The oxidation state of copper in bimetallic (Pt–Cu, Pd–Cu) catalysts during water denitration
Jacinto
Sá
*ab,
Noelia
Barrabés
*c,
Evgeny
Kleymenov
a,
Chen
Lin
a,
Karin
Föttinger
c,
Olga V.
Safonova
a,
Jakub
Szlachetko
ad,
Jeroen A.
van Bokhoven
ab,
Maarten
Nachtegaal
a,
Atsushi
Urakawa
e,
Gastón A.
Crespo
f and
Günther
Rupprechter
c
aPaul Scherrer Institut, 5232 Villigen, Switzerland
bETH Zurich, Institute for Chemical and Bioengineering, 8093 Zurich, Switzerland. E-mail: jacinto.sa@psi.ch; Tel: +41-56310-2910
cInstitute of Materials Chemistry, Vienna University of Technology, Getreidemarkt 9, A-1060 Vienna, Austria. E-mail: noe.barrabes@gmail.com; Tel: +43-1-58801-165110
dInstitute of Physics, University of Kielce, Poland
eInstitute of Chemical Research of Catalonia (ICIQ), Av. Països Catalans 16, 43007 Tarragona, Spain
fDep. of Inorganic, Analytical and Applied Chemistry, University of Geneva, Quai E.-Ansermet 30, CH-1211 Geneva, Switzerland
First published on 19th December 2011
Abstract
Catalytic denitration of water with bimetallic systems has emerged as a viable solution for removal of nitrates from drinking water. Despite the progress in process development during the last two decades, only a few studies were performed to determine catalyst structure under working conditions. Herein, we determined the relative population of Cu oxidation states in Pt–Cu and Pd–Cu bimetallic catalysts by in situ high resolution X-ray absorption spectroscopy in combination with principal component analysis. The initial state of the catalyst was a Pt–Cu or Pd–Cu alloy. Segregation of the metal components occurred under reaction conditions especially for a Pt–Cu system. The active oxidation states of copper were metallic and alloy, and their concentration was highly dependent on the amount of hydrogen in the feed. Initial alloy phase of the catalysts ensures close proximity between Cu and the noble metals after segregation, essential to maintain catalysts activity.
Introduction
Fulfilling the demands for drinking water and wastewater treatment has become a major social, technological, economical and political predicament. The Nitrate Directive (EC, 1991) is the European legislation dealing with protection of water assets from agricultural pollution and in particular pollutants containing nitrogen. The directive established strict limits for nitrogen containing pollutants, namely 50, 0.1 and 0.5 ppm for NO3−, NO2− and NH4+, respectively.1Fertilisers and waste effluents from certain industries cause nitrate excess in water. Nitrate pollution can cause water reservoirs eutrophication and serious human health problems such as cancer and the blue-baby disease. In 1989, Vorlop and Tacke published a pioneering work describing how to remove nitrates from water.2 They demonstrated the necessity for a bimetallic system to reduce nitrates, composed of a precious metal (Pt or Pd) and a promoter (Cu, Ni, Fe, Sn, In or Ag).3,4
The nitrate reduction implies a stepwise mechanism with nitrate reduction to nitrite as the rate limiting step and NO hydrogenation as the key stage with respect to process selectivity, avoiding the formation of undesired ammonium.5,6 The mechanism for nitrate hydrogenation has been widely accepted, in contrast to the nature of the active sites, which is highly debated. In the bi-functional mechanism reported by Epron et al.,7 nitrate hydrogenation occurs firstly on copper sites through a direct redox mechanism and copper is brought back to the metallic state by hydrogen spill over from palladium. Alternatively, Gao et al.8 proposed that Cu2O may be involved in the nitrate-to-nitrite conversion on a Pd–Cu/TiO2 catalyst, considering that negatively charged nitrate ions are more easily adsorbed on the positively charged surface of Cu2O instead of Cu. Edelmann et al.9 performed in situ X-ray absorption spectroscopy (XAS) on a conventional Pd–Cu/Al2O3 catalyst. They concluded based on XAS experimental results that Cu and Pd oxidation state is metallic. However due to the low time and spectral resolution one cannot discard CuxO being formed on the surface and quickly reduced by hydrogen spill over from the noble metal.
We determined the oxidation state of copper on commonly used Pt–Cu/Al2O3 and Pd–Cu/Al2O3 measured by operando high-energy resolution fluorescence detection (HERFD) XAS.10,11 The advantage of recording a high resolution XAS spectrum becomes obvious if considering pre-edges of the third-row transition metals, which arise from quadrupole transitions to unoccupied 3d states.12 These small peaks located a few eV below the K-edge become clearly resolved and serve as a direct probe of the chemical state. In particular, it allows us to distinguish Cu+ and Cu2+ unambiguously.13,14 The results suggest a direct redox mechanism in which Cu0 is oxidized to CuO due to the reduction of NO3− to NO2−, and regenerated by the noble metal. The efficiency of the regeneration step depends on the availability of hydrogen.
Experimental
Catalyst preparation
First, monometallic 2% Pd/Al2O3 and 2% Pt/Al2O3 were prepared by impregnation using an aqueous solution of the respective precursor salts, Pd(NO3)2·2H2O (∼40% Pd basis, Aldrich) and H2PtCl6 (99.995% Aldrich). After drying at 120 °C, the monometallic catalysts were directly impregnated with the copper salt precursor (Cu(NO3)2·3H2O, Alfa Aesar) solution to obtain 1 wt% Cu. The samples were dried and finally calcined at 300 °C for 2 h in air.Ex situ characterization
Temperature-programmed reduction (TPR) studies were performed using a TPD/R/O 1100 (Thermo Finnigan) equipped with a thermal conductivity detector (TCD). Prior to TPR, the sample (around 50 mg) was dried under helium at 120 °C for 24 h, then cooled down to room temperature. TPR was carried out from room temperature up to 800 °C at a heating rate of 10 °C min−1 in 5% H2 in argon (20 mL min−1 total flow).High-resolution transmission electron microscopy (HRTEM) was performed with a JEOL 2010F instrument equipped with a field emission source. The point-to-point resolution was 1.9 Å and the resolution between lines was 1.4 Å. Samples were ground and deposited on holey-carbon coated grids from ultrasonicated alcohol suspensions.
In situ characterization
In situ HERFD XAS studies were performed at the SuperXAS beamline of the Swiss Light Source. The beamline provides an X-ray flux of 6 × 1011 photon s−1 and an energy bandwidth of 1 eV at the Cu K-edge. The spot size at the sample was 0.1 mm (vertical) by 0.2 mm (horizontal). Emitted X-ray radiation was registered with a Johann-type spectrometer equipped with five spherically curved Si(111) crystals and a PILATUS 100 K detector.15 A bag with kapton windows filled with helium was placed between the sample, crystals, and detector to minimize absorption of X-rays by air. The energy resolution of the spectrometer at the Cu Kα1 emission line was below 1 eV.The catalysts were tested in a flow reactor composed of a thin 3 mm outer diameter kapton capillary (Fig. 1). Degassed water containing NO3− as reactant was co-fed with H2 or N2 by means of a pneumatic pump and a mass flow controller, respectively. The catalytic performance was evaluated offline by UV-Vis measurements using Merck colorimetric tests. In a typical run, 20–30 mg of catalysts was loaded into the kapton capillary, kept in place with two glass wool plugs (one at each end) and the catalyst was reduced in situ for 1 h under 15 mL min−1 of pure H2 at 300 °C. After reduction, the catalyst was cooled down in H2 to room temperature and a reaction mixture composed of 100 ppm of NO3− and H2 or N2 were passed through the catalytic bed. The liquid flow rate was constant (2 mL min−1) and the gas flow rate was varied between 2 and 6 mL min−1.
 | ||
| Fig. 1 Schematic representation of a setup used for in situ HERFD XAS experiments | ||
The oxidation state of copper during catalytic reaction was determined by linear combination fitting (LCF) of the HERFD XAS spectra with references measured with the same spectrometer parameters, upon background subtraction and spectra normalization.16 The Cu K-edge positions of the HERFD XAS spectra of the catalysts after calcination and reduction were used as references for oxidic and alloy phases, respectively.
Results and discussion
Fig. 2 shows the TPR spectra of calcined Pd–Cu/Al2O3 and Pt–Cu/Al2O3. A sharp feature, associated with the reduction of the metals, dominated both profiles. The main reduction peak of Pd–Cu was centered at 130 °C, whereas that of Pt–Cu was positioned at 225 °C. The single-peak feature suggests that the metals are in close contact, which leads to a shift of the noble metal reduction to higher temperature, and copper to lower temperature.17 Some minor features in the spectra are most likely related to the reduction of isolated copper oxide particles, i.e., not in the vicinity of the noble metal. Featureless TPR spectra were measured when the catalysts were pre-reduced at 300 °C for 1 h, suggesting that reduction at 300 °C for 1 h is sufficient to fully reduce the catalysts. Thus, reduction of the catalysts prior to reaction at 300 °C for 1 h will ensure that metals are reduced and in alloy form. | ||
Fig. 2 Temperature-programmed reduction of bimetallic catalysts. ( ) Pd–Cu/Al2O3 calcined; (—) Pt–Cu/Al2O3 calcined; and ( ) Pd–Cu/Al2O3 calcined; (—) Pt–Cu/Al2O3 calcined; and ( ) Pt–Cu/Al2O3 after reduction at 300 °C for 1 h. ) Pt–Cu/Al2O3 after reduction at 300 °C for 1 h. | ||
Fig. 3a shows a TEM image of Pd–Cu/Al2O3 after reduction. The image is dominated by highly dispersed metal particles with an average size of ca. 5.0 nm in diameter. Fig. 3b shows a HRTEM image of an individual particle, with spots in the Fourier transformed image at 2.2 Å, which is characteristic of a Pd–Cu alloy.18 The resolution was insufficient to detect surface segregation by one of the metals. TEM and HRTEM images of Pt–Cu/Al2O3 (not shown) were very similar, though the particle size was slightly smaller, ca. 4.0 nm in diameter.
 | ||
| Fig. 3 TEM image of Pd–Cu/Al2O3 after reduction at 300 °C for 1 h. (a) High magnification image; (b) HR-TEM 5 nm Pd–Cu particle. | ||
Fig. 4 shows the Cu K-edge HERFD XAS spectra of Pt–Cu/Al2O3 during the hydrogenation of nitrates. With increasing hydrogen flow, the Cu K-edge shifted to lower energy, the whiteline intensity decreased, and a feature in the edge became more noticeable. By comparison with the references depicted in Fig. 4 inset, it is obvious that copper reduces further with the increase of hydrogen in the stream. This observation indicates that the system is dynamic, and adjusts itself to the conditions applied. More importantly, HERFD XAS is sensitive to those changes in the Cu oxidation state, and structure. The changes can be quantified by using linear combinations of the spectra of reference compounds measured under similar conditions. The data were fitted with references for Cu2+ (CuO), Cu+ (Cu2O), Cu0 (Cu foil), and Cu alloy (catalyst after reduction). To determine the number of spectral components in each spectrum, principal component analysis (PCA)19 was used. PCA is routinely used to determine the number of independent variables, i.e., how many species are significantly represented in each spectrum. Fig. 5 depicts an example of such fitting. The spectrum of Pt–Cu/Al2O3 after exposure to the reaction mixture could be fitted using only three components, namely Cu2+, Cu0, and Cu alloy. Using this approach all data were fit and the fractional oxidation states of Cu were determined and correlated with the catalytic measurements.
 | ||
Fig. 4 Normalized HERFD XAS spectra of Pt–Cu/Al2O3 after exposure to H2 in the presence of a solution containing 100 ppm of NO3−. Catalyst exposed to H2 ( ) 2 mL min−1 for 10 min; ( ) 2 mL min−1 for 10 min; ( ) 4 mL min−1 for 10 min; and (▲) 6 mL min−1 for 10 min. The inset shows the reference spectra of alloy, Cu0, and CuO. ) 4 mL min−1 for 10 min; and (▲) 6 mL min−1 for 10 min. The inset shows the reference spectra of alloy, Cu0, and CuO. | ||
 | ||
| Fig. 5 Linear combination fitting of the normalized HERFD XAS spectrum of Pt–Cu/Al2O3 after exposure to 6 mL min−1 H2 in the presence of a solution containing 100 ppm of NO3− after 75 min on stream. | ||
Fig. 6 represents the fraction of metallic, alloy, and oxidic Cu phases of Pt–Cu/Al2O3 exposed to a stream of 100 ppm nitrates under different hydrogen or nitrogen flows. At time 0 min, the copper phase of the catalyst is fully fitted with a single component, namely Cu alloy. Nitrate conversion was roughly 40%, which is one of the highest detected throughout the experiment. The alloy phase decreased to about 18% within the first 5 min, yielding increases of both Cu0 and Cu2+. Conversion also dropped to about 20%. The next 25 min revealed further increase of Cu2+ due to oxidation of the alloy phase, since the fraction of metallic phase remained constant. However, the loss of alloy was not reflected in activity loss, which suggests bulk oxidation. After 30 min on stream, the copper phase in the catalyst was composed of roughly 50% of Cu0 and 50% of Cu2+, and most importantly no alloy phase was observed. The results of the first 30 min show that when hydrogen concentration is low, copper oxidation occurs with the oxygen extracted from nitrate during its reduction to nitrite. Segregation of Pt and Cu is paralleled by oxidation of Cu to CuO. Segregation likely leads to Cu and Pt species on the alumina in close contact. This is confirmed by hydrogen activation on the noble metal, which interacts with the copper oxide and reduces it.
 | ||
Fig. 6 Deconvolved HERFD XAS spectra of Pt–Cu/Al2O3 (top), and nitrate conversion (bottom) as a function of time on stream after exposure to various flows of H2 or N2 in the presence of a solution containing 100 ppm of NO3−. Top: concentration of Cu species ( ) Pt–Cu alloy; (■) Cu0 and ( ) Pt–Cu alloy; (■) Cu0 and ( ) CuO; and (—) gas flow. Bottom: ( ) CuO; and (—) gas flow. Bottom: ( ) nitrate conversion; and (—) gas flow. ) nitrate conversion; and (—) gas flow. | ||
To avoid irreversible oxidation of copper, the H2 flow was increased from 2 to 4 mL min−1 after 30 min, which resulted in a steady increase of the fraction of Cu0 and alloy, at the cost of the copper oxide phase. Catalytic conversion increased from 20% to about 32%. The rather small amount of copper alloy phase suggests that the alloy is being formed at the surface, which is expected since bulk alloy formation requires high temperatures. A further increase in conversion was witnessed when the H2 flow was increased from 4 to 6 mL min−1, with the correspondent increase in the metallic and alloy phase fractions. The result suggests that copper oxide is not active and it is a reaction intermediate, i.e., it gets formed during reduction of nitrates to nitrites, and converted to Cu0 by hydrogen spilled over the Pt. The amounts of Cu0 and Cu2+ phases are therefore controlled by the amount of hydrogen in the feed.
After reaching a steady value for nitrate conversion and nitrite formation, H2 flow was decreased from 6 to 2 mL min−1, which resulted in a slight increase of the alloy phase. More significantly, catalytic conversion dropped to about 10%. Since the oxide phase did not change significantly, the alloy phase was regenerated at the cost of the metallic phase. The catalytic activity drop advocates for preferential reduction of the CuO phase versus nitrogen containing species. As a reminder, the noble metal has twofold role; maintains copper in a low oxidation state and reduces nitrite to nitrogen, both achieved by hydrogen spillover. This was further corroborated when the gas feed was switched from hydrogen to nitrogen. The switch led to steep increase in copper oxide phase and decrease of catalytic conversion to 0%. The catalytic conversion reached zero when the copper alloy was still being formed. Preferential standard reduction of CuO with respect to nitrates is expected based on the reduction potentials, summarized below.20
| NO3−(aq) + 2H+(aq) + e− → NO2(g) + H2O(l) E0 = +0.7 V |
| NO3−(aq) + 4H+(aq) + 3e− → NO(g) + 2H2O(l) E0 = +0.96 V |
| Cu2+(aq) + e− → Cu+(aq) E0 = +0.16 V |
| Cu2+(aq) + 2e− → Cu0(aq) E0 = +0.34 V |
| Cu+(aq) + e− → Cu0(aq) E0 = +0.52 V |
When the flow was reverted to hydrogen, both metallic phase and catalytic activity were recovered however not to their original values. This is most likely due to the loss of contacts between Cu and Pt phases. Further increase of hydrogen led to an increase of copper metal but not to a great extent. The catalytic conversion does not recover as fast as the metallic phase because the activated hydrogen is preferentially used in the reduction of CuO.
The major challenge for the implementation of catalytic systems to remove nitrates from water is to suppress the formation of nitrites (intermediate) and ammonia (byproduct). In the case of the Pt–Cu catalyst, the ammonia concentration was always below 0.5 ppm (Fig. 7), which is within the EU directive. The amount of ammonia formed is dependent on the activity of the catalyst, inferring that ammonia is formed from the over-hydrogenation of NO intermediate when its surface concentration is low,21 rather than at a different active site. Nitrite concentration, on the other hand, was always above the legal limit (0.1 ppm), except when no conversion was detected. This is characteristic of a consecutive reaction intermediate that is leached from the surface to the effluent before further transformation into reaction products could occur.
 | ||
Fig. 7 Pt–Cu/Al2O3 nitrite formation (top) and ammonia formation (bottom), as a function of time on stream after exposure to various flows of H2 or N2 in the presence of a solution containing 100 ppm of NO3−. Top: ( ) nitrite conversion; and (—) gas flow. Bottom: ( ) nitrite conversion; and (—) gas flow. Bottom: ( ) ammonia concentration; and (—) gas flow. ) ammonia concentration; and (—) gas flow. | ||
Pd–Cu/Al2O3 is often considered a better catalyst for the removal of nitrates from water. In the Pd–Cu/Al2O3, copper was found primarily in alloy form under hydrogen flow (Fig. 8). This did not change significantly when hydrogen flow was increased from 2 to 4 mL min−1. This represents a clear difference between the two systems since in the Pt–Cu/Al2O3, the copper alloy phase was rapidly converted into metallic and oxide. Another important difference was the overall conversion, which was much higher in the Pd system. The increase of hydrogen flow did not affect the oxidation state of copper but changed drastically the catalytic conversion from 70% to 20%. This suggests reactive surface poisoning by hydrogen, i.e., hydrogen blocks adsorption of nitrates. For the first time Cu+ phase was detected (high conversion) suggesting its involvement as intermediate at least for the Pd system. However it does not seem to affect the overall performance of the catalyst.
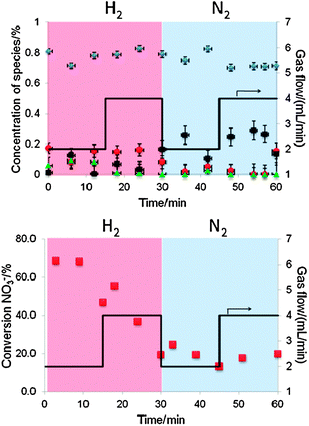 | ||
Fig. 8 Deconvolved HERFD XAS spectra of Pd–Cu/Al2O3 (top), and nitrate conversion (bottom) as a function of time on stream after exposure to various flows of H2 or N2 in the presence of a solution containing 100 ppm of NO3−. Top: concentration of Cu species ( ) Pt–Cu alloy; (■) Cu0; ( ) Pt–Cu alloy; (■) Cu0; ( ) CuO and ( ) CuO and ( ) Cu2O; and (—) gas flow. Bottom: ( ) Cu2O; and (—) gas flow. Bottom: ( ) nitrate conversion; and (—) gas flow. ) nitrate conversion; and (—) gas flow. | ||
When the gas feed was switched from hydrogen to nitrogen the alloy phase decreased slightly and metallic phase increased. As in the case of Pt, the result suggests copper oxidation, followed by its segregation and reduction of this phase by spilled over hydrogen. What is striking is that nitrate reduction remains taking place after 30 min in nitrogen. The rather surprising result points to a contribution of a source of hydrogen that maintains the copper primarily in the metallic state. A possible source of hydrogen is the Pd β-hydride phase, which acts as a sink for hydrogen that can be released when hydrogen is required. To our knowledge this is the first evidence for the active role of hydride phase in the denitration process. Hydride was suggested to decrease the selectivity to N2 since it promotes overhydrogenation reactions22 but to our knowledge no evidence was given in the role of hydride to stabilize metallic/alloy phase of copper. The nitrates were converted to non-aqueous nitrogen species, such as NO, N2O or N2, since neither nitrites or ammonium were detected (Fig. 9). The increase of the metallic fraction at the cost of the alloy suggests oxidation of copper and segregation followed by quick reduction by the hydrogen stored in the hydride phase.
 | ||
Fig. 9 Pd–Cu/Al2O3 nitrite formation (top) and ammonia formation (bottom), as a function of time on stream after exposure to various flows of H2 or N2 in the presence of a solution containing 100 ppm of NO3−. Top: ( ) nitrite conversion; and (—) gas flow. Bottom: ( ) nitrite conversion; and (—) gas flow. Bottom: ( ) ammonia concentration; and (—) gas flow. ) ammonia concentration; and (—) gas flow. | ||
The present observations suggest that the Pd system is better since it is able to maintain copper in its active form (metallic and/or alloy) more efficiently than its Pt counterpart.
Conclusion
We demonstrated that HERFD XAS can be used to monitor in situ the oxidation state of the copper phase of bimetallic catalysts used in the removal of nitrates from water. The copper was found to go over a redox cycle, in which copper gets oxidized when it converts nitrates to nitrites and subsequently regenerated to the metallic state (active form) by activated hydrogen spilled over from the noble metal. Hydrogen concentration plays a key role in the catalyst activity since it determines the reduction of CuO and therefore the amount of the active copper phase.Acknowledgements
We thank Erich de Boni (PSI) for help in development of the in situ catalytic setup. We thank the Swiss Light Source for providing the beam time. Noelia Barrabés thanks Marie Curie project (PIEF-GA-2009-236741—ENVIROCATHYDRO) for financial support.Notes and references
- http://ec.europa.eu/environment/water/water-nitrates/index_en.html .
- K. D. Vorlop and T. Tacke, Chem. Ing. Tech., 1989, 61, 836–837 CrossRef CAS.
- J. A. Anderson and M. Fernández-García, Catalytic and photocatalytic removal of inorganic and organic pollutants from aqueous sources, RSC-Specialist Periodical Reports.
- N. Barrabés and J. Sá, Appl. Catal., B, 2011, 104, 1–5 CrossRef.
- T. Tacke and K. D. Vorlop, Chem. Ing. Tech., 1993, 65, 1500–1502 CrossRef CAS.
- J. Wärna, I. Turunen, T. Salmi and T. Maunula, Chem. Ing. Tech., 1994, 49, 5763–5773 Search PubMed.
- F. Epron, F. Gauthard, C. Pineda and J. Barbier, J. Catal., 2001, 198, 309–318 CrossRef CAS.
- W. L. Gao, N. J. Guan, J. X. Chen, X. X. Guan, R. C. Jin, H. S. Zeng, Z. G. Liu and F. X. Zhang, Appl. Catal., B, 2003, 46, 341–351 CrossRef CAS.
- A. Edelmann, W. Schiesser, H. Vinek and A. Jentys, Catal. Lett., 2000, 69, 11–16 CrossRef CAS.
- J. A. van Bokhoven, C. Louis, J. T. Miller, M. Tromp, O. V. Safonova and P. Glatzel, Angew. Chem., Int. Ed., 2006, 45, 4651–4654 CrossRef CAS.
- O. V. Safonova, M. Tromp, J. A. van Bokhoven, F. M. F. de Groot, J. Evans and P. Glatzel, J. Phys. Chem. B, 2006, 110, 16162–16164 CrossRef CAS.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 1987, 109, 6433–6442 CrossRef CAS.
- E. Kleimenov, E. Alayon, J. van Bokhoven, A. Eliseev, M. Janousch, N. Verbitskij, A. Vinogradov and M. Nachtegaal, Chimia, 2010, 64, 572 Search PubMed.
- E. Kleymenov, J. A. van Bokhoven, C. David, P. Glatzel, M. Janousch, R. Alonso-Mori, M. Studer, M. Willimann, A. Bergamaschi, B. Henrich and M. Nachtegaal, Rev. Sci. Instrum., 2011, 82, 065107 CrossRef.
- J. Sá, C. Kartusch, M. Makosch, C. Paun, J. A. van Bokhoven, E. Kleymenov, J. Szlachetko, M. Nachtegaal, H. G. Manyar and C. Hardacre, Chem. Commun., 2011, 47, 6590–6592 RSC.
- F. Epron, F. Gauthard and J. Barbier, Appl. Catal., A, 2002, 237, 253–261 CrossRef CAS.
- N. Barrabés, D. Cornado, K. Foettinger, A. Dafinov, J. Llorca, F. Medina and G. Rupprechter, J. Catal., 2009, 263, 239–246 CrossRef.
- K. Pearson, Philos. Mag., 1901, 2, 559 Search PubMed.
- http://www.siliconfareast.com/ox_potential.htm .
- J. Sá and J. A. Anderson, Appl. Catal., B, 2008, 77, 409–417 CrossRef.
- C. M. Mendez, H. Olivero, D. E. Damiani and M. A. Volpe, Appl. Catal., B, 2008, 84, 156–161 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |