Experimental and computational studies of nitrogen doped Degussa P25 TiO2: application to visible-light driven photo-oxidation of As(III)
Xiaoling
Wang
a,
Simo O.
Pehkonen
*b,
Jaakko
Rämö
c,
Marja
Väänänen
d,
James G.
Highfield
e and
Kari
Laasonen
f
aDivision of Environmental Science and Engineering, NUS, 119260, Singapore
bMasdar Institute of Science and Technology, P.O.Box 54224, Abu Dhabi, UAE. E-mail: spehkonen@masdar.ac.ae
cCewic, Thule Institute, Typpitie 1, FI-90650 Oulu, Finland
dLaboratory of Chemical Process Engineering, University of Oulu, FI-90014, Finland
eInstitute of Chemical Engineering Sciences, 627833, Singapore
fDept. of Chemistry, University of Oulu, Finland
First published on 19th December 2011
Abstract
Degussa P25 TiO2 doped with nitrogen via NH3 treatment at 400–600 °C was found to be capable of catalyzing the photo-oxidation of aqueous As(III) to As(V) under visible light. Substitutional doping of N for O creates N–Ti–O surface linkages, as confirmed by XPS. Kinetic studies have been performed in batch systems with 40 μM As(III) and 0.05 g L−1 TiO2. Photocatalytic oxidation of As(III) in the presence of N-doped TiO2 annealed at 500 °C was complete within 4 hours of irradiation at λ > 435 nm. The presence of titanium oxynitride (or similar surface moieties) is linked with the good photooxidation catalytic activity toward As(III). Zeta potentials of the N-doped TiO2 showed negative shifts at low pH values, which became more pronounced when measured after photocatalytic testing, suggesting at least partial retention of As(V). First principles DFT calculations were also carried out for a model anatase (101) surface, in both the pristine and N-doped condition. Dissociation of water is energetically favoured over the N-TiO2 surface. This surface activation mechanism provides the rationale for increased As(V) retention. Enhanced adsorptive properties in TiO2 would render it a more versatile remediation agent.
Introduction
Titanium dioxide (TiO2) is an excellent base material for photocatalysis due to its high oxidizing power, stability, non-toxic quality, and low price. Unfortunately, it suffers from one key limitation in the pristine form. Its rather high intrinsic band gap (∼3.2 eV) results in a photo-response restricted to the UV region of the electro-magnetic spectrum. In order to use solar irradiation or indoor lighting to drive photo-processes more efficiently over this powdered semiconductor, there have been various attempts to confer visible-light responsiveness by chemical modification. Transition metal (cationic) dopants have been extensively investigated.1–4 However, this approach is often double-edged due to the tendency to introduce localized d-levels deep in the band gap, which often serve as recombination centres for the photogenerated charge carriers. Recently, substitutional doping using anionic species, e.g., B, C, N or S, to replace lattice oxygen, has been a more promising alternative approach,5–8 inspired by the first report of high visible-light-driven activity for N-doped TiO2 by Asahi et al.9 Since then, a great deal of attention has been paid to this form of chemical modification. Nitrogen-doped TiO2 (denoted N-TiO2 hereafter) has been produced either by treating pre-formed TiO2 or by direct wet chemical (hydrothermal) synthesis starting from soluble Ti complexes. In the former methodology, TiO2 is thermally treated with NH3 gas,9,10 or sputtered with nitrogen gas.9,11 In the latter, N-TiO2 is obtained by solution chemical syntheses,7,12 followed by calcination of the dried precipitate. Elsewhere, a method using TiCl4 with O2 and NH3 as gas precursors to fabricate N-TiO2 by atmospheric pressure chemical vapor deposition (APCVD) has also been reported.13The effectiveness of doping TiO2 with N as a sensitizer for visible-light response has now been confirmed in a variety of studies,14–17 and attempts to extend its performance by further modification have recently been reviewed.18 It has been reported that visible-light-driven N-TiO2 photocatalysts are able to bleach methylene blue (MB),9,19,20 to oxidatively degrade compounds such as 2-propanol,10,21 acetone vapour,7 stearic acid,22 and even azo dyes.23 However, its potential for removal of typical aqueous inorganic contaminants is little explored, although promising results for As(III) photo-oxidation have been reported, in which superior adsorptive properties attributed to doping were especially notable.24
Arsenic occurs (in its inorganic form) as oxyanions of trivalent arsenite, As(III), or pentavalent arsenate, As(V), in the aquatic environment. The distribution between As(III) and As(V) in water depends on the redox potential and pH. Under typical anoxic groundwater conditions at a pH of ∼7, As(III) is the predominant form of arsenic, which is more toxic and mobile than As(V), while in oxic groundwater, As(V) is dominant. Arsenite has low affinity to mineral surfaces, while arsenate adsorbs easily to them. The most common method of arsenic removal from water currently is coagulation with iron salts and alum, followed by microfiltration. However, the oxidation of As(III) to As(V) is needed to achieve an effective removal of arsenic from water through adsorption of As(V) onto various metal oxyhydroxides. The development of more robust As(III) oxidation processes to meet the new stringent arsenic drinking water standards is required to overcome the disadvantages of presently known As(III) oxidation methods.
The TiO2 sample of interest is Degussa P25, a de facto “standard” in photocatalysis, whose superior charge separation properties are linked to its intra-particle multi-phase structure.25,26 In the present study, the nitrogen-doped variant is obtained from simple annealing in ammonia vapour. SEM is used to show that some aggregation occurs, but with little loss in surface area, as measured by N2 physisorption. The electronic states (N 1s and O 1s) of the doped semiconductor are investigated by X-ray photoelectron spectroscopy to verify the doping type as substitutional, and to shed light on the local bonding configuration. The obtained N-TiO2 is further tested in the photocatalytic oxidation of aqueous As(III). First principles DFT calculations for the most stable and abundant (anatase 101) surface in Degussa P25 TiO2 show the effect of doping with N.
