Towards engineering O2-tolerance in [Ni–Fe] hydrogenases
Pierre-Pol
Liebgott
,
Sébastien
Dementin
,
Christophe
Léger
and
Marc
Rousset
*
CNRS, Laboratoire de Bioénergétique et Ingénierie des Protéines, Institut de Microbiologie de la Méditerranée, 31 chemin Joseph Aiguier, 13402, Marseille, France. E-mail: rousset@ifr88.cnrs-mrs.fr; Tel: +33 4 91164393
First published on 26th October 2010
Abstract
Hydrogenases catalyze the conversion between 2H+ + 2e− and H2. Most of these enzymes are inhibited by O2, which represents a major drawback for their use in biotechnological applications. Improving hydrogenase O2 tolerance is therefore a major contemporary challenge to allow the implementation of a sustainable hydrogen economy. A few bacteria, however, contain hydrogenases that activate H2 even in the presence of O2. Intriguingly, kinetic and spectroscopic studies lead to assuming that different mechanisms might be responsible for the resistance, depending on the enzyme type. The various hypotheses that emerged from these studies are still a matter of debate. In order to better understand the molecular bases of resistance to O2 inhibition, we explored different methods to improve the O2-tolerance of the O2-sensitive [Ni–Fe] hydrogenase from Desulfovibrio fructosovorans. A whole bunch of mutants has been studied and fully characterized, which revealed that actually, different mechanisms can lead to O2 tolerance. These mechanisms are described in this review and compared to the current hypothesis of O2 tolerance.
 Pierre-Pol Liebgott | Pierre-Pol Liebgott was born in 1977 and received his Master's Degree in Biochemistry in 2003. He received his Ph.D. in Microbiology and Molecular Biology in 2007 from the University of Provence in Marseille, where he studied the transformation of phenolic compounds present in the olive-mill wastewaters, in identifying the enzymes involved in this process. He is currently a post-doctoral fellow in the Bioenergetics and Protein Engineering Laboratory (BIP) from CNRS in Marseille. Under the supervision of Marc Rousset and Christophe Léger, his post-doctoral research focuses on improving the oxygen tolerance of [Ni-Fe] hydrogenases. |
 Sébastien Dementin | Sébastien Dementin obtained his Ph.D. in Biochemistry in 2001 from the Paris XI University. His thesis research focussed on mechanistic studies of the Mur synthetases. Then his interest turned toward enzymology of metalloproteins (focussing on hydrogenases and nitrate reductases) during postdoctoral fellowships with Dr Marc Rousset at the BIP Laboratory in Marseilles and with Dr David Pignol at the Cellular Bioenergetics Laboratory at the CEA in Cadarache. From 2007 he is a permanent researcher in the group of Marc Rousset where he continues studying hydrogenases in order to improve their properties by protein engineering. |
 Christophe Léger | Christophe Léger obtained his Ph.D. in Physical Chemistry at the University of Bordeaux I, France, where he studied electrodeposition. In 1999, he joined Fraser Armstrong at the University of Oxford, U.K., to study redox enzymes by electrochemistry. In 2002, he was hired by the French National Centre for Scientific Research (CNRS) in Marseilles. He is interested in bioinorganic chemistry, enzyme kinetics, and studying the mechanism of multicenter redox enzymes. |
 Marc Rousset | Marc Rousset did his graduate work in the Laboratory of Bacterial Chemistry from CNRS in Marseille (PhD 1991). After a postdoctoral fellowship in bacterial genetics at the University of Missouri-Columbia, he joined CNRS in 1993 as a staff scientist. From 2001 he is the leader of the Molecular Ecology and Hydrogen Metabolism team. In 2006 he founded the Group of Research on BioHydrogen, an interdisciplinary nationwide federation of forty four laboratories dedicated to biological hydrogen research. He studies hydrogenases, from their catalytic mechanism to their cellular functions. He is also interested in Bioenergy production and implementation. |
Broader contextAn option attracting great attention for replacing greenhouse gas-producing fossil fuels is molecular hydrogen, provided that it is produced in a clean and renewable way. Many organisms including bacteria, Archaea and some unicellular eukaryotes have an active hydrogen metabolism, but the most promising, which possess the potential to meet the future energy demand, are surely photosynthetic microbes such as the green algae Chlamydomonas or the cyanobacterium Synechocystis. Biological conversion of light energy into dihydrogen is potentially very efficient in terms of energy conservation, as 10% of the incident light energy can theoretically be recovered into hydrogen. For example, an average sunlight flux of 46Mwh/ha/day can be converted with a 10% yield in 1650 m3 of hydrogen per hectare per day, which represents 145 TOE (ton of oil equivalent) per hectare per year. One of the main bolts in direct biological photoproduction of hydrogen is the inevitable exposure of hydrogenase to O2, a strong inhibitor of the enzyme. Improving hydrogenase resistance to O2 is therefore a major challenge. In our group, we study the molecular bases of O2-inhbition with the aim to determine ways to improve hydrogenases in organisms of interest. We have recently determined that different mechanisms can lead to O2 tolerance and that depending on the mutations, different types of improvement can be reached. |
I. Introduction
An attracting option for replacing greenhouse gas-producing fossil fuels in the future is molecular hydrogen, provided that it is produced using a clean and renewable method. The development of new biotechnological processes, designed to meet the future energy demand, may take advantage of microbes that have been using H2 from very early in the evolution of life.1,2 A large number of microorganisms including bacteria, Archaea and unicellular eukaryotes have an active hydrogen metabolism, in which hydrogen can be used as an energy source or produced to release reducing power. In biological systems, the reaction is catalyzed by metal-containing enzymes called hydrogenases.Three classes of hydrogenases can be distinguished based on their metal content:3 (i) The [Ni–Fe] hydrogenases (EC 1.12.2.1) harbor a hetero di-nuclear nickel–iron active site (Fig. 1A). These hydrogenases are the most widespread in nature. (ii) The [Fe–Fe] hydrogenases (EC 1.12.7.2) contain a di-iron active site (Fig. 1B). (iii) The members of the third hydrogenase class function as H2-forming methylenetetrahydromethanopterine dehydrogenases (EC 1.12.98.2) and are found only in a small group of methanogenic Archaea.4,5 It is important to note that this later type of hydrogenase does not carry out electrochemical processes. What makes hydrogenases special, when compared to other enzymes, is that a gas, H2, is the substrate (see ref. 6 for reviewing gas processing enzymes). It is believed that this small molecule does not diffuse freely in the enzyme but is transported within a specific hydrophobic channel, the location of which has been determined in both [Fe–Fe]7,8 or [Ni–Fe] hydrogenases.9,10 However, site directed mutagenesis and kinetic studies have confirmed the existence of this channel in [Ni–Fe] hydrogenases,11,12 but not in [Fe–Fe] hydrogenases.13
![Structure details of the active sites from [Ni–Fe] hydrogenase (A) and [Fe–Fe] hydrogenase (B). The hetero atoms from the prosthetic groups and the protein ligands are represented as sticks. Nickel is in cyan, iron is in orange, sulfur is in yellow, oxygen is in red, nitrogen is in blue, and carbon is in grey. DTMA: dithiomethylamine.](/image/article/2011/EE/c0ee00093k/c0ee00093k-f1.gif) | ||
| Fig. 1 Structure details of the active sites from [Ni–Fe] hydrogenase (A) and [Fe–Fe] hydrogenase (B). The hetero atoms from the prosthetic groups and the protein ligands are represented as sticks. Nickel is in cyan, iron is in orange, sulfur is in yellow, oxygen is in red, nitrogen is in blue, and carbon is in grey. DTMA: dithiomethylamine. | ||
The biological conversion of light energy into dihydrogen by photosynthetic microorganisms is potentially very efficient in terms of energy conservation, as 10% of the incident light energy can theoretically be converted into hydrogen.14 For example, an average sunlight flux of 46 Mwh ha−1 day−1 can be converted with a 10% yield in 1650 m3 of hydrogen per hectare per day, which represents 145 TOE (ton of oil equivalent) per hectare per year. However, the photosynthetic production of dihydrogen is only a transient phenomenon under natural conditions,15 because of hydrogenase inhibition by the dioxygen produced during water photolysis.16 As a result, actual dihydrogen production efficiencies obtained in laboratory experiments are lower than 1%.17,18 Improving hydrogenase O2-tolerance is then a major challenge for a broad range of biotechnological applications. Microorganisms harboring optimized hydrogenases may play a major role in hydrogen generation for fuels, but also as a constituent of biofuel cells and biosensors, which also represent an important potential application for hydrogenases in vitro.
II. Hydrogenase structures
The core of [Ni–Fe] hydrogenases consists of two subunits; the large subunit is approximately 60 kDa and houses the Ni–Fe-active site, whereas the small subunit, which can be of variable size, harbors from one to three iron–sulfur clusters (Fig. 2). In certain enzymes, additional subunits enable the interaction of these clusters with physiological electron carriers such as quinones, pyridine nucleotides, ferredoxins, and cytochromes.19 Crystal structure analysis of heterodimeric [Ni–Fe] hydrogenases from Desulfovibrio species10,20–22 (Fig. 2), now taken as the prototypical [Ni–Fe] hydrogenases, revealed that the Ni–Fe cofactor is deeply buried in the large subunit. The Ni is coordinated to the protein via four thiol groups from conserved cysteine residues; two of these are bridging ligands that coordinate both Fe and Ni (Fig. 1A).10,20–22 Infrared spectroscopy revealed that the Fe coordination sphere also possesses three diatomic ligands: one CO and two CN molecules.23,24 The sixth iron coordination position is assumed to be occupied by a bridging hydride between iron and nickel.25,26![Crystallographic structure of the [Ni–Fe] hydrogenase from D. fructosovorans at 1.8 Å resolution (PDB: 1YQW). The gas channel network is depicted in grey. It was calculated with a probe radius of 0.8 Å. The position of the valine 74 and leucine 122 are highlighted in the inset.](/image/article/2011/EE/c0ee00093k/c0ee00093k-f2.gif) | ||
| Fig. 2 Crystallographic structure of the [Ni–Fe] hydrogenase from D. fructosovorans at 1.8 Å resolution (PDB: 1YQW). The gas channel network is depicted in grey. It was calculated with a probe radius of 0.8 Å. The position of the valine 74 and leucine 122 are highlighted in the inset. | ||
Regarding [Fe–Fe] hydrogenases, the active site consists of a ferredoxin-like [4Fe-4S] cluster connected via one cysteine residue to a binuclear Fe2 cluster.3,27,28 Both Fe ions have one CO and one CN− ligand. One bridging CO also coordinates the two metals. In addition, there is an unusual dithiolate ligand (Fig. 1B) that is bound to the irons. It is now commonly accepted that this non protein ligand is a dithiomethylamine molecule.29,30 The Fe ion distal to the [4Fe-4S] cluster has a vacant coordination site that is believed to be the reactive centre.
III. Reaction with O2 and CO
It can be stated, as a general rule, that hydrogenases of either type are inhibited by O2, but individual sensitivities can vary to a wide extent. For example, the reaction of the [Fe–Fe] enzymes with O2 is diverse. The O2-inactivation rate can considerably vary: the [Fe–Fe] enzyme from Desulfovibrio desulfuricans is rapidly inhibited compared to the enzyme from Clostridium acetobutylicum.31,32 The observation that the competitive inhibitor CO protects the enzymes from C. acetobutylicum demonstrates that dioxygen targets the active site.31 In a study of the enzyme from Chlamydomonas reinhardtii, it has recently been suggested that the reaction starts with the binding and partial reduction of dioxygen at the di-iron centre, followed by the destruction of the H-cluster's active site [4Fe-4S] center.33In the case of [Ni–Fe] hydrogenases, O2 has also been shown to oxidize directly the bimetallic active site.34 However the difference with the previous enzyme is that [Ni–Fe] hydrogenases are not damaged by dioxygen as they can be reactivated by reduction. The oxidized enzyme is inactive. EPR studies revealed that it may exist under two different states named Ni–A and Ni–B, the structures of which are still a matter of debate. X-Ray diffraction,10,22 EPR,34 ENDOR35,36 and EXAFS37 data indicate that in both states an oxygen species is bridging nickel and iron atoms (for review see ref. 38 and 39). Most of the studies agree with the presence of a hydroxide in the Ni–B sate.37,40–43 The nature of the oxygen species in Ni-A is more controversial, either oxo,35 hydroxo37,39–41 or peroxo42,44 species have been proposed. Ni-A requires the presence of dioxygen to be formed, whereas Ni–B can also be obtained in anaerobiosis under oxidative conditions.45–47
Beside the structural differences, there is another very important feature that differentiates Ni-A and Ni–B: it is their reactivation kinetics.48,49 Ni–B is called the ready state because it quickly becomes active upon reduction, while Ni-A is called the unready state because it needs a long period of incubation under reducing conditions before becoming active. This is illustrated by electrochemical experiments conducted on the Allochromatium vinosum [Ni–Fe] hydrogenase. After inactivation by O2, the enzyme is reactivated by reduction at low potential under H2.50 For example at −208 mV and pH 6, the reactivation process occurs in two phases: a fast (instantaneous) phase corresponding to the reactivation of the Ni–B state and a slower phase (several hundreds of seconds) assigned to Ni-A. FTIR-spectroelectrochemical studies of different hydrogenases indicated that one-electron reduction of Ni-A and Ni–B leads to two different states: Ni-A leads to the Ni-SU state, and Ni–B leads to Ni-SI.38 The enzyme in the Ni-SI state is active, whereas the Ni-SU state is still inactive.38 The rate-limiting step of the reactivation process is the gradual and spontaneous conversion of the Ni-SU to the active Ni-SI state.44
X-Ray diffraction studies showed that the main structural difference between oxidized and reduced states of the active site is the absence of the oxygen species that bridges the metals.51,52 This was confirmed by an 17O ENDOR study in which the signal due to Ni-A labelled with 17O was lost upon reductive activation.35 Therefore, reduction of the oxidized states is triggered by the removal of the bridging oxygen species, which allows H2 to bind to the active site and catalytic turnover.
