souza, F. Dauzenberg and J. Purmova, EP2183236, 2010, to Furanix Technologies BV.
- H. Mehdi, A. Bodor, D. Lantos, I. T. Horvát, D. E. D. Vos and K. Binnemans, J. Org. Chem., 2007, 72, 517 CrossRef.
- I. Gamba, M. Lapis and J. Dupont, Adv. Synth. Catal., 2008, 350, 160 CrossRef CAS.
- I. Zhang, M. Xian, Y. He, L. Li, J. Yang, S. Yu and X. Xu, Bioresour. Technol., 2009, 100, 4368 CrossRef.
- T. Heinze and A. Koschella, Polímeros, 2005, 15, 84 CAS; A. F. Turbak, R. B. Hammer, R. E. Davies and H. L. Hergert, Chem. Technol., 1980, 10, 51 CAS.
- R. Lungwitz and S. Spange, New J. Chem., 2008, 32, 392 RSC; R. Lungwitz, M. Friedrich, W. Linert and S. Spange, New J. Chem., 2008, 32, 1493 RSC.
- Y. Fukaya, A. Sugimoto and H. Ohno, Biomacromolecules, 2006, 7, 3295 CrossRef CAS.
- S. Köhler, T. Liebert, M. Schöbitz, J. Schaller, F. Meister, W. Günther and Th. Heinze, Macromol. Rapid Commun., 2007, 28, 2311 CrossRef.
- Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44 RSC.
- J. Wu, J. Zhang, H. Zhang, J. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266 CrossRef CAS.
- S. Barthel and Th. Heinze, Green Chem., 2006, 8, 301 RSC.
- Th. Heinze, K. Schikal and S. Barthel, Macromol. Biosci., 2005, 5, 520 CrossRef CAS.
- S. A. Forsyth, D. R. MacFarlane, R. J. Thomson and M. von Itzstein, Chem. Commun., 2002, 714 RSC.
- L. Feng and Z.-L. Chen, J. Mol. Liq., 2008, 142, 1 CrossRef.
- (a) T. G. A. Youngs, J. D. Holbrey, M. Deetlefs, M. Nieuwenhuyzen, M. F. C. Gomes and C. Hardacre, ChemPhysChem, 2006, 7, 2279 CrossRef CAS; (b) T. G. A. Youngs, C. Hardacre and J. D. Holbrey, J. Phys. Chem. B, 2007, 111, 13765 CrossRef CAS.
- (a) R. C. Remsing, R. P. Swatloski, R. D. Rogers and G. Moyna, Chem. Commun., 2006, 1271 RSC; (b) R. C. Remsing, G. Hernandez, R. P. Swatloski, W. W. Massefski, R. D. Rogers and G. Moyna, J. Phys. Chem. B, 2008, 112, 11071 CrossRef CAS.
- J. S. Moulthrop, R. P. Swatloski, G. Moyna and R. D. Rogers, Chem. Commun., 2005, 1557 RSC.
- M. Sellin, B. Ondruschka and A. Stark, in Cellulose solvents: for analysis, Shaping and Chemical Modification, ed. T. Liebert, Th. Heinze and K. Edgar, ACS Symp. Series, Washington, DC, 2010, vol. 1033, p. 121 Search PubMed.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672 RSC.
- (a) M. B. Turner, S. K. Spear, J. D. Holbrey and R. D. Rogers, Biomacromolecules, 2004, 5, 1379 CrossRef CAS; (b) M. B. Turner, S. K. Spear, J. D. Holbrey, D. T. Daly and R. D. Rogers, Biomacromolecules, 2005, 6, 2497 CrossRef CAS.
- G. D'Andola, L. Szarvas, K. Massonne and V. Stegmann, WO2008043837, 2008, to BASF SE.
- F. Wendler, B. Kosan, M. Krieg and F. Meister, Macromol. Symp., 2009, 280, 112 CrossRef CAS.
- Y. Cao, J. Wu, J. Zhang, H. Li, Y. Zhang and J. He, Chem. Eng. J., 2009, 147, 13 CrossRef CAS.