Experimental section
Preparation of photocatalysts
P25 TiO2 (ca. 70% anatase, 30% rutile; BET area ca. 55 m2 g−1) was kindly supplied by Degussa Co. The TiO2 powders were annealed under pure NH3 flow at 400, 500, and 600 °C for 3 hours. The discharged products were subjected to manual grinding before further use.Characterization of photocatalysts
The as-modified powders were characterized by X-ray diffraction (Shimadzu XRD-6000 operating with a Cu Kα radiation source filtered with a graphite monochromator at λ = 1.5406 Å), X-ray photoelectron spectroscopy (Kratos Analytical Ltd., UK), and diffuse reflectance UV-visible absorption spectroscopy (Jasco V-550 UV/Vis spectrophotometer). All XPS data presented here were acquired using Al Kα X-rays (1486.6 eV) at a constant dwell time of 100 ms and a pass energy of 40 eV. The binding energies of Ti 2p, N 1s, and O 1s peaks were calibrated with respect to the C 1s peak at 284.6 eV due to adventitious hydrocarbon contamination. A small portion of the powders was diluted in ethanol and ultrasonically dispersed, followed by examination using scanning electron microscopy (SEM, Jeol Co., Japan, Model JSM-5600) to reveal the size and the shape of individual particles. Specific surface area and pore diameter of powders were measured by N2 physisorption at 77 K in a Micromeritics ASAP 2010 analyzer.Photocatalytic activity
Photocatalytic oxidation tests on As(III) were made in a Teflon cylindrical reactor of 200 mL capacity. For a typical experiment, access to the reactants by ambient air was assured via a pre-drilled sampling hole. TiO2 suspensions were dispersed in a standard ultrasonic cleaning bath for 10 minutes and then continuously stirred throughout the reaction. Dilute HCl and NaOH were used to adjust the pH of the suspension. Testing was done under acidic conditions (pH = 2). For experiments conducted under N2, the TiO2 suspension was sparged with N2 for 20 minutes prior to the experiment and maintained under N2 throughout the experiment. The dissolved oxygen level in these experiments was monitored by a Hanna Instruments Model HI 9146 dissolved oxygen probe. As(III) was added (as NaAsO2) at the desired concentration, and the suspension was stirred in the dark for 30 minutes to allow for an adsorption–desorption equilibrium to be established. Aliquots of the irradiation solutions were withdrawn periodically and passed through 0.45 μm syringe filters before analysis. Arsenate(V) concentrations were determined by UV-vis spectrophotometry (Shimadzu UV-1601) using the molybdenum blue method.27The light source was an ozone-free 450 W xenon arc lamp (Oriel) equipped with a liquid water filter to remove IR radiation. The light flux measured by a radiometer (UVItec RX-003) at 365 nm was 1.56 mW cm−2. For the wavelength selection, Oriel cut-off filters (λ >309 nm and λ > 435 nm) were used.
Zeta potential
Zeta potential measurements were made on pristine P25 and N-TiO2 (annealed at 500 °C) at a dispersion concentration of 0.1 g L−1 using a Coulter DELSA 440SX system. Both pH and ionic strength were investigated at three initial values: pH = 3, 5.5 and 9; and concentrations of 0.001, 0.01 and 0.1 M. Analytical grade sodium hydroxide, perchloric acid and sodium perchlorate were used for this purpose. The same measurements were also performed after light exposure from a 15 W Hg light source (Philips F15T8), positioned 5.5 cm above the surface of the TiO2 dispersions. This resulted in optical power densities in the range 930–1006 μW cm−2. Fluxes were determined at specific wavelengths using a UDT 80X Opt-o-meter with appropriate interference filters. At 372 nm these ranged from 47.7 to 54.1 μW cm−2; and at 353 nm from 2.28 to 2.60 μW cm−2. Thus, the majority of the lamp output was in the visible region. Dispersions were irradiated for three hours and allowed to re-equilibrate for 24 hours, after which the zeta potentials were determined. When studying the influence of As(III) oxidation on zeta potential, 80 μM NaAsO2 was added to the 0.1 g L−1 TiO2 dispersions. The irradiations and zeta potential measurements were performed likewise.Computational details
The (101) surface of anatase was modeled with a slab thickness of ca. 8.5 Å and an intervening vacuum layer of 6.6 Å. The surface unit cell was 1 × 2 and the simulation cell dimensions were 15.14, 7.59, and 10.24 Å. This cell contains 24 Ti and 44 O atoms. All calculations were done with the VASP code (version 4.6.36). PAW pseudo-potentials were used for all the atoms28,29 and the plane wave energy cut-off was 250 eV. A 1 × 2 × 2 Monkhorst–Pack k-point set was used and no symmetry constraints were used in the calculations. The exchange and correlation was approximated with the PW91 model and a spin-unrestricted formalism was used.To test the computational scheme, the bulk lattice parameters were optimized using the anatase unit cell30 and 6 × 6 × 6 Monkhorst–Pack k-points. The a and b parameters were optimized together and c separately. The minimum lattice parameters were a = b = 3.82 Å (experimental = 3.7842 Å30) and c = 9.78 Å (experimental = 9.5146 Å). The calculated lattice parameters were in very good agreement with experiments and independently reported computations.31
The clean (101) anatase surface was studied and relatively small atomic relaxations were observed. On comparing the energy difference between the ideal and relaxed surface, as reported in ref. 31, the relaxation energy was found to be identical (0.79 J m−²), thereby providing a high degree of confidence to this modeling work.