Most [Ni–Fe] hydrogenases are inhibited by CO in a competitive manner.16,53 It reaches the active site using the same gas channel as H2 and O2 (Fig. 2).12 It binds weakly to the Ni ion at the active site,38,54–56 only after reductive activation of the enzyme to the Ni-SI forms, presumably when the bridging oxygen species has been removed from the active site. No binding of CO occurs when the enzyme is in the inactive states Ni-A, Ni–B, and Ni-SU. In addition, CO-inhibition blocks electron and proton transfer at the active site, although reduction at the proximal [4Fe-4S] cluster is detected.55 The kinetics of CO inhibition can be probed by PFV, a technique where the enzyme is adsorbed onto an electrode that acts as a sink or a source of electrons. The measured current is proportional to the enzyme activity. This makes it possible to measure turnover rates with high temporal resolution and potential control.45,57 The kinetics of CO binding is fast, about 108 s−1 M−1. Considering that diffusion is the rate-limiting step of CO-inhibition,11,12 we used this inhibitor to probe gas diffusion in hydrogenases.
IV. Specific characteristics of the naturally occurring O2-tolerant [Ni–Fe] hydrogenases
[Ni–Fe] hydrogenases are considered to be more robust than [Fe–Fe] hydrogenases because they can be totally reactivated after inhibition by O2. Moreover, there are even a few examples in nature of relatively O2-tolerant enzymes. It should be noted, however, that their number is very low considering the ubiquity of hydrogenases in the biodiversity. O2-tolerance defines one hydrogenase that retains some activity in the presence of O2. The level of residual activity can vary depending on the enzyme, but it should be remembered that trace amounts (a few μM) of O2 readily inhibit standard O2-sensitive [Ni–Fe] hydrogenases. Most of the O2-tolerant hydrogenases known today are gathered in the very intriguing bacterium Ralstonia eutropha, a bacterium that lives in soil and water and was originally isolated from sludge. R. eutropha is one of the very rare organisms able to grow chemolithoautotrophically using hydrogen as the sole energy source and dioxygen as terminal electron acceptor. This is called the Knallgas (detonating gas) reaction. Three distinct O2-tolerant [Ni–Fe] hydrogenases, that each serve unique physiological roles, are present in this bacterium: a membrane-bound hydrogenase (ReMBH) coupled to the respiratory chain, a cytoplasmic soluble hydrogenase (ReSH) able to generate reducing equivalents by reducing NAD+ at the expense of hydrogen, and a regulatory hydrogenase (ReRH) which acts in a signal transduction cascade to control transcription of hydrogenase genes.58 These O2-tolerant hydrogenases are usually less active than standard O2-sensitive enzymes. Their H2 oxidation activities are reduced by a factor of about 5 for the ReSH,59 about 50 for the ReMBH and about 500 for the ReRH.60 The H2 production activities are usually considerably weak, especially because of the strong inhibitory effect of H2.61 A few other organisms also contain O2-tolerant hydrogenases. The hyperthermophilic, hydrogen-oxidizing bacterium Aquifex aeolicus contains three characterized hydrogenases.62–64 Two of them, hydrogenases I and II, are connected to the membrane by a membrane-integral cytochrome b. Whereas hydrogenase I (AaHyd-1), which is homologous to the ReMBH, is rather involved in a dihydrogen-dioxygen pathway. Hydrogenase II, isolated as a multiprotein complex associated with a sulfur reductase, appears to be involved in sulfur respiration.64Rubrivivax gelatinosus contains one membrane-bound hydrogenase.65 Regulatory hydrogenases (homologous to the ReRH) are present in Rhodobacter capsulatus (RcRH)66 and Bradyrhizobium japonicum (BjRH).43E. coli also contains several membrane-bound hydrogenases. One of them, hydrogenase I (EcHyd-1), which is homologous to ReMBH is O2-tolerant.67 For yet unknown reasons, all the O2-tolerant hydrogenases studied so far have been shown to be resistant to CO inhibition.43,68,69Three strategies seem to have been developed through evolution to allow [Ni–Fe] hydrogenases to be catalytically active in the presence of dioxygen. In the case of the ReSH, the O2 resistance is assumed to be due to the presence of extra CN ligands at the active site70 that might be incorporated by a specific maturation protein HypX.71 Deletion of hypX led to a complete knock-out of hydrogenase activity in R. leguminosarum72 and, in R. eutropha, affected the O2 resistance of the ReSH enzyme73 and the activity of ReMBH74 as well, while the hydrogen-sensor (ReRH) remained unaffected.75 However, the presence of a Ni-bound cyanide under native conditions is now questioned (Zebger, private communication).
In the case of the group of the O2-tolerant membrane-bound hydrogenases (MBH), which includes R. eutrophaReMBH, A. aeolicus hydrogenase I (AaHyd-1) and E. coli hydrogenase I (EcHyd-1), the O2 tolerance seems to originate from unusual redox properties and kinetic behaviours. In PFV experiments, the activity of the ReMBH and AaHyd-1 recovers extremely fast after dioxygen exposure.76,77 The reactivation kinetics is characterized by a parameter called Eswitch, which defines the electrode potential at which the enzyme switches form inactive to active.78 This parameter has no thermodynamic significance because it varies with the electrode scan rate. It can be used, however, as an indicator of the activation kinetics, the faster the activation rate, the higher Eswitch potential.79 The Eswitch value for the O2-tolerant MBH group is consistently found to lie within the high redox potential range: + 150 mV for EcHyd-1,67 + 130 mV for ReMBH,80 and +77 mV in the case of AaHyd-1,77 which is between 110 and 220 mV higher than that of standard hydrogenases.80
These exceptional kinetic properties might be related in some fashion to the unusual redox properties of the prosthetic groups. A complex and split EPR signal attributed to the [3Fe-4S]1+ cluster is observed for the ReMBH and AaHyd-1.62 This signal has been attributed to a [3Fe-4S]1+ cluster magnetically coupled to another paramagnetic centre and suggests that the proximal [4Fe-4S] possesses an exceptionally high redox potential of +40 mV.81 This feature favours a fast electron transfer from the active site towards the proximal centre, which is in agreement with the low H2 production activity of the enzyme. It has been assumed that this modified proximal (Fe–S) cluster might contribute to the O2 tolerance properties, but the molecular mechanism is still unknown. Remarkably, there are two additional cysteine residues in the close vicinity of the proximal [4Fe-4S] cluster that are absent in O2-sensitive standard [Ni–Fe] hydrogenases. These two additional cysteines may be responsible for tuning this unusual redox potential, or they might provide additional ligands for an atypical proximal cluster structurally more complex than a standard cubane. Interestingly, these two additional cysteines are conserved in O2-tolerant membrane-bound hydrogenases.81
The spectroscopic analysis of the active site performed by EPR and FTIR revealed that, although the regular signatures of the active states are present, essentially Ni–B is formed after exposure to O2. Ni-A signal never appears with the ReMBH whatever the condition tested.81 In the case of AaHyd-1, a weak Ni-A signal has been reported to appear after O2 exposure,82 while only Ni–B was detected in a recent study.77 The EcHyd-1 exhibits a Ni-A signal upon aerobic isolation but this signal is then barely detectable when the enzyme is exposed to O2 after activation.67 The presence of the usual nickel signatures indicates that the chemistry at the active site is identical to that catalysed by standard hydrogenases. The O2 tolerance properties of these enzymes are therefore likely to be due to a fast reactivation rate, as shown in recent electrochemical studies76,77,79 and by the lack of the Ni-A signal after aerobic inactivation.
The RH and related enzymes adopted apparently another strategy consisting in reducing the gas channel size at the interface with the active site cavity. At the end of the hydrophobic channel, near the active site, two hydrophobic residues, usually a valine and a leucine that are conserved in O2-sensitive hydrogenases, are replaced by larger residues, respectively isoleucine and phenylalanine, in the O2-tolerant hydrogen-sensors.22 It has therefore been suggested that increasing the bulk of residues occupying these two positions may act like a molecular sieve, reducing the channel diameter at that point, thereby preventing efficient dioxygen access to the active site. This hypothesis was supported by two experiments in which the bulky amino-acids from ReRH and RcRH were substituted by valine and leucine. In both cases, determination of the inactivation kinetics in the presence of dioxygen revealed that the mutated enzymes were inactivated after prolonged incubation and required a reductive activation to reach the maximum activity.83,84 Even though the mutated enzymes became more sensitive to dioxygen than the wild-type, it should be noted that they retain a significant level of activity after prolonged dioxygen exposure, and therefore still belong to the O2-tolerant group of hydrogenases.
V. Understanding the molecular bases of O2-tolerance
It would be particularly fruitful to take advantage of the properties of these O2-tolerant enzymes by cloning their corresponding genes into organisms of biotechnological interest. Unfortunately, cloning [Ni–Fe] hydrogenases is still very difficult and the progresses realised recently remains very limited.59,85 Even though [Ni–Fe]-hydrogenase operons are highly conserved and exhibit a high degree of similarity, each maturation system is specific to the corresponding structural subunits, probably because of tight protein–protein interactions occurring during processing.86 This specificity barrier is responsible for the general failure of hydrogenases to be matured in heterologous hosts.87 Nevertheless, these O2-tolerant enzymes represent precious inspiration sources for the study of the molecular bases of O2 inhibition.Understanding the resistance to O2 inhibition is therefore a key issue, because this knowledge is needed to improve the resident hydrogenases in organisms of biotechnological interest. One major difficulty, however, lies in the complexity associated with the apparent diversity of the mechanisms that lead to O2 tolerance, as described above. Our research strategy is to study, at the molecular level, the reaction mechanisms of O2 and other inhibitors in a model O2-sensitive hydrogenase; our approach being inspired by some key features observed in the O2 tolerant enzymes. This approach offers the possibility to take advantage of the high H2-production activity of the O2-sensitive hydrogenases. Our model enzyme is the O2-sensitive [Ni–Fe] hydrogenase from Desulfovibrio fructosovorans. This enzyme is soluble, highly produced, genetically accessible, and can be crystallized without much difficulties. All the mutants of interests can be fully characterized, at the structural, spectroscopic and kinetic levels.
VI. Role of the polarity of the side chain in the diffusion rate
As described above, the structure of the hydrophobic channel that conducts gases to the active site has been proposed to participate to the O2 tolerance in the RH-type of enzyme.22,83,84 As a result, it was important to understand how the structure of the tunnel in hydrogenases determines the diffusion rate and possibly the selectivity of the enzymes with respect to substrates and inhibitors of similar sizes.Being inspired by hydrogen-sensors, substitutions of the Val74 and Leu122 with isoleucine and phenylalanine, respectively, were carried out in the [Ni–Fe] hydrogenase from D. fructosovorans (Fig. 2). Surprisingly, these substitutions did not improve O2 tolerance and did not significantly modify the catalytic properties of the enzyme under anoxic conditions.88 As indicated above, the reverse experiment conducted on the ReRH and RcRH actually decreased their O2 tolerance.83,84 Consequently, we concluded that the residue bulkiness at these positions was not the only parameter affecting O2 tolerance. The orientation or chemical nature of the side chain is probably also crucial.11,88
This observation led us to analyse in detail the influence of the lateral chain that points to the gas channel. In order to explore the respective roles of the bulk, hydrophobicity, charge and polarity, we constructed and purified a number of single and double mutants at positions 122 and/or 74: Val74 was replaced with aspartate, asparagine, tryptophan, glutamate, methionine, glutamine, isoleucine, and phenylalanine; Leu122 was replaced with methionine and phenylalanine.12 The mutants were screened using PFV to estimate the rate of inhibition by CO (kinCO), the rate of CO release (koutCO), the rate of O2 inhibition (kinO2) and the Michaelis constant Km. The diffusion rate can be directly deduced from kinCO because CO is a competitive inhibitor and because it reacts so rapidly with the active site that the limiting step is the diffusion rate.11,12 The effect of the mutations that keeps the side chain hydrophobic indicates a simple correlation between bulk and diffusion rate. Compared to the wild-type (WT), the diffusion rate is reduced by about two orders of magnitude for the V74F and about three orders of magnitude for the V74W. The increase of the molecular volume (calculated according to ref. 89) of the amino-acid side chains lining the channel has a strong influence on the diffusion rate. In this set of experiments, the effect observed is only due to the steric hindrance that shrivels the tunnel.
In the case of polar amino-acids, two levels of repercussion were observed on diffusion, involving both bulk and charge. The influence of changing the size and polarity of the residue at position 74 was analysed by comparing the V74E, V74D, V74Q and V74N mutants two at a time. Increasing the length of the side chain by one carbon (V74D to V74E, or V74N to V74Q) slows the diffusion rate about 30-fold. The magnitude of this effect lies within the same range as that observed with hydrophobic residues. Nevertheless, charge also matters as the substitution V74 to D74 slows the diffusion by a factor 40, while the molecular volume of aspartate is 40% smaller than that of valine.89 Polarity has an even stronger effect as the substitution V74 to Q74 reduces the diffusion by about four orders of magnitude, while the molecular volumes of these two amino acids are quite similar.89 Within the polar amino acids, replacing a carboxylic acid with an amide, keeping the van der Waals volume constant (V74E to V74Q, or V74D to V74N), slows diffusion by a factor of about 12. The two contributions, size and polarity, are independent of each other, and therefore the combination of the two (V74D to V74Q) decreases the rate of diffusion by more than two orders of magnitude. The electrostatic interaction of the amino acid side chains lining the tunnel is therefore very unfavourable to CO diffusion. This could be due to a direct interaction of the polar group with the gas molecule, or to the stabilization of a water molecule that would be part of the barrier to ligand entry, as observed in certain myoglobin mutants.90
VII. Slowing diffusion along the gas channel increases resistance to O2 and Km for H2
In [Ni–Fe] hydrogenase, the rate of inhibition by CO is about four orders of magnitude faster than the rate of aerobic inhibition (3.104 s−1 M−1).12,16 Considering that CO and O2 diffuse within the protein at about the same rate, this observation implies that the rate of inhibition by O2 is limited by the reaction at the active site. Mutations such as V74N or V74W decrease the rate of intramolecular diffusion by blocking the tunnel, but this has no effect on the overall reaction with O2 because the diffusion process does not limit the inhibition rate (Fig. 3). However, other mutations decrease the rate of diffusion by so much (three orders of magnitude for V74E or four orders of magnitude for V74Q) that this step becomes slower than the reaction of O2 at the active site, thus decreasing the overall rate of inhibition by O2 (Fig. 3 and 4).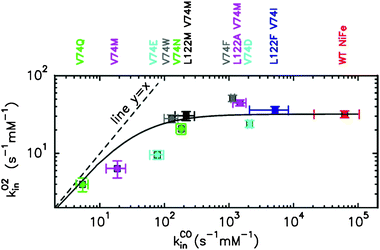 | ||
| Fig. 3 Rate of inhibition by O2 (kO2in) plotted against the rate of binding of CO (kCOin). The dashed line depicts y = x for which O2-inhibition rate would be a linear function of the diffusion rate; the plain line is the best fit to equation: 1/kO2in = 1/kCOin + 1/kO2inmax, with kO2inmax = 32 s−1 mM(O2)−1.12 Error bars represent either the deviation from the average of three to five independent determinations or the estimated error introduced from the extrapolation to 40 °C. | ||
![The inhibition by O2 of D. fructosovorans [Ni–Fe] hydrogenase selected mutants. (a) The change in dioxygen concentration plotted against time, reconstructed from the amount of O2 injected and the time constant of the exponential decay; the latter is calculated from fitting the change in current. (b) The plain lines show the change in [Ni–Fe] hydrogenase activity (current i normalized by its value i(0) just before the inhibitor is added); the dashed lines are the fits to the equations derived in ref. 31; (c,d) Enlarged views of the data in b, showing the decreases in current after the first exposure to O2 (c) and the partial reactivation of the L122M-V74M mutant (d). E = +200 mV versus SHE, T = 40 °C, pH 7, electrode rotation rate w = 2 kr.p.m.](/image/article/2011/EE/c0ee00093k/c0ee00093k-f4.gif) | ||
| Fig. 4 The inhibition by O2 of D. fructosovorans [Ni–Fe] hydrogenase selected mutants. (a) The change in dioxygen concentration plotted against time, reconstructed from the amount of O2 injected and the time constant of the exponential decay; the latter is calculated from fitting the change in current. (b) The plain lines show the change in [Ni–Fe] hydrogenase activity (current i normalized by its value i(0) just before the inhibitor is added); the dashed lines are the fits to the equations derived in ref. 31; (c,d) Enlarged views of the data in b, showing the decreases in current after the first exposure to O2 (c) and the partial reactivation of the L122M-V74M mutant (d). E = +200 mV versus SHE, T = 40 °C, pH 7, electrode rotation rate w = 2 kr.p.m. | ||
We concluded that modifying the tunnel changes the rate of transport of all diatomic molecules, including H2. In the mutant V74Q, in which diffusion rate is the most affected, Km is increased by two orders of magnitude (Fig. 5), while at the same time the enzyme activity under 1 atm of H2 (kcatapp), which incorporates H2 binding, is only reduced by a factor three. An explanation for this discrepancy is found in the analysis of the relation between Km and kinCO, which shows that H2 diffuses about 30 times faster than CO.12 Therefore, even though substrate diffusion limits the rate of H2 oxidation in this mutant, diffusion of H2 remains fast enough to maintain a high turnover rate.
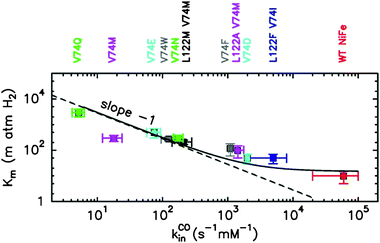 | ||
Fig. 5 The Michaelis constant (Km) plotted against the rate of binding of CO (kinCO). The dashed line has slope −1; the plain line is the best fit to the equation: 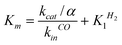 , with α = k+1H2/k+1CO, derived from the kinetic model in ref. 12: , with α = k+1H2/k+1CO, derived from the kinetic model in ref. 12: . . | ||
VIII. Methionine provides O2-tolerance without direct relation with diffusion
We also tested the effects of replacing Val74 and Leu122 with methionines.88 Methionine should not only be bulky enough to slow dioxygen diffusion, but it is also known to participate in oxidative stress responses and protection in several proteins.91–93 The sulfur atom of this amino acid has a high affinity for oxygen, which results in a strong reactivity with reactive oxygen species, or in the ability to form weak S–O interactions.94 We made the hypothesis that methionines placed at the entrance of the active site cavity at positions M74 and M122 may protect the (Ni–Fe) site from oxidation, either by reacting or at least by interacting with the oxygen species present at the active site under oxidizing conditions.42 The goal of that substitution was twofold: slowing diffusion and modifying the reactivity with O2.The molecular volume of methionine is 30% larger than that of valine and about the same as leucine.89 The diffusion rate in the case of the V74M-L122M (MM mutant) is decreased by more than two orders of magnitude and even three orders of magnitude for the V74M (M mutant) (Fig. 3 and 5), which goes far beyond the expected effect of the volume increase on the diffusion. The interaction of gases with methionine is therefore stronger than simple steric obstruction.
We studied hydrogenase oxidation kinetics in the presence of O2 using two methods: PFV and the proton-deuterium exchange (HDE).95 Using these techniques, we clearly showed that the MM and M mutated hydrogenases became O2 tolerant. HDE revealed that the presence of MV further increased O2 tolerance, as the M mutant is able to consume D2 in the presence of 150 μM of O2, which is close to the O2 concentration of 200 μM in air-equilibrated solutions (Fig. 6). The D2-oxidation activity determined under these conditions is about 10% of the activity measured in standard anaerobic assays. This low activity level should be kept in perspective, as the trace amounts of methyl viologen (MV) present in the essay (≈1μM) are far below KmMV = 9 mM.96 On the other hand, an interesting property of the MM mutant was detected by PFV: unlike the WT enzyme, it partly reactivates in the presence of H2 even under very oxidizing conditions (Fig. 4).88 The M mutant also reactivates under H2 at high potential but to a lesser extent. This process is slow and has a small amplitude, but is significant because under the very oxidizing conditions used in the experiments, one would expect nothing but the inactivation of the enzyme.45,57
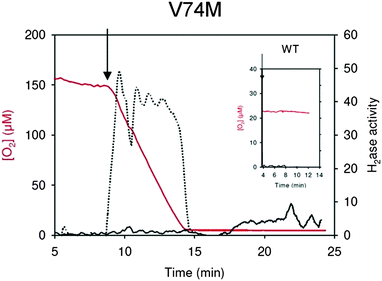 | ||
| Fig. 6 HDE kinetics for the V74M mutated enzymes in the presence of O2. The arrow indicates the injection of the activated enzyme. Red lines: dioxygen concentration, black lines: hydrogenase mediated isotope exchange rates, dotted lines: D2-consumption rate. The hydrogenase activities (exchange or consumption) are expressed in μmol min−1 mg−1. The control experiment with the wild-type enzyme is shown as the inset. In the presence of only 23 μM of O2 the wild-type enzyme is totally inactivated within 1 min. | ||
Crystallographic studies showed that methionines are not modified in the oxidized enzymes, and especially that methionine sulfoxides are not formed.88 Spectroscopic studies conducted on these mutants revealed that methionine interacts with the active site and modifies its environment.88 FTIR studies determined that the mutants were inactivated more slowly and reactivated more rapidly than the native enzyme. This slower inactivation is attributed to a reduced active site accessibility that is due to partial tunnel obstruction by the mutations.11 However the faster activation necessarily involves a quicker removal of the bound oxygen species. This was assumed to involve methionine that would stabilize the rearrangement of the oxygen species that is necessary to allow its protonation, facilitating its escape from the oxidized enzyme. As a result, the phenotype of the V74M and L122M-V74M mutants is not a consequence of a modification of the structure of the active site, but rather reveals subtle changes in the kinetics of the reaction with O2.
IX. Concluding remarks
The analysis of the naturally O2-tolerant hydrogenases revealed that different mechanisms have been developed through evolution to allow H2 catalysis in aerobiosis. Besides, our mechanistic studies also revealed that different effects lead to an improved resistance to O2. On the one hand, blocking the gas tunnel to slow dioxygen access (this is operational for the V74Q, V74M and V74E mutants) may improve O2 tolerance, provided that the diffusion rate becomes slower than the reaction of O2 at the active site. This approach can be compared to the ReRH mode of action. On the other hand, modifying the enzyme kinetics, as in the case of the V74M mutation, in which methionines both slow diffusion and influence the reactivity of the enzyme with O2, is another option. Accelerating reactivation rate appears to be more efficient and probably more promising than reducing diffusion rate. This approach can be compared to SH and or MBH, even though their mechanisms are still unknown. Combining beneficial modifications will certainly be the key to designing O2-tolerant hydrogenases.The main outcome of these studies is the demonstration that it is possible to induce O2-tolerance in a [Ni–Fe] hydrogenase, in such a way that modified enzymes are active in the presence of O2 at concentrations close to that in air-equilibrated solutions. The fact that the substitutions tested are located in conserved regions makes it possible to engineer [Ni–Fe] hydrogenases in a wide range of organisms, as a heterologous expression of [Ni–Fe] hydrogenase is still difficult. This achievement opens the way for future development in the field of biological hydrogen production or utilization.
Acknowledgements
This work was funded by the Centre National de la Recherche Scientifique, l'Agence Nationale de la Recherche, and the University of Provence.References
- W. Martin and M. Muller, Nature, 1998, 392, 37–41 CrossRef CAS.
- C. Rotte, K. Henze, M. Muller and W. Martin, Curr. Opin. Microbiol., 2000, 3, 481–486 CrossRef CAS.
- J. C. Fontecilla-Camps, A. Volbeda, C. Cavazza and Y. Nicolet, Chem. Rev., 2007, 107, 4273–4303 CrossRef CAS.
- S. Shima and R. K. Thauer, Chem. Rec., 2007, 7, 37–46 CrossRef CAS.
- S. Shima, O. Pilak, S. Vogt, M. Schick, M. S. Stagni, W. Meyer-Klaucke, E. Warkentin, R. K. Thauer and U. Ermler, Science, 2008, 321, 572–575 CrossRef CAS.
- J. C. Fontecilla-Camps, P. Amara, C. Cavazza, Y. Nicolet and A. Volbeda, Nature, 2009, 460, 814–822 CrossRef CAS.
- J. Cohen, K. Kim, P. King, M. Seibert and K. Schulten, Structure, 2005, 13, 1321–1329 CrossRef CAS.
- Y. Nicolet, B. J. Lemon, J. C. Fontecilla-Camps and J. W. Peters, Trends Biochem. Sci., 2000, 25, 138–143 CrossRef CAS.
- Y. Montet, P. Amara, A. Volbeda, X. Vernede, E. C. Hatchikian, M. J. Field, M. Frey and J. C. Fontecilla-Camps, Nat. Struct. Biol., 1997, 4, 523–526 CrossRef CAS.
- A. Volbeda, M. H. Charon, C. Piras, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, Nature, 1995, 373, 580–587 CrossRef CAS.
- F. Leroux, S. Dementin, B. Burlat, L. Cournac, A. Volbeda, S. Champ, L. Martin, B. Guigliarelli, P. Bertrand, J. C. Fontecilla-Camps, M. Rousset and C. Léger, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11188–11193 CrossRef CAS.
- P. P. Liebgott, F. Leroux, B. Burlat, S. Dementin, C. Baffert, T. Lautier, V. Fourmond, P. Ceccaldi, C. Cavazza, I. Meynial-Salles, P. Soucaille, J. C. Fontecilla-Camps, B. Guigliarelli, P. Bertrand, M. Rousset and C. Leger, Nat. Chem. Biol., 2010, 6, 63–70 CrossRef CAS.
- T. Lautier, P. Ezanno, C. Baffert, V. Fourmond, L. Cournac, J. C. Fontecilla-Camps, P. Soucaille, P. Bertrand, I. Meynial-Salles and C. Léger, Faraday Discuss., 2010 Search PubMed in press.
- R. C. Prince and H. S. Kheshgi, Crit. Rev. Microbiol., 2005, 31, 19–31 CrossRef CAS.
- L. Cournac, G. Guedeney, G. Peltier and P. M. Vignais, J. Bacteriol., 2004, 186, 1737–1746 CrossRef CAS.
- C. Leger, S. Dementin, P. Bertrand, M. Rousset and B. Guigliarelli, J. Am. Chem. Soc., 2004, 126, 12162–12172 CrossRef CAS.
- A. Melis, L. Zhang, M. Forestier, M. L. Ghirardi and M. Seibert, Plant Physiol., 2000, 122, 127–136 CrossRef CAS.
- S. Fouchard, A. Hemschemeier, A. Caruana, J. Pruvost, J. Legrand, T. Happe, G. Peltier and L. Cournac, Appl. Environ. Microbiol., 2005, 71, 6199–6205 CrossRef CAS.
- P. M. Vignais and A. Colbeau, Curr. Issues Mol. Biol., 2004, 6, 159–188 Search PubMed.