- H. Xie, P. Jarvi, M. Karesoja, A. King, I. Kilpelainen and D. S. Argyropoulos, J. Appl. Polym. Sci., 2009, 111, 2468 CrossRef CAS.
- H. Zhang, Z. Wang, Z. Zhang, J. Wu, J. Zhang and J. He, Adv. Mater., 2007, 19, 698 CrossRef CAS.
- N. S. Venkataramanan, K. Matsui, H. Kawanami and Y. Ikushima, Green Chem., 2007, 9, 18 RSC.
- O. A. El Seaud, A. Koschella, L. C. Fidale, S. Dorn and Th. Heinze, Biomacromolecules, 2007, 8, 2627 CrossRef.
- T. Liebert and Th. Heinze, BioResources, 2008, 3, 576 Search PubMed.
- N. Kamiya, Y. Matsushita, M. Haaki, K. Nakashima, M. Narita, M. Goto and H. Takahashi, Biotechnol. Lett., 2008, 30, 1037 CrossRef CAS.
- A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904 CrossRef CAS.
- T. Balensiefer, WO2008090156, to BASF SE.
- H. Zhao, C. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, J. Biotechnol., 2009, 139, 47 CrossRef CAS.
- (a) K. Massonne, G. D'Andola, V. Stegmann, W. Mormann, M. Wezstein and W. Leng, WO2007101811, 2007, to BASF SE; (b) K. Massonne, G. D'Andola, V. Stegmann, W. Mormann, M. Wezstein and W. Leng, WO2007101812, 2007, to BASF SE; (c) K. Massonne, G. D'Andola, V. Stegmann, W. Mormann, M. Wezstein and W. Leng, WO2007101813, 2007, to BASF SE.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS.
- R. Rinaldi and F. Schüth, ChemSusChem, 2010, 3, 266 CrossRef CAS.
- N. Villandier and A. Corma, Chem. Commun., 2010, 46, 4408 RSC.
- H. Watanabe, Carbohydr. Polym., 2010, 80, 1168 CrossRef CAS.
- Z. H. Zhang and Z. B. K. Zhao, Carbohydr. Res., 2010, 344, 2069.
- C. Sievers, M. B. Valenzuela-Olarte, T. Marzialetti, I. Musin, P. K. Agawal and Ch. W. Jones, Ind. Eng. Chem. Res., 2009, 48, 1277 CrossRef CAS.
- R. Rinaldi and F. Schüth, Energy Environ. Sci., 2009, 2, 610 RSC.
- T. C. R. Brennan, S. Datta, H. W. Blanch, B. A. Simmons and B. M. Holmes, BioEnergy Res., 2010, 3, 123 CrossRef.
- C. Moreau, M. N. Belacem and A. Gandini, Top. Catal., 2004, 27, 11 CrossRef CAS.
- Top Value Added Chemicals from Biomass: Vol. 1—Results of Screening for Potential Candidates from Sugars and Synthesis Gas, ed. T. Werpy and G. Petersen, National Renewable Energy Laboratory, 2004 Search PubMed.
- Y. Román-Leshkov, J. N. Chheda and J. A. Dumesic, Science, 2006, 312, 1933 CrossRef CAS.
- J. N. Chheda, Y. Román-Leshkov and J. A. Dumesic, Green Chem., 2007, 9, 342 RSC.
- J. A. Dumesic, Y. Roman-Leshkov and J. N. Chheda, WO2007146636, to Wisconsin Alumni Research Foundation.
- K. M. Rapp, EP0230250, to Sueddeutsche Zucker AG.
- Q. Bao, K. Qiao, D. Tomida and C. Yokoyama, Catal. Commun., 2008, 9, 1383 CrossRef CAS.
- B. F. M. Kuster and H. S. van der Baan, Carbohydr. Res., 1977, 54, 165 CrossRef CAS.
- J. Zimmermann, B. Ondruschka and A. Stark, Org. Process Res. Dev., 2010 DOI:0.1021/op100055f.
- Y. Su, H. M. Brown, X. Huang, X.-D. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117 CrossRef CAS.
- G. Wegener, Ind. Crops Prod., 1992, 1, 113 CrossRef CAS.
- V. Myllymäki and R. Aksela, WO2005017001, to Kemira OYJ.
- L. A. Edye and W. O. S. Doherty, WO2008095252, to Queensland University of Technology.
- R. Rinaldi and F. Schüth, ChemSusChem, 2009, 2, 1096 CrossRef CAS.
- T. Balensiefer, J. Brodersen, G. D'Andola, K. Massonne, S. Freyer and V. Stegmann, WO2008090155, to BASF SE.
- W. L. Faith, Ind. Eng. Chem., 1995, 27, 9 Search PubMed.
- F. Bergius, Ind. Eng. Chem., 1937, 29, 247 CrossRef CAS.
- (a) J. Upfal, D. R. MacFarlane and S. A. Forsyth, WO2005017252, to Viridian Chemical Pty Ltd; (b) S. S. Y. Tan, R. D. MacFarlane, J. Upfal, L. A. Edye, W. O. Doherty, A. F. Patti, J. M. Pringle and J. L. Scott, Green Chem., 2009, 11, 339 RSC.
- M. Zavrel, D. Bross, M. Funke, J. Buchs and A. C. Spiess, Bioresour. Technol., 2009, 100, 2580 CrossRef CAS.
- W.-R. Pitner, E. F. Aust, M. Schulte and U. Schmid-Grossmann, WO201000357, to Merck Patent GmbH.
- (a) W. Arlt, M. Seiler, C. Jork and Th. Schneider, WO02074718, to BASF AG; (b) C. Jork, M. Seiler, Y. A. Beste and W. Arlt, J. Chem. Eng. Data, 2004, 49, 852 CrossRef CAS.
- A. J. Ragaukas, Ch. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, Ch. A. Eckert, W. J. Frederick, Jr, J. P. Hallett, D. J. Leak, Ch. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef.
- X. G. Liu, X. R. Ju, X. D. Mao and Z. Zhang, Chem. Ind. Forest Prod., 2005, 25(3), 111 Search PubMed.
- L.-L. Xie, A. Favre-Reguillon, X.-X. Wang, X. Fu, S. Pellet-Rostaing, G. Toussaint, Ch. Geantet, M. Vrinat and M. Lemaire, Green Chem., 2008, 10, 524 RSC.
Footnotes |
| † It should be noted that some derivatisation has been noticed: T. Heinze, S. Dorn, M. Schoebitz, T. Liebert, S. Koehler and F. Meister, Macromol. Symp., 2008, 262, 8; T. Liebert, Macromol. Symp., 2008, 262, 28; G. Ebner, S. Schiehser, A. Potthast and T. Rosenau, Tetrahedron Lett., 2008, 49, 7322. |
| ‡ Detailed solubility data of various carbohydrates in ionic liquids are available: M. E. Zakrezewska, E. Bogel-Lukasik and R. Bogel-Lukasik, Energy Fuels, 2010, 24, 737; A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712. |
| § It should be noted that enzymatic reactions with ionic liquids are not per se ineffective, e.g. S. Park and R. J. Kazlauskas, Curr. Opin. Biotechnol., 2003, 13, 432; S. Murugesan and R. J. Linhardt, Curr. Org. Synth., 2005, 2, 437; F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757. |
| ¶ The synthesis of furfural from xylose has been likewise investigated in a few publications. For example S. Lima, P. Neves, M. M. Antunes, M. Pillinger, N. Ignatiyev and A. A. Valente, Appl. Catal., A, 2009, 363, 93; C. Sievers, I. Musin, T. Marzialetti, M. B. V. Olarte, P. K. Agrawal and C. W. Jones, ChemSusChem, 2009, 2, 665. |
| This journal is © The Royal Society of Chemistry 2011 |