Results and discussion
1. Sample characterization
Degussa P25 TiO2 is a white powder in the pristine (as-supplied) form. However, after annealing in NH3 at 400, 500, and 600 °C, samples assumed pale yellow to greenish-yellow hues. X-Ray diffraction (XRD) analysis (not shown) verified that the samples did not undergo phase transformation or any significant redistribution between anatase and rutile. Just as in the virgin state, nitrogen-doped P25 (N-TiO2) remained predominantly anatase (over rutile) in an approximate ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, consistent with independent reports for similar annealing temperatures.19,21,32Fig. 1a and b show the morphology of P25 and its N-doped variant obtained after annealing at 600 °C. Both powders were of near-spherical shape, but the doped samples had a mean particle diameter of 0.45–0.73 μm, somewhat larger than the value for P25 (d ≈ 0.34 μm). It was found that the BET (specific surface) area also decreased slightly with an increase in the annealing temperature, as shown in Table 1. One common feature was the presence of agglomerates in the doped samples (Fig. 1c), consistent with the trend that a higher average pore diameter is often associated with agglomeration.33
:1, consistent with independent reports for similar annealing temperatures.19,21,32Fig. 1a and b show the morphology of P25 and its N-doped variant obtained after annealing at 600 °C. Both powders were of near-spherical shape, but the doped samples had a mean particle diameter of 0.45–0.73 μm, somewhat larger than the value for P25 (d ≈ 0.34 μm). It was found that the BET (specific surface) area also decreased slightly with an increase in the annealing temperature, as shown in Table 1. One common feature was the presence of agglomerates in the doped samples (Fig. 1c), consistent with the trend that a higher average pore diameter is often associated with agglomeration.33
 | ||
| Fig. 1 SEM images of (a) P25 TiO2, (b) N-TiO2 annealed at 600 °C, and (c) agglomerates of N-TiO2 samples (600 °C). | ||
The UV-Vis diffuse reflectance spectra in Fig. 2 revealed that nitrogen-doped TiO2 powders have new absorption features in the visible light region from ∼420 to 520 nm, whereas P25 does not absorb in this spectral region. Visible light absorption also appears to develop with increasing annealing temperature. The more pronounced absorption tail for the N-doped samples extending towards the near-IR (Fig. 2) is reminiscent of Ti3+ formation,53 but there is no corroborative XPS signal (vide infra), so its origin is unclear. It is also noteworthy that the absorption tail is only significant for the 500 °C sample (also the best photocatalyst toward As(III) oxidation, vide infra) and that generally the absorption tails herein are much smaller than for example those found by Balcerski et al.17 for oxidized TiN, those measured for nitrogen doped TiO2 films prepared under plasma processing by Pulsipher et al.34 or N-doped nano-titanias and titanium nitride samples prepared in an ammonia/argon atmosphere under conditions similar to the current study.38
 | ||
| Fig. 2 UV-Vis absorption spectra of N-doped and Degussa P25 TiO2. | ||
For the chemical identification of the valence states of N, O and Ti, N-TiO2 and P25 samples were examined by XPS. Fig. 3a shows the XPS spectra in the Ti 2p electron binding energy region. For P25, peaks at 458.7 and 464.5 eV correspond to 2p3/2 and 2p1/2 core levels, respectively, of Ti(IV) bound to oxygen. These are in agreement with the published literature values.35 It should be noted that binding energies of the Ti 2p core levels shift to lower energies upon N-doping. This suggests that the chemical properties of the TiO2 powder have been modified by the nitrogen atoms introduced into the lattice. Regardless of the type of doping of the nitrogen atoms (i.e., substitutional or interstitial), the effect of doping is to render the Ti ions more electronegative.
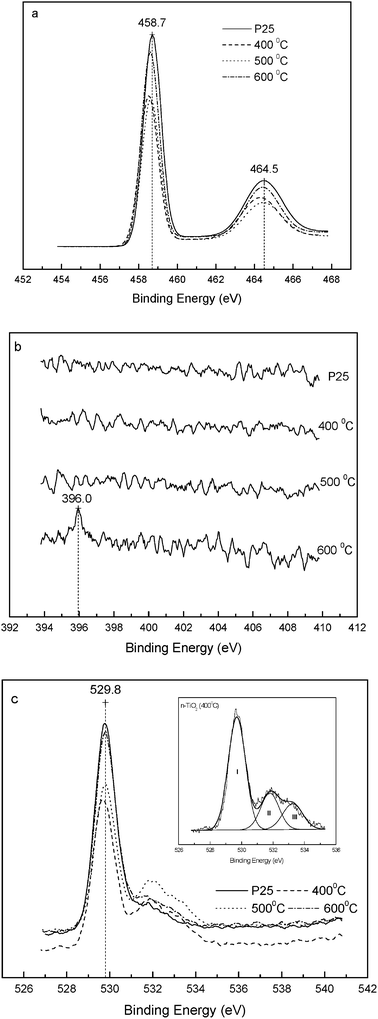 | ||
| Fig. 3 XPS spectra from P25 and N-TiO2 samples annealed at 400, 500, and 600 °C. (a) Ti 2p, (b) N 1s, and (c) O 1s, inset: fitting curves of N-TiO2 (400 °C). | ||
XPS showed the clear absence of a N 1s peak in P25 as expected. However, it was also absent from the two doped samples annealed at 400 and 500 °C, probably due to the low nitrogen content.36,37 Moreover, it is often observed that the N 1s peaks are smaller than the O 1s peaks for the same samples.38 For N-TiO2 annealed at 600 °C a weak peak at 396 eV characteristic of N 1s was observed (Fig. 3b). The N 1s XPS results are consistent with the observed UV-VIS absorption spectra (Fig. 2), wherein the 600 °C annealed sample had a much higher visible light absorbance at wavelengths of 420 to 500 nm as compared to the other two lower temperature samples. Ar+ sputtering on the surface of the sample to expose the subsurface region for XPS analysis21,37,39 was not used here due to concerns of induced chemical change.39 The single peak of N 1s at 396 eV demonstrates the presence of a Ti–N nitridic N atom within the TiO2 lattice.40 N introduced in the form of chemisorbed NHx species would be expected to show a N 1s peak above 398.6 eV.41 Similarly, the presence of the N–O linkage could also be ruled out, since N 1s binding energies in NOx species were found to be above 399 eV.42–44 Any interaction between N and O would result in a less negative charge on nitrogen, as compared to a nitridic nitrogen and hence be expected to cause a shift toward a higher binding energy. Location of the N 1s core level at 396 eV also rules out interstitial N-doping. Thus, the nitrogen atoms introduced into the as-prepared N-TiO2 are almost certainly substitutionally doped into the oxygen sites in P25 TiO2 and form an N–Ti–O bond. A similar conclusion has been reached by Miyauchi et al.21 and Sathish et al.19
The O 1s peaks are shown in Fig. 3c. The major oxygen peak in all samples is centered at 529.8 eV, which may be assigned to O2− bound to Ti(IV).40 It should be noted that the peak in the P25 sample is accompanied with a small shoulder at about 531.5 eV (Table 2). This may be due to adsorbed water from the surrounding environment. However, compared to P25, the binding energy of the shoulder in N-TiO2 is shifted to 531.8 eV with the appearance of a minor shoulder at about 533.2 eV (inset in Fig. 3c). The O 1s peak for chemically modified titanium dioxide samples at around 532 eV is typically assigned to titanium oxynitride.34,38 As mentioned above, the O 1s peaks are normally larger than the N 1s peaks for the same sample due to the higher XPS elemental sensitivity toward O 1s as compared to N 1s, and from Fig. 3c and Table 2, it is clear that the nitrogen has been incorporated to all the samples from 400 °C to 600 °C as evidenced by the sizable peaks at around 532 eV and 533 eV as compared to Degussa P25. It is also noteworthy that the 500 °C sample with the largest contribution from 532 eV (i.e., titanium oxynitride) to the total oxygen peak area is the most photocatalytically active (vide infra). The O/Ti ratio calculated by integration of the O 1s and Ti 2p bands (corrected for the respective elemental sensitivity factors) is higher than that of stoichiometric TiO2. This can be attributed to the surface excess of oxygen.35 It is generally believed that the adsorptivity of TiO2 decreases after high temperature calcination due to the decrease in the specific surface area.45 However, the O 1s XPS features observed here for the N-TiO2 samples annealed at 400 and 500 °C indicate that they have a stronger ability to adsorb oxygen than P25. Formal charge balance requires that oxygen vacancies are also created by N-doping, and these interact strongly with oxygen molecules.35,46,47 Thermal treatment will result initially in oxygen deficiency on the TiO2 surface, but the density of bulk oxygen vacancies would also be expected to increase with increasing level of N-doping, which in turn increases with the annealing temperature.7,11 The lower surface O excess in the sample annealed at 600 °C may be due to redistribution of predominantly surface vacancies into the bulk. The fraction of each O state listed in Table 2 was determined from the ratio of areas obtained by deconvolution of their respective contributions to the overall spectra (Fig. 3c, inset). It is not clear what species of oxygen are adsorbed. The superoxide ion, O2−, the peroxide ion, O22−, and “on-top” O2− are all possible adsorbates.46 The electron density on each oxygen atom of these species is lower than that of lattice O (O coordinated to two Ti neighbors), thus giving rise to a shift in the O 1s electron core level to higher binding energy (see Table 2).
| Catalysts | I | II | III | O/Ti | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BEa/eV | FWHMb/eV | Area (%) | BE/eV | FWHM/eV | Area (%) | BE/eV | FWHM/eV | Area (%) | ||
| a Binding energy. b Full width at half of the maximum height of peaks. | ||||||||||
| Degussa P25 | 529.8 | 1.07 | 85.0 | 531.5 | 1.77 | 15.1 | — | — | — | 2.4 |
| N-TiO2 (400 °C) | 529.8 | 1.29 | 62.2 | 531.8 | 1.37 | 21.4 | 533.2 | 1.50 | 16.5 | 3.2 |
| N-TiO2 (500 °C) | 529.8 | 1.16 | 62.5 | 531.9 | 1.66 | 30.2 | 533.2 | 1.14 | 7.3 | 3.2 |
| N-TiO2 (600 °C) | 529.8 | 1.20 | 80.1 | 531.8 | 1.62 | 14.4 | 532.8 | 1.50 | 5.5 | 2.5 |
Zeta potentials are shown as a function of pH (at different ionic strengths), for P25 and N-doped TiO2 annealed at 500 °C in Fig. 4a and b, respectively, for both dark and UV-pre-irradiated samples. At the lowest pH investigated, values for the P25 control ranged from +20 to +40 mV, whereas the corresponding values for the N-doped sample were somewhat lower, ranging from roughly +10 to +30 mV. At pH 5, a similar trend was evident, but under alkaline conditions, any differences were obscured by the spread of final pH values. This tends to support the earlier claim by Miyauchi et al.21 that N-doping lowers zeta potentials at acidic pH. Differences in the actual potentials probably derive from the different methods employed and sample types, i.e, particulates vs. films.48 The effect of pre-irradiation by UV+VIS light did not show any systematic trends for either sample.
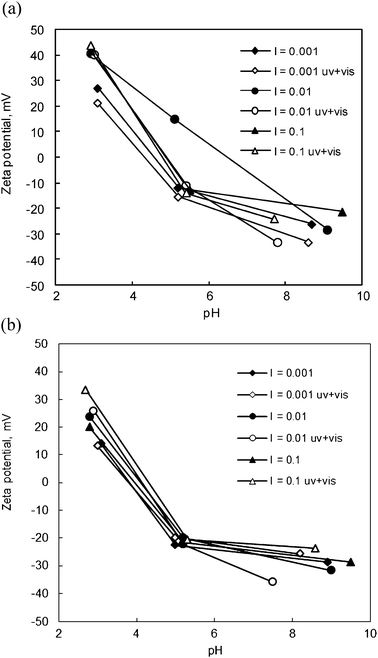 | ||
| Fig. 4 Zeta potentials of the (a) Degussa P25 and (b) N-doped TiO2 (annealed at 500 °C) as a function of pH. I refers to ionic strength in units of M, and uv + vis for irradiated samples. | ||
2. As(III) oxidation on the surface of nitrogen doped TiO2
 | ||
| Fig. 5 Growth of As(V) in the presence of N-TiO2 (vs. P25) under (a) λ > 435 nm; (b) λ > 309 nm irradiation. The initial concentration of As(III) was 40 μM, the photocatalyst dosage was 0.05 g L−1 | ||
The N-TiO2 powders exhibited a higher activity than the undoped P25 control, the one annealed at 500 °C being the most active of the three. It should be noted that the same sample also contained the highest level of surface O, as measured in terms of the O 1s XPS peak intensity at 531.9 eV (see Table 2). This peak has been identified elsewhere (see, e.g., Rogers et al.,49 Campbell50) as the superoxide ion, O2−, which is an efficient electron scavenger. Furthermore, Ferguson et al.51 and Lee and Choi52 have reported that O2− plays a major role in the TiO2 photocatalyzed oxidation of As(III) to As(V). However, according to Fig. 2, the N-TiO2 sample annealed at 600 °C was the strongest absorber of visible light, so its lower photoactivity must be associated with overriding compositional and/or textural changes (other than sintering) induced by annealing. Consistent with this deduction, there is ample evidence in the literature to show that annealing generally results in a loss of photocatalytic activity in N-TiO2.10,19,21 One possibility is that, assuming the N atom is genuinely “nitridic” in the sense of carrying formally a triple-negative charge, the doping process introduces O vacancies to maintain charge neutrality according to the formula TiO2−3xN2x. An increase in O vacancies is potentially deleterious, as these are known to be efficient centres for electron/hole recombination.10 However, in view of the fact that the main absorption band is still located below 500 nm, this view does not seem tenable, because the associated optical absorption (due to excitation of a Ti3+ centre) is known to peak in the near infrared.53,54 No evidence is seen for this by diffuse reflectance UV-Vis. Although Di Valentin et al.15 have rationalized its possible absence as due to charge transfer from Ti3+ to bulk N (to form a diamagnetic N− centre), they also show that the formation of O vacancies only becomes energetically favourable during heating in vacuo, conditions not employed in this work. Furthermore, XPS has already shown a surface O excess in the most active N-TiO2 photocatalyst (i.e., the one prepared at 500 °C). With regard to compositional changes, the more severe (600 °C) thermal treatment will promote ionic diffusion and presumably result in a more homogeneous (bulk) distribution of dopant N. This would suggest that the surface most favourable for photo-activity is N-rich, a state that must inevitably result from the preparation method utilized here. What may be special about the surface N atom with respect to (photo)-catalytic functionality is considered in the modeling section (vide infra).
Two control experiments under dark conditions, also shown in Fig. 5a, were performed on P25 and N-TiO2 annealed at 500 °C. Curiously, although the rate of oxidation in both was very slow as compared to that under visible light, it was still measurable, suggesting the occurrence of a weak sacrificial (non-catalytic) reduction process based on As(III). Under “UV + vis” radiation (λ > 309 nm) the photocatalytic activity of the N-TiO2 samples were initially somewhat lower than pristine P25 TiO2, as shown in Fig. 5b. However, the reaction appeared to be complete for all samples within 30 minutes. Comparison of the data from Fig. 5a (visible-only) and Fig. 5b (UV + vis) suggests that, for the N-TiO2, visible-light-driven photo-oxidation was roughly a factor of 5 lower than under full (UV + vis) irradiation. It can obviously be inferred that the “UV-only” response had deteriorated for the doped samples, otherwise they should have been superior to the undoped control. As shown in Fig. 2, the absorbance of N-TiO2 remains comparable to that of P25 in the UV region. Once again, this can be taken as evidence that although the annealing process develops the desired visible absorption, it simultaneously induces deleterious effects, such that a working compromise must be reached. While the known issue of aggregation per se does not appear to affect surface areas significantly, other desirable surface properties are evidently lost. Similar observations reported elsewhere were attributed to a loss of crystallinity.17 Thus, in order to produce a visible-light-sensitized TiO2 that is superior under all (and especially solar-) light conditions to the undoped analogue, greater insights are needed into the physical chemistry of the doping process and the precise catalytic role of the dopant employed.
To address the role of surface excess of oxygen in the photocatalytic oxidation of As(III) by TiO2, experiments were also carried out under N2 purging, as shown in Fig. 6. Under air-equilibrated conditions, dissolved oxygen was sufficient (the dissolved oxygen was measured to be at around 6.0−6.5 mg L−1) for quite a rapid photo-oxidation in the presence of P25 and N-TiO2. However, under continuous N2 purge of 30 mL min−1 (where the dissolved oxygen was measured to be less than 0.1 mg L−1), the relative decrease in the rate over P25 was much greater than that over N-TiO2. This observation reinforces the view from XPS that there is a stronger interaction between O2 and N-TiO2. The more sustained activity in the doped sample can be attributed to the higher level of pre-sorbed oxygen. N-TiO2 may even have the ability to capture and utilize any trace O2 contaminant under N2 purging, which augurs well for applications under oxygen-deficient conditions.
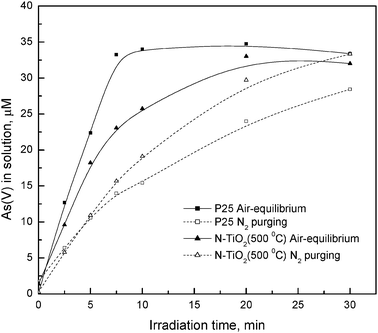 | ||
| Fig. 6 Effect of oxygen on the photocatalytic oxidation of As(III). The initial concentration of As(III) was 40 μM, the photocatalyst dosage was 0.05 g L−1, and the incident light λ > 309 nm. | ||
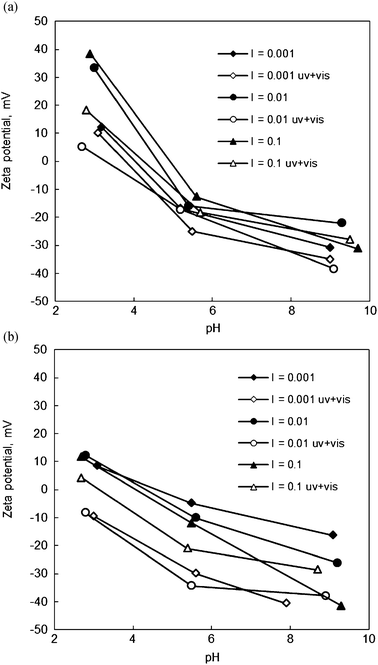 | ||
| Fig. 7 Influence of photocatalytic oxidation of As(III) on the final zeta potentials of (a) Degussa P25 TiO2 and (b) N-doped TiO2 (annealed at 500 °C) as a function of initial pH. I refers to ionic strength in units of M, and uv + vis for irradiated samples. | ||
In separate experiments, the pH decrease by the increased acidity of the oxidation product As(V) and likely by the consumption of the hydroxide ion in the oxidation reaction was observed as a lowered final pH of the suspension in the zeta potential measurements: ΔpH −0.1, −1.5 and −3.2 for the pH levels 3, 5.5 and 10, respectively.
 | ||
| Fig. 8 Model of the anatase (101) surface: red spheres = O, green spheres = Ti. | ||
Water dissociation on the N doped anatase (101) surface was studied by comparing the energies for the following 3 configurations:
(a) 2 Ti–OH groups—1 OH on Ti, 1 H on Os (dissociated);
(b) 1 Ti–OH group, 1 Ti–NH group—1 OH on Ti, 1 H on Ns (dissociated, see Fig. 9);
 | ||
| Fig. 9 Model of the anatase (101) surface with NH and OH groups: red spheres = O, green spheres = Ti, blue sphere = N, grey spheres = H. Note that the modifications are added to Fig. 8 for ease of representation. However, the calculations were performed with a smaller unit cell. | ||
(c) 1 Ti–OH2 group—coordinated molecular water (non-dissociated control),
where Os and Ns denote surface oxygen and surface nitrogen, respectively.
The reaction energy difference between cases (c) and (a) is +2.45 kcal mol−1, i.e., slightly in favour of molecular adsorption. In sharp contrast, the energy difference between cases (c) and (b) is −16.2 kcal mol−1, i.e., strongly in favour of dissociation. The first reaction can be compared to independent results for the rutile (101) surface, where a very similar reaction energy of +2.54 kcal mol−1 was obtained,58 or to the anatase (001) surface, where the reaction energy was −17 kcal mol−1.59 Clearly the dissociation energy varies a lot from surface to surface, but our calculations demonstrate that the surface N favors the dissociation significantly.
The surface charge is most likely related to the proton abstraction from the surface resulting in a negative O- or N- group on the surface. Proton abstraction from the hydrated N-doped anatase (101) surface was studied. According to these calculations, the proton is much more readily abstracted from the H–Os or HO–Ti group than from the H–Ns group. The energy difference was –20.4 kcal mol−1 for HOs and –11.7 kcal mol−1 for HO–Ti. This result is in contradiction with the model proposed by Miyauchi et al.21 They suggest that the H–Ns bond should be weaker than the H–O bond and deprotonate more easily. Our calculations do not support this model, thus an alternative explanation is needed to rationalize the observed shift to negative zeta potentials observed in Fig. 4.
The foregoing calculations all reinforce the view that substitutional N at the anatase (101) surface is invariably protonated on exposure to water vapour. Not only is dissociative adsorption favoured in the vicinity of Ns, but deprotonation of the resulting H–Ns surface species is strongly disfavoured relative to that of a hydroxyl (HOs) group.
Of the two aforementioned OH groups, the HO–Ti is more relevant to the proposed model, since it is formed from water dissociation in the vicinity of Ns. It can be safely concluded that there is a HO–Ti group near the N. Now, if this OH group is acidic (i.e., it will easily deprotonate) then one can explain the more negative zeta potential.
The proton dissociation energy in the HO–Ti group is 134 kcal mol−1. This can be compared to the dissociation energies of HCl and HF, viz., 103 and 135 kcal mol−1, respectively.60 HCl is a strong Brønsted acid and it dissociates fully in water, whereas HF is a rather weak acid, with a pKa = 3.20.60 The acidity of the OH group cannot be determined without including the overlying aqueous phase in the calculations, but one can exclude the acidity of the NH group and the HO–Ti probably behaves as a rather strong acid.
To summarize, a model for the surface of the N doped TiO2 surface has been proposed in which the substitutional nitrogen will cause a water molecule to dissociate. The nitrogen is protonated and an OH group is attached to a nearby Ti atom. This OH group is very likely acidic and it will deprotonate easily leaving a negatively charged O–Ti at the surface. The model assumes that the water is not significantly dissociated on the undoped TiO2 surface since the forming HOs group is more acidic than the HO–Ti group. Furthermore, Tilocca and Selloni61 have reported the anatase (101) surface to be ineffective in water dissociation unless a significant density of surface O vacancies is present. Finally, at the molecular level, the water–TiO2 interphase is very complex and a detailed study of it would require several demanding calculations beyond the scope of the current study. One particular aim here was to see whether the predictions from the calculations would rationalize and/or corroborate the observed trends in the zeta potentials between the pristine and N-TiO2. The zeta-potential is sensitive to surface charge and we showed that the N-doped TiO2 surface can dissociate water molecules and the Ti bound OH group is rather acidic.
In conclusion, doping Degussa P25 TiO2 with nitrogen confers good visible activity in photo-oxidation of As(III). However, the doping process also suppresses UV photo-activity relative to pristine P25, and requires further development. The most active N-TiO2 shows a surface excess in oxygen by XPS, attributed to photosorbed anionic oxidant(s). This provides a rationale for its better photo-oxidative properties under oxygen-deficient conditions as compared to pristine P25. Modelling studies have revealed that N-doping of the anatase (101) surface favours dissociative adsorption of water with the formation of a N–H bond stable against deprotonation. Zeta potential measurements suggest that it may also promote the retention of the oxidized product, As(V), at the TiO2 surface, possibly by activation of the anatase (101) surface for coverage by (exchangeable) OH groups. This combination of features is beneficial in a photocatalyst and encourages further studies of N-doped TiO2 in the field of environmental remediation.
Appendix
Estimation of adsorption capacity of TiO2 in relation to total As concentration
A square nanometre of the titanium dioxide used in these experiments (anatase:rutile of 3:1) includes around 10 OH groups in an aqueous environment,55 although lower values for some other types of TiO2 have also been reported. Since the specific surface area was almost invariant at ∼50 m2 g−1 (Table 1), a dispersion of 0.1 g l−1 exposes ∼5 m2 of surface in one litre of aqueous suspension. Then the total number of surface OH groups, x, can be obtained from the simple relation: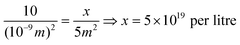
Assuming an adsorption stoichiometry of 1 OH:1 As(V) complex, this corresponds to the same number of adsorbed arsenic species, or 8.3 × 10−5 moles (83 μmol) of surface adsorption sites in one litre. By coincidence, this is close to the initial arsenic(III) concentration in these experiments.
Acknowledgements
The VALOKATA (funded by TEKES (Finland)) project is thanked for the financial support of the zeta potential study. Prof. Matti Weckström and Dr Kyösti Heimonen are warmly acknowledged for expert support in light intensity measurements, Lic. (Tech) Kukka Rämö for improving the manuscript graphically. We would like to thank the CSC—Scientific Computing (Espoo, Finland) for the computer time and KL would like to thank Dr Giorgio Lanzani for help related to the VASP code and PAW pseudo potentials.References
- E. Borgarello, J. Kiwi, M. Graetzel, E. Pelizzetti and M. Visca, J. Am. Chem. Soc., 1982, 104, 2996 CrossRef CAS.
- J. M. Herrmann, J. Disdier and P. Pichat, Chem. Phys. Lett., 1984, 108, 618 CrossRef CAS.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem. B, 1994, 98, 13669 CrossRef.
- X. Li and F. Li, Environ. Sci. Technol., 2001, 35, 2381 CrossRef CAS.
- S. Sakthivel and H. Kisch, Angew. Chem., Int. Ed., 2003, 42, 4908 CrossRef CAS.
- S. U. M. Khan, M. Al-Shahry and W. B. J. Ingler, Science, 2002, 297, 2243 CrossRef CAS.
- T. Ihara, M. Miyoshi, Y. Iriyama, O. Matsumoto and S. Sugihara, Appl. Catal., B, 2003, 42, 403 CrossRef CAS.
- T. Umebayashi, T. Yamaki, H. Itoh and K. Asai, Appl. Phys. Lett., 2002, 81, 454 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS.
- H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483 CrossRef CAS.
- T. Ihara, M. Miyoshi, M. Ando, S. Sugihara and Y. Iriyama, J. Mater. Sci., 2001, 36, 4201 CrossRef CAS.
- D. W. Bahnemann, A. Henglein, J. Lilie and L. Spanel, J. Phys. Chem., 1984, 88, 709 CrossRef CAS.
- Y. Guo, X. Zhang and G. Han, Mater. Sci. Eng., B, 2006, 135, 83 CrossRef CAS.
- T. L. Thompson and J. T. Yates, Jr., Chem. Rev., 2006, 106, 4428 CrossRef CAS.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44 CrossRef CAS.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS.
- W. Balcerski, S. Y. Ryu and M. R. Hoffmann, J. Phys. Chem. C, 2007, 111, 15357 CAS.
- J. Zhang, Y. Wu, M. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715 CAS.
- M. Sathish, B. Viswanathan, R. P. Viswanath and C. S. Gopinath, Chem. Mater., 2005, 17, 6349 CrossRef CAS.
- T. Matsumoto, N. Iyi, Y. Kaneko, K. Kitamura, S. Ishihara, Y. Takasu and Y. Murakami, Catal. Today, 2007, 120, 226 CrossRef CAS.
- M. Miyauchi, A. Ikezawa, H. Tobimatsu, H. Irie and K. Hashimoto, Phys. Chem. Chem. Phys., 2004, 6, 865 RSC.
- M. Maeda, T. Yamada and T. Watanabe, J. Electrochem. Soc., 2007, 154, 29 CrossRef.
- Y. Liu, X. Chen, J. Li and C. Burda, Chemosphere, 2005, 61, 11 CrossRef CAS.
- Q. Li, N. J. Easter and J. K. Shang, Environ. Sci. Technol., 2009, 43, 1534 CrossRef CAS.
- D. C. Hurum, K. A. Gray, K. Tijana and M. C. Thurnauer, J. Phys. Chem. B, 2005, 109, 977 CrossRef CAS.
- G. H. Li, S. Ciston, Z. V. Saponjic, L. Chen, N. M. Dimitrijevic, R. Tijana and K. A. Gray, J. Catal., 2008, 253, 105 CrossRef CAS.
- V. Lenoble, V. Deluchat, B. Serpaud and J.-C. Bollinger, Talanta, 2003, 61, 267 CrossRef CAS.
- P. Blöhl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Kresse and J. Joubert, Phys. Rev. B: Condens. Matter, 1999, 59, 1758 CrossRef CAS.
- M. Horn, C. F. Schwerdtfeger and E. P. Meagher, Z. Kristallogr., 1972, 136, 273 CrossRef CAS.
- M. Lazzeri, A. Vittadini and A. Selloni, Phys. Rev. B: Condens. Matter, 2001, 63, 155409 CrossRef.
- H. Irie, S. Washizuka, N. Yoshino and K. Hashimoto, Chem. Commun., 2003, 1298 RSC.
- D. Liu and J. Lin, J. Mater. Sci., 1999, 34, 1959 CrossRef CAS.
- D. J. V. Pulsipher, I. T. Martin and E. R. Fisher, ACS Appl. Mater. Interfaces, 2010, 2(6), 1743–1753 CAS.
- S. A. Bilmes, P. Mandelbaum, F. Alvarez and N. M. Victoria, J. Phys. Chem. B, 2000, 104, 9851 CrossRef CAS.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2004, 108, 10617 CrossRef CAS.
- M. Mrowetz, W. Balcerski, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2004, 108, 17269 CrossRef CAS.
- Z. Zhang, J. B. M. Goodall, D. J. Morgan, S. Browna, R. J. H. Clark, J. C. Knowles, N. J. Mordand, J. R. G. Evans, A. F. Carley, M. Bowker and J. A. Darr, J. Eur. Ceram. Soc., 2009, 29, 2343–2353 CrossRef CAS.
- O. Diwald, T. L. Thompson, T. Zubkov, E. G. Goralski, S. D. Walck and J. T. Yates, Jr., J. Phys. Chem. B, 2004, 108, 6004 CrossRef CAS.
- N. C. Saha and H. G. Tompkins, J. Appl. Phys., 1992, 72, 3072 CrossRef CAS.
- S. Souto and F. Alvarez, Appl. Phys. Lett., 1997, 70, 1539 CrossRef CAS.
- C. H. Cardinaud, G. Lemperier, M. C. Peignon and P. Y. Jouan, Appl. Surf. Sci., 1993, 68, 595 CrossRef.
- A. S. Vanini, J. P. Audouard and P. Marcus, Corros. Sci., 1994, 36, 1825 CrossRef.
- G. Latha, N. Rajendran and S. Rajeswari, J. Mater. Eng. Perform., 1997, 6, 743 CrossRef CAS.
- L. Jing, Z. Xu, X. Sun, J. Shang and W. Cai, Appl. Surf. Sci., 2001, 180, 308 CrossRef CAS.
- G. Lu, A. Linsenbigler and J. T. Yates, J. Chem. Phys., 1995, 102, 3005 CrossRef CAS.
- M. A. Henderson, W. S. Epling, C. L. Perkins, H. F. Peden and U. Diebold, J. Phys. Chem. B, 1999, 103, 5328 CrossRef CAS.
- A. Hoizumi, H. Sugihara, Y. Yokogawa, T. Kameyama and O. Takai, Colloids Surf., 2001, 182, 257 CrossRef.
- J. W. Rogers, Jr., N. D. Shinn, J. E. Schirber, E. L. Venturini, D. S. Ginley and B. Morosin, Phys. Rev. B: Condens. Matter, 1988, 38, 5021 CrossRef.
- C. Campbell, Surf. Sci., 1986, 173, L641 CrossRef CAS.
- M. A. Ferguson, M. R. Hoffmann and J. G. Hering, Environ. Sci. Technol., 2005, 39, 1880 CrossRef CAS.
- H. Lee and W. Choi, Environ. Sci. Technol., 2002, 36, 3872 CrossRef CAS.
- J. Highfield and M. Graetzel, J. Phys. Chem., 1988, 92, 464 CrossRef CAS.
- Z. Lin, A. Orlov, R. M. Lambert and M. C. Payne, J. Phys. Chem. B, 2005, 109, 20948 CrossRef CAS.
- C. H. Shin, G. Bugli and G. Djega-Mariadassou, J. Solid State Chem., 1991, 95, 145 CrossRef CAS.
- P. K. Dutta, A. K. Ray, V. K. Sharma and F. J. Millero, J. Colloid Interface Sci., 2004, 278, 270 CrossRef CAS.
- A. Vittadini, M. Casarin and A. Selloni, Theor. Chem. Acc., 2007, 117, 663–671 CrossRef CAS.
- H. Perron, et al. , Surf. Sci., 2007, 801, 518 CrossRef.
- R. Erdogan, O. Ozbek and I. Onal, Surf. Sci., 2010, 604, 1029 CrossRef CAS.
- P. Atkins and J. de Paula, Atkins' Physical Chemistry, Oxford University Press, 7th edn, 2002, Tables 14.3a and 9.1 Search PubMed.
- A. Tilocca and A. Selloni, J. Phys. Chem. B, 2004, 108, 4743 CrossRef CAS.
Footnote |
| † See Appendix 1. |
| This journal is © The Royal Society of Chemistry 2012 |