- P. M. Matias, C. M. Soares, L. M. Saraiva, R. Coelho, J. Morais, J. Le Gall and M. A. Carrondo, J. Biol. Inorg. Chem., 2001, 6, 63–81 CrossRef CAS.
- Y. Higuchi, T. Yagi and N. Yasuoka, Structure, 1997, 5, 1671–1680 CrossRef CAS.
- A. Volbeda, Y. Montet, X. Vernede, C. E. Hatchikian and J. C. Fontecilla-Camps, Int. J. Hydrogen Energy, 2002, 27, 1449–1461 CrossRef CAS.
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, C. E. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS.
- A. J. Pierik, W. Roseboom, R. P. Happe, K. A. Bagley and S. P. Albracht, J. Biol. Chem., 1999, 274, 3331–3337 CrossRef CAS.
- A. L. De Lacey, V. M. Fernandez, M. Rousset and R. Cammack, Chem. Rev., 2007 Search PubMed.
- A. Pardo, A. L. De Lacey, V. M. Fernandez, H. J. Fan, Y. Fan and M. B. Hall, J. Biol. Inorg. Chem., 2006, 11, 286–306 CrossRef CAS.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS.
- Y. Nicolet, C. Piras, P. Legrand, C. E. Hatchikian and J. C. Fontecilla-Camps, Structure, 1999, 7, 13–23 CrossRef CAS.
- U. Ryde, C. Greco and L. De Gioia, J. Am. Chem. Soc., 2010, 132, 4512–4513 CrossRef CAS.
- A. Silakov, B. Wenk, E. Reijerse and W. Lubitz, Phys. Chem. Chem. Phys., 2009, 11, 6592–6599 RSC.
- C. Baffert, M. Demuez, L. Cournac, B. Burlat, B. Guigliarelli, P. Bertrand, L. Girbal and C. Leger, Angew. Chem., Int. Ed., 2008, 47, 2052–2054 CrossRef CAS.
- G. Goldet, C. Brandmayr, S. T. Stripp, T. Happe, C. Cavazza, J. C. Fontecilla-Camps and F. A. Armstrong, J. Am. Chem. Soc., 2009, 131, 14979–14989 CrossRef CAS.
- S. T. Stripp, G. Goldet, C. Brandmayr, O. Sanganas, K. A. Vincent, M. Haumann, F. A. Armstrong and T. Happe, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17331–17336 CrossRef CAS.
- J. W. van der Zwaan, J. M. Coremans, E. C. Bouwens and S. P. Albracht, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1990, 1041, 101–110 Search PubMed.
- M. Carepo, D. L. Tierney, C. D. Brondino, T. C. Yang, A. Pamplona, J. Telser, I. Moura, J. J. Moura and B. M. Hoffman, J. Am. Chem. Soc., 2002, 124, 281–286 CrossRef CAS.
- M. van Gastel, M. Stein, M. Brecht, O. Schroder, F. Lendzian, R. Bittl, H. Ogata, Y. Higuchi and W. Lubitz, J. Biol. Inorg. Chem., 2006, 11, 41–51 CrossRef.
- G. Davidson, S. B. Choudhury, Z. Gu, K. Bose, W. Roseboom, S. P. Albracht and M. J. Maroney, Biochemistry, 2000, 39, 7468–7479 CrossRef CAS.
- A. L. De Lacey, V. M. Fernandez, M. Rousset and R. Cammack, Chem. Rev., 2007, 107, 4304–4330 CrossRef CAS.
- M. E. Pandelia, H. Ogata and W. Lubitz, ChemPhysChem, 2010, 11, 1127–1140 CrossRef CAS.
- M. Stein, E. van Lenthe, E. J. Baerends and W. Lubitz, J. Am. Chem. Soc., 2001, 123, 5839–5840 CrossRef CAS.
- C. Stadler, A. L. de Lacey, Y. Montet, A. Volbeda, J. C. Fontecilla-Camps, J. C. Conesa and V. M. Fernandez, Inorg. Chem., 2002, 41, 4424–4434 CrossRef CAS.
- A. Volbeda, L. Martin, C. Cavazza, M. Matho, B. W. Faber, W. Roseboom, S. P. Albracht, E. Garcin, M. Rousset and J. C. Fontecilla-Camps, J. Biol. Inorg. Chem., 2005, 10, 239–249 CrossRef CAS.
- L. K. Black, C. Fu and R. J. Maier, J Bacteriol, 1994, 176, 7102–7106 CAS.
- S. E. Lamle, S. P. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 6595–6604 CrossRef CAS.
- K. A. Vincent, A. Parkin and F. A. Armstrong, Chem. Rev., 2007, 107, 4366–4413 CrossRef CAS.
- A. L. de Lacey, E. C. Hatchikian, A. Volbeda, M. Frey, J. C. Fontecilla-Camps and V. M. Fernandez, J. Am. Chem. Soc., 1997, 119, 7181–7189 CrossRef CAS.
- A. K. Jones, S. E. Lamle, H. R. Pershad, K. A. Vincent, S. P. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2003, 125, 8505–8514 CrossRef CAS.
- R. Cammack, V. M. Fernandez and K. Schneider, Biochimie, 1986, 68, 85–91 CrossRef CAS.
- V. M. Fernandez, K. K. Rao, M. A. Fernandez and R. Cammack, Biochimie, 1986, 68, 43–48 CrossRef CAS.
- S. E. Lamle, S. P. Albracht and F. A. Armstrong, J. Am. Chem. Soc., 2004, 126, 14899–14909 CrossRef CAS.
- E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–566 CrossRef CAS.
- A. Volbeda and J. C. Fontecilla-Camps, Coord. Chem. Rev., 2005, 249, 1609–1619 CrossRef CAS.
- M. Teixeira, G. Fauque, I. Moura, P. A. Lespinat, Y. Berlier, B. Prickril, H. D. Peck, Jr., A. V. Xavier, J. Le Gall and J. J. Moura, Eur. J. Biochem., 1987, 167, 47–58 CAS.
- S. P. Albracht, Biochim. Biophys. Acta, Bioenerg., 1994, 1188, 167–204 CrossRef.
- A. L. De Lacey, C. Stadler, V. M. Fernandez, E. C. Hatchikian, H. J. Fan, S. Li and M. B. Hall, J. Biol. Inorg. Chem., 2002, 7, 318–326 CrossRef CAS.
- W. Lubitz, E. Reijerse and M. van Gastel, Chem. Rev., 2007, 107, 4331–4365 CrossRef CAS.
- C. Léger and P. Bertrand, Chem. Rev., 2008, 108, 2379–2438 CrossRef CAS.
- T. Burgdorf, O. Lenz, T. Buhrke, E. van der Linden, A. K. Jones, S. P. Albracht and B. Friedrich, J. Mol. Microbiol. Biotechnol., 2005, 10, 181–196 CrossRef CAS.
- M. Ludwig, J. A. Cracknell, K. A. Vincent, F. A. Armstrong and O. Lenz, J. Biol. Chem., 2008, 284, 465–477 CrossRef.
- P. M. Vignais and B. Billoud, Chem. Rev., 2007, 107, 4206–4272 CrossRef CAS.
- G. Goldet, A. F. Wait, J. A. Cracknell, K. A. Vincent, M. Ludwig, O. Lenz, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2008, 130, 11106–11113 CrossRef CAS.
- M. Brugna-Guiral, P. Tron, W. Nitschke, K. O. Stetter, B. Burlat, B. Guigliarelli, M. Bruschi and M. T. Giudici-Orticoni, Extremophiles, 2003, 7, 145–157 CAS.
- M. Guiral, C. Aubert and M. T. Giudici-Orticoni, Biochem. Soc. Trans., 2005, 33, 22–24 CrossRef CAS.
- M. Guiral, P. Tron, C. Aubert, A. Gloter, C. Iobbi-Nivol and M. T. Giudici-Orticoni, J. Biol. Chem., 2005, 280, 42004–42015 CrossRef CAS.
- P. C. Maness, S. Smolinski, A. C. Dillon, M. J. Heben and P. F. Weaver, Appl. Environ. Microbiol., 2002, 68, 2633–2636 CrossRef CAS.
- S. Elsen, A. Colbeau, J. Chabert and P. M. Vignais, J Bacteriol, 1996, 178, 5174–5181 CAS.
- M. J. Lukey, A. Parkin, M. M. Roessler, B. J. Murphy, J. Harmer, T. Palmer, F. Sargent and F. A. Armstrong, J. Biol. Chem., 2010, 285, 3928–3938 CrossRef CAS.
- K. A. Vincent, J. A. Cracknell, O. Lenz, I. Zebger, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 16951–16954 CrossRef CAS.
- M. Bernhard, T. Buhrke, B. Bleijlevens, A. L. De Lacey, V. M. Fernandez, S. P. Albracht and B. Friedrich, J. Biol. Chem., 2001, 276, 15592–15597 CrossRef CAS.
- R. P. Happe, W. Roseboom, G. Egert, C. G. Friedrich, C. Massanz, B. Friedrich and S. P. Albracht, FEBS Lett., 2000, 466, 259–263 CrossRef CAS.
- B. Bleijlevens, T. Buhrke, E. van der Linden, B. Friedrich and S. P. Albracht, J. Biol. Chem., 2004, 279, 46686–46691 CrossRef CAS.
- L. Rey, D. Fernandez, B. Brito, Y. Hernando, J. M. Palacios, J. Imperial and T. Ruiz-Argueso, Mol Gen Genet, 1996, 252, 237–248 CAS.
- B. Bleijlevens, F. A. van Broekhuizen, A. L. De Lacey, W. Roseboom, V. M. Fernandez and S. P. Albracht, J. Biol. Inorg. Chem., 2004, 9 Search PubMed.
- T. Buhrke and B. Friedrich, Arch. Microbiol., 1998, 170, 460–463 CrossRef CAS.
- T. Buhrke, B. Bleijlevens, S. P. Albracht and B. Friedrich, J. Bacteriol., 2001, 183, 7087–7093 CrossRef CAS.
- J. A. Cracknell, A. F. Wait, O. Lenz, B. Friedrich and F. A. Armstrong, Proc. Natl. Acad. Sci. U. S. A., 2009 Search PubMed.
- M. E. Pandelia, V. Fourmond, P. Tron-Infossi, E. Lojou, P. Bertrand, C. Leger, M. T. Giudici-Orticoni and W. Lubitz, J. Am. Chem. Soc., 2010 Search PubMed.
- K. A. Vincent, N. A. Belsey, W. Lubitz and F. A. Armstrong, J. Am. Chem. Soc., 2006, 128, 7448–7449 CrossRef CAS.
- V. Fourmond, P. Infossi, M. T. Giudici-Orticoni, P. Bertrand and C. Leger, J. Am. Chem. Soc., 2010, 132, 4848–4857 CrossRef CAS.
- K. A. Vincent, A. Parkin, O. Lenz, S. P. Albracht, J. C. Fontecilla-Camps, R. Cammack, B. Friedrich and F. A. Armstrong, J. Am. Chem. Soc., 2005, 127, 18179–18189 CrossRef CAS.
- O. Lenz, M. Ludwig, T. Schubert, I. Burstel, S. Ganskow, T. Goris, A. Schwarze and B. Friedrich, ChemPhysChem, 2010, 11, 1107–1119 CrossRef CAS.
- M. Guiral, P. Tron, V. Belle, C. Aubert, C. Léger, B. Guigliarelli and M.-T. Giudici-Orticoni, Int. J. Hydrogen Energy, 2006, 31, 1424–1431 CrossRef CAS.
- T. Buhrke, O. Lenz, N. Krauss and B. Friedrich, J. Biol. Chem., 2005, 280, 23791–23796 CrossRef CAS.
- O. Duche, S. Elsen, L. Cournac and A. Colbeau, FEBS J., 2005, 272, 3899–3908 CrossRef.
- O. Lenz, A. Gleiche, A. Strack and B. Friedrich, J. Bacteriol., 2005, 187, 6590–6595 CrossRef CAS.
- M. R. Leach and D. B. Zamble, Curr. Opin. Chem. Biol., 2007, 11, 159–165 CrossRef CAS.
- L. Casalot and M. Rousset, Trends Microbiol., 2001, 9, 228–237 CrossRef CAS.
- S. Dementin, F. Leroux, L. Cournac, L. De Lacey, A. Volbeda, C. Léger, B. Burlat, N. Martinez, S. Champ, L. Martin, O. Sanganas, M. Haumann, V. M. Fernández, B. Guigliarelli, J. C. Fontecilla-Camps and M. Rousset, J. Am. Chem. Soc., 2009, 131, 10156–10164 CrossRef CAS.
- M. Hackel, H. J. Hinz and G. R. Hedwig, Biophys. Chem., 1999, 82, 35–50 CrossRef CAS.
- K. Nienhaus, P. Deng, J. S. Olson, J. J. Warren and G. U. Nienhaus, J. Biol. Chem., 2003, 278, 42532–42544 CrossRef CAS.
- E. R. Stadtman, J. Moskovitz, B. S. Berlett and R. L. Levine, Mol. Cell. Biochem., 2002, 234–235, 3–9 CrossRef.
- E. R. Stadtman, Arch. Biochem. Biophys., 2004, 423, 2–5 CrossRef CAS.
- E. R. Stadtman, Free Radical Res., 2006, 40, 1250–1258 CrossRef CAS.
- D. Pal and P. Chakrabarti, J Biomol Struct Dyn, 2001, 19, 115–128 CAS.
- Y. Berlier, P. A. Lespinat and B. Dimon, Anal. Biochem., 1990, 188, 427–431 CAS.
- S. Dementin, V. Belle, P. Bertrand, B. Guigliarelli, G. Adryanczyk-Perrier, A. L. De Lacey, V. Fernandez, M. Rousset and C. Léger, J. Am. Chem. Soc., 2006, 128, 5209–5218 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |