Ag nanoparticles decorated polyaniline nanofibers: synthesis, characterization, and applications toward catalytic reduction of 4-nitrophenol and electrochemical detection of H2O2 and glucose†
Received
10th November 2011
, Accepted 28th December 2011
First published on 5th January 2012
Abstract
Polyaniline nanofibers (PANINFs) have been facilely prepared by electrochemical polymerization of aniline monomers in acidic aqueous media without using any templates and surfactants. The subsequent treatment of such nanofibers with a AgNO3 aqueous solution leads to in situ chemical reduction of Ag+ on them to form Ag nanoparticles decorated PANINFs (AgNPs/PANINFs) nanocomposites. We investigated the catalytic activity and electrochemical properties of these nanocomposites. It is found that such nanocomposites exhibit excellent catalytic activity toward reduction of 4-nitrophenol to 4-aminophenol by NaBH4 and exhibit remarkable catalytic performance for H2O2 reduction. The enzymeless H2O2 sensor constructed using the nanocomposites shows a fast amperometric response time of less than 3 s. The linear range and detection limit are estimated to be from 0.1 mM to 60 mM (r = 0.998) and 1.7 μΜ at a signal-to-noise ratio of 3, respectively. We have fabricated a glucose biosensor by immobilizing glucose oxidase into the AgNPs/PANINFs-modified glassy carbon electrode for glucose detection. This sensor exhibits good response to glucose. The linear response range is estimated to be from 1 mM to 12 mM (r = 0.997) at −0.58 V. The detection limit is estimated to be 0.25 mM at a signal-to-noise ratio of 3.
1. Introduction
In recent years, electrically conducting polymers (CPs) have been paid considerable attention due to their combination of unique mechanical, optical, and electronic properties1 and their potential applications in many fields.2 Among the CPs, polyaniline (PANI) has been paid considerable attention due to its facile synthesis, environmental stability, tunable electrical and optical properties, and simple doping/dedoping chemistry.3 PANI has been widely used in a variety of applications such as batteries, molecular electronic devices, chemical sensors, electrodes for light-emitting diodes, capacitors and antistatic and anticorrosion coatings.4 Much effort has been put in synthesizing nanostructured PANI, especially one-dimensional (1D) nanostructures owing to their novel physical properties and potential application.5 PANI 1D nanostructures can be synthesized using hard and soft template methods. In the hard template method characteristic of synthesizing the desired material using aluminium oxide or track-etch membrane as a template, polymerization occurs within the pores of the membrane.6 However, a rather tedious post-synthesis process is often required to remove the template used. Additionally, the 1D structures preformed may be destroyed or form undesirable aggregates when released from the template. In the soft template method, the micelles, emulsions, liquid crystals, and surfactant gel have been used to synthesize PANI 1D nanostructures.7 However, this method suffers from that soft templates limit the range of chemicals that can be used. Moreover, up to date, some novel templateless methods have also been developed to shape PANI into nanofibers, such as electrospinning, mechanical stretching, interfacial polymerization, dilute polymerization, gamma or ultraviolet irradiation, ultrasonic irradiation, radiolysis, and nanofiber or nanoparticle seeding etc.8 On the other hand, much effort has been put into synthesis and characterization of metal nanoparticles (NPs) due to their particular optical, electronic, and catalytic properties. Functionalizing PANI 1D nanostructures with metal NPs can combine the properties of two nanomaterials, such as high conductivity and surface area of 1D nanomaterials and unique catalytic properties of metals, which may play an important role in a catalysis field like methanol oxidation.9 For example, Gao et al.10 and Hosseini and Momeni11 have reported that Ag/PANI nanocomposites exhibit high electrocatalytic activity toward reduction of dopamine and oxidation of hydrazine, respectively.11
In this study, we develop a new templateless and surfactantless route for the facile synthesis of PANI nanofibers (PANINFs), carried out by electrochemical polymerization of aniline monomers in acidic aqueous media without the use of any templates and surfactants. We further fabricated Ag nanoparticles decorated PANINFs (AgNPs/PANINFs) nanocomposites via in situ chemical reduction of Ag+ by PANINFs, carried out by treating PANINFs with a AgNO3 aqueous solution at room temperature. The nanocomposites were found to exhibit excellent catalytic activities for both reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by NaBH4 and electrochemical H2O2 reduction. An enzymeless H2O2 sensor was constructed using the nanocomposites as sensing interface. This sensor shows a fast amperometric response time of less than 3 s and has a linear range of 0.1–60 mM (r = 0.998) and a detection limit of 1.7 μΜ at a signal-to-noise ratio of 3. The glucose sensor fabricated by immobilizing glucose oxidase (GOD) into the AgNPs/PANINFs-modified glassy carbon electrode (GCE) shows good response to glucose and has a linear response range of 1–12 mM (r = 0.997) and a detection limit of 0.25 mM at a signal-to-noise ratio of 3.
2. Experimental
2.1 Materials
Aniline, hydrochloric acid (HCl), H2O2 (30 wt%), sodium citrate, glucose, Na2HPO4, and NaH2PO4 were purchased from Aladin Ltd. (Shanghai, China). 4-NP, sodium borohydride (NaBH4), glucose oxidase (GOD) and AgNO3 were purchased from Aldrich Chemical Comp. All chemicals were used as received without further purification. The water used throughout all experiments was purified through a Millipore system. Phosphate buffer saline (PBS) was prepared by mixing stock solutions of NaH2PO4 and Na2HPO4 and a fresh solution of H2O2 was prepared daily.
2.2 Preparation of PANINFs
The indium-tin oxide (ITO) coated glass (resistance 25 Ω sq−1), defined as the ITO electrode in this work, was pretreated using the following process. First, the ITO glass was sonicated in neat ethanol, soap solution, and water for ∼15 min each and then dried in air. The electropolymerization was performed on a CHI660D electrochemical workstation (Shanghai, China) in a three-electrode cell, with the use of a Ag/AgCl (saturated KCl) as the reference electrode, a platinum coil as the auxiliary electrode, and an ITO electrode (geometric area = 2 cm2) as the working electrode. PANINFs were fabricated on the ITO electrode by electropolymerization of 0.1 M aniline in 1 M HCl at an applied potential of 2.5 V vs. Ag/AgCl over 1800 s period. Then, the PANINFs-coated ITO electrode was transferred into a centrifuge tube and rinsed with 2 mL of doubly distilled water. The PANINFs completely fell off the ITO surface. After that, a 30 s sonication was used to obtain an aqueous dispersion of PANINFs.
2.3 Synthesis of AgNPs/PANINFs nanocomposites
In a typical experiment, 0.45 mL of the as-formed aqueous dispersion of PANINFs was added into 4.5 mL water. After that 0.05 mL of 0.5 M AgNO3 was added into the above mixture under stirring and the reaction mixture was kept at room temperature for 2 h. Then, the products were collected by centrifugation at 8000 rpm for 5 min and washed with distilled water several times, and then redispersed into 1 mL of water for further characterization and use.
2.4 Preparation of AgNPs
In brief, 0.45 mL of 0.5 M sodium citrate was added into 4.5 mL water, and then 0.05 mL of 0.5 M AgNO3 aqueous solution was added slowly into the above solution at room temperature under vigorous stirring. After that, 5 μL of 5 mM NaBH4 aqueous solution was added slowly into the solution under vigorous stirring.
2.5 Catalytic reduction of 4-NP experiments
2.7 mg of AgNPs/PANINFs composites was added to 2 mL of aqueous 4-NP solution (5 mM). Subsequently, the above solution was mixed with 2 mL fresh NaBH4 solution (0.16 M). The molar ratio of AgNP![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4-NP:BH4−1 in the catalytic reaction was about 1:20:640. The reaction was carried out at room temperature with continuous stirring. Parts of the mixture were taken out at different time intervals for the determination with UV-Vis absorption spectra.
:4-NP:BH4−1 in the catalytic reaction was about 1:20:640. The reaction was carried out at room temperature with continuous stirring. Parts of the mixture were taken out at different time intervals for the determination with UV-Vis absorption spectra.
2.6 Electrocatalytic experiments
The modified electrodes were prepared by a simple casting method. Prior to the surface coating, the GCE was polished with 1.0 and 0.3 μm alumina powder, respectively, and rinsed with doubly distilled water, followed by sonication in ethanol solution and doubly distilled water successively. Then, the electrode was allowed to dry in a stream of nitrogen. For the cyclic voltammetry experiment, 6 μL of the AgNPs/PANINFs nanocomposites dispersion was dropped on the clean surface of GCE and left to dry at room temperature. Finally, the AgNPs/PANINFs/GCE was obtained. For the control experiments, 6 μL of PANINFs dispersion was also dropped on the clean surface of GCE to obtain PANINFs/GCE. Next, for the experiments of glucose detection, the GOD/AgNPs/PANINFs/GCE was also prepared by the same method except for an extra dropping of 4 μL of 40 mg mL−1 GOD, and then the electrode was dried at 4 °C for 2 h. For current time experiment, 3 μL of chitosan (0.5%) solution was additionally cast on the surface of the above-modified GCE and dried before electrochemical experiments.
2.7 Instruments
Scanning electron microscopy (SEM) measurements were made on a XL30 ESEM FEG scanning electron microscope at an accelerating applied potential of 20 KV. The sample for SEM characterization was prepared by placing a drop of the dispersion on a bare ITO substrate and air-dried at room temperature. Transmission electron microscopy (TEM) measurements were made on a HITACHI H-8100 EM (Hitachi, Tokyo, Japan) with an accelerating voltage of 200 kV. The sample for TEM characterization was prepared by placing a drop of the dispersion on a carbon-coated copper grid and dried at room temperature. UV-visible absorption spectra were obtained on a UV-5800 Spectrophotometer. Electrochemical measurements are performed with a CHI 660D electrochemical analyzer (CH Instruments, Inc., Shanghai). A conventional three-electrode cell was used, including a GCE (geometric area = 0.07 cm2) as the working electrode, a Ag/AgCl (saturated KCl) electrode as the reference electrode, and platinum foil as the counter electrode. The potentials are measured with a Ag/AgCl electrode as the reference electrode. All the experiments were carried out at ambient temperature.
3. Results and discussion
3.1 SEM, TEM, and EDS characterization of PANINFs and AgNPs/PANINFs nanocomposites
Fig. 1a and b show typical SEM images of the products thus formed, indicating that they consist exclusively of a large quantity of fibers with a length of more than 2 μm and diameters in the range of 100–200 nm. The fibrillar morphology is further evidenced by TEM observation, as shown in Fig. 1c. The high magnification TEM image shown in Fig. 1d reveals that the nanofiber is assembled from smaller nanofibers. The chemical composition of these fibers was determined by the energy-dispersive spectrum (EDS), as shown in Fig. 1e. The presence of C and N elements shows that the nanofibers are PANI formed from aniline monomers by electropolymerization. The observation of the peak of the Cl element can be attributed to the fact that the electrochemical polymerization of aniline in HCl yields cationic polymer PANI due to the proton doping effect, the Cl− as counter-ions, however, will diffuse into the PANI nanostructures for charge compensation.12 The other peaks of Si, Na, Al, In, and Sn elements can be assigned to the ITO substrate used for SEM characterization. Fig. S1 (ESI†) shows a schematic to illustrate the chemical structure and electropolymerization process of PANI. It is of importance to point out that although conventional electrochemical polymerization of conjugated monomers generally produces many islands or forests of conjugated polymers on the ITO electrode,13 PANI nanofibers can be reproducibly obtained in our present study. Investigation of the effect of applied potential on the morphology of PANI further reveals that different applied potentials of 1.0 V and 1.5 V also lead to nanofibers, as shown in Fig. S2 (ESI†). However, the exact nanofiber formation mechanism is not completely understood at present time and requires further study.
 |
| | Fig. 1 (a) Low, (b) high magnification SEM images, (c) low, (d) high magnification TEM images and (e) the corresponding EDS of PANINFs thus formed. | |
Fig. 2a shows the low magnification TEM image of the products formed by treating PANINFs with a AgNO3 aqueous solution at room temperature, indicating that a large amount of nanoparticles are generated on PANINFs. The high magnification TEM image further reveals that most of the nanoparticles are spherical in shape and have diameters ranging from 20 to 100 nm, as shown in Fig. 2b. The chemical composition of the composites was determined by EDS, as shown in Fig. 2c. Compared to the EDS of pure PANINFs shown in Fig. 1e, an additional peak of the Ag element is observed, indicating that these particles are AgNPs. All these observations indicate the formation of AgNPs/PANINFs nanocomposites. The content of Ag in the composites was measured to be approximately 38.6% (by weight). Li et al. have reported that poly(o-phenylenediamine) (POPD) microparticles can effectively adsorb Ag+ ions due to that many amino and imino groups located adjacent to each other in the POPD chains provide abundant coordination sites for Ag+ ions.14 It was found that POPD has the ability to effectively reduce the as-absorbed Ag+ ions to form AgNPs. Such observation was further verified by the same group in an aromatic diamine polymers-based system.15 More recently, we have also demonstrated the in situ growth of AgNPs on POPD nanofibers and poly(m-phenylenediamine) structures.16 Similarly, we can attribute the spontaneous formation of AgNPs in our present study to that PANINFs can effectively adsorb Ag+ ions via coordination and then in situ reduce the as-absorbed Ag+ ions to form AgNPs. Note that the AgNPs/PANINFs composites can be well dispersed in water and acetone, as shown in Fig. S3 (ESI†).
 |
| | Fig. 2 (a) Low, (b) high magnification TEM images, and (c) the corresponding EDS of the as-prepared AgNPs/PANINFs composites. | |
We also investigated the influence of the relative amount of Ag+ on the formation of AgNPs/PANINFs. When the amount of Ag+ was increased up to 4-fold, AgNPs with diameters ranging from 5 to 20 nm were obtained as the main products, as shown in Fig. S4a and b (ESI†). When the amount of Ag+ was further increased up to 8-fold, smaller AgNPs with higher loading were obtained, as shown in Fig. S4c and d (ESI†). These observations indicate that both size and loading of AgNPs on PANINFs can be tuned by changing the amount of Ag+ used.
3.2 Catalytic reduction of 4-NP
It is known that 4-AP is very useful and important in many applications including analgesic and antipyretic drugs, photographic developer, corrosion inhibitor, anticorrosion lubricant, and so on.17 The reduction of 4-NP by NaBH4 in the presence of noble metal nanoparticles as catalysts has been largely studied for the efficient production of 4-AP. Therefore, the reduction of 4-NP to 4-AP with an excess amount of NaBH4 was used as a model system to quantitatively evaluate the catalytic activities of the resultant AgNPs/PANINFs nanocomposites (designated as catalysts). As shown in Fig. 3a, pure 4-NP shows a distinct spectral profile with an absorption maximum at 316 nm. In the absence of the AgNPs/PANINFs nanocomposites, the mixture of 4-NP and NaBH4 shows a strong adsorption peak at 400 nm, corresponding to the 4-NP ions under alkaline conditions. After AgNPs/PANINFs nanocomposites were added into the reaction system, the reduction process was monitored by measuring the time-dependent adsorption spectra of the reaction mixture solution. It was found that the absorption intensity of 4-NP at 400 nm decreases quickly with elapsed time, accompanied by the appearance of the new peak of 4-AP at 300 nm, indicating successful conversion of 4-NP to 4-AP. It should be noted that the reduction of 4-NP by NaBH4 was completed within 3 minutes upon the addition of AgNPs/PANINFs composites (Fig. 3b), with the observation of a fading and ultimate bleaching of the yellow-green color of the reaction mixture in aqueous solution. In contrast, the 4-NP absorption peak at 400 nm was almost unchanged and the 4-NP absorption peak at around 300 nm did not appear for 2 h in the presence of the PANINF and NaBH4, as shown in Fig. S5 (ESI†). We also investigated the effects of the PANINF support on the catalytic activity. A reaction time of 20 minutes was required to achieve the full reduction of 4-NP by NaBH4 using AgNPs alone as catalysts (Fig. 3c). Fig. S6 (ESI†) shows the TEM image of the citrate-stabilized AgNPs. Fig. S7 (ESI†) shows the TEM images of AgNPs/PANINFs composites after the reduction reaction, suggesting that NaBH4 has small influence on the AgNPs and the fibrous material. Due to the presence of large excess NaBH4 compared to 4-NP, the rate of reduction is independent of the concentration of NaBH4, and the reaction could be considered pseudo-first-order with respect to the concentration of 4-NP. Hence, ln(Ct/C0) versus time can be obtained based on the absorbance as the function of time, and good linear correlations are observed, as shown in Fig. 3d, suggesting that the reactions follow pseudo-first-order kinetics. Then, the kinetic reaction rate constants (defined as kapp) are estimated from the slopes of the linear relationship to be 21.39 × 10−3 s−1 and 3.28 × 10−3 s−1 for AgNPs/PANINF composites and AgNPs, respectively. These results clearly indicate that the AgNPs/PANINFs composites have higher catalytic activity for the reduction of 4-NP than the AgNPs, due to the PANINF support and may play an active part in the catalysis, yielding a synergistic effect. The synergistically enhanced catalytic activity may be explained as follows: it is expected that 4-NP can be adsorbed onto PANINFs via π–π stacking interactions as PANINF is π-rich in nature. Such adsorption provides a high concentration of 4-NP near to the AgNPs on PANINF, leading to highly efficient contact between them, as shown in Scheme 1a. In contrast, without a highly adsorbent PANINF support, 4-NP must collide with AgNPs by chance, and remains in contact for the catalysis to proceed. When this is not achieved, 4-NP will pass back into solution and can only react further when it collides with AgNPs again, as shown in Scheme 1b.18
![UV-vis absorption spectra of 4-NP (a) and time dependent absorption spectra for the catalytic reduction of 4-NP by NaBH4 in the presence of AgNPs/PANINFs nanocomposites (b) and AgNPs alone (c). (d) Plot of ln(Ct/C0) of 4-NP against time for the catalysts. Conditions: [4-NP] = 5 mM; [catalysts] = 0.675 mg mL−1; [NaBH4] = 160 mM.](/image/article/2012/CY/c2cy00454b/c2cy00454b-f3.gif) |
| | Fig. 3 UV-vis absorption spectra of 4-NP (a) and time dependent absorption spectra for the catalytic reduction of 4-NP by NaBH4 in the presence of AgNPs/PANINFs nanocomposites (b) and AgNPs alone (c). (d) Plot of ln(Ct/C0) of 4-NP against time for the catalysts. Conditions: [4-NP] = 5 mM; [catalysts] = 0.675 mg mL−1; [NaBH4] = 160 mM. | |
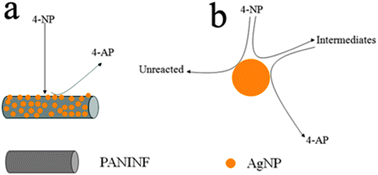 |
| | Scheme 1 A schematic diagram (not to scale) to illustrate the proposed mechanism of reduction of 4-NP to 4-AP using (a) AgNPs/PANINFs nanocomposites and (b) AgNPs alone as catalysts. | |
3.3 Enzymeless H2O2 sensing
To testify the sensing application of the resultant AgNPs/PANINFs, we designed an enzymeless H2O2 sensor by direct deposition of the AgNPs/PANINFs on a bare GCE surface. Fig. 4a shows the cyclic voltammograms (CVs) of bare GCE, PANINFs/GCE, AgNPs/PANINFs/GCE toward the reduction of H2O2 in 0.2 M PBS at pH 7.4. It is obviously seen that the responses of both the bare GCE and PANINFs/GCE toward the reduction of H2O2 are quite weak in the potential range between −0.8 to 0.2V. In contrast, the AgNPs/PANINFs/GCE exhibits a remarkable catalytic reduction current peak about 20 μA centered at −0.450 V vs. Ag/AgCl. It is also important to note that the AgNPs/PANINFs/GCE exhibits no electrochemical response in the absence of H2O2. All the above observations indicate that the AgNPs contained in the nanocomposites exhibit notable catalytic performance for H2O2 reduction and the PANINFs exhibit no catalytic activity. In addition, the relative standard deviation (RSD) of the amperometric response to 1 mM H2O2 was 3.2% for 8 successive measurements, indicating the good reproducibility of the AgNPs/PANINFs/GCE. The stability of the sensor is tested by measuring response currents of the electrode. After 7 days, only 5.6% signal of current was lost, indicating the good stability for H2O2 detection. Fig. 4b shows a typical current–time plot of the AgNPs/PANINFs/GCE in N2-saturated 0.2 M PBS buffer (pH: 7.4) on consecutive step change of H2O2 concentrations under optimized conditions. When an aliquot of H2O2 was dropped into the stirring PBS solution, the reduction current rose steeply to reach a stable value. The sensor could accomplish 97% of the steady state current with 3 s, indicating a fast amperometric response behavior. The inset in Fig. 4b shows the calibration curve of the sensor. The linear detection range is estimated to be from 0.1 mM to 60 mM (r = 0.998), and the detection limit is estimated to be 1.7 μΜ at a signal-to-noise ratio of 3.
 |
| | Fig. 4 (a) CVs of different electrodes in N2-saturated 0.2 M PBS at pH = 7.4 in the presence of 1 mM H2O2 (scan rate: 50 mV s−1). (b) Typical steady-state response of the AgNPs/PANINFs/GCE to successive injection of H2O2 into the stirred N2-saturated 0.2 M PBS at pH = 7.4. Inset: the corresponding calibration curve (applied potential: −0.3 V). | |
We further investigated the catalytic activity of AgNPs/PANINFs/GCE for H2O2 in the presence of interference species such as uric acid (UA) and dopamine (DA), as shown in Fig. S8 (ESI†). It is observed that the current response of UA and DA was much lower than that of H2O2, indicating that UA and DA had no interference on the detection of H2O2 at the studied concentration.
It is well known that diabetes mellitus is a worldwide public health problem and thus the detection of glucose in blood serum is particularly important.19 Based on the high electrocatalytic activity of AgNPs/PANINFs/GCE toward H2O2, a glucose sensor was further developed by immobilizing GOD into the AgNPs/PANINFs nanocomposites film on a GCE surface. The sensing mechanism is that GOD can selectively catalyze the oxidation of glucose in the presence of oxygen to form H2O2, which can be electrochemically detected.20Fig. 5a shows CVs of the GOD/AgNPs/PANINFs/GCE in PBS solution (pH: 7.4) in the presence of varied concentrations of glucose saturated with O2. The signal response of the GOD/AgNPs/PANINFs/GCE on glucose with different concentrations was recorded in the solution of 0.2 M PBS (pH: 7.4) via increasing glucose concentration step by step. When the concentration of glucose increases from 1 mM to 12 mM, the amount of H2O2 generated by the GOD catalyzing the oxidation of glucose increase quickly, and the current of H2O2 reduction peaks at about −0.58 V presents a continuous enhancement. Fig. 5b shows the catalytic current versus glucose concentration relationship. A good linear relationship is found between the catalytic current and glucose concentration at a range from 1 to 12 mM (r = 0.98). The detection limit of glucose is 0.25 mM. The relative standard deviation (RSD) of the current response to 8 mM glucose is 4.5% for 5 successive measurements, indicating that the glucose biosensor has good reproducibility. The interference from common interference species such as uric acid (UA) dopamine (DA), and ascorbic acid (AA) was also investigated by using their relevant physiological levels (0.1 mM).21 Fig. S9 (ESI†) shows the amperometric response of AgNPs/PANINFs/GCE toward 2 mM glucose in the absence and presence of the interference species at −0.58 V. It is seen that the injections of these interference into 2 mM glucose show negligible responses.
 |
| | Fig. 5 (a) CVs of the GOD/AgNPs/PANINFs/GCE in O2-saturated PBS solution (pH: 7.4) in the presence of varied glucose concentrations: 1, 2, 4, 6, 8, 10, and 12 mM from inner to outer (scan rate: 50 mV s−1). (b) The calibration curve (r = 0.997) corresponding to amperometric responses at the reduction peak. | |
The long-term stability of the prepared glucose biosensor is a critical factor in practical application. To evaluate the stability of the glucose biosensor, the GOD/AgNPs/PANINFs/GCE was stored at 4 °C when not in use. The storage stability was examined by periodical measurements of the biosensor response to 4 mM glucose in PBS solution (pH: 7.4) at a scan rate of 50 mV s−1 everyday and keeping the modified electrode at 4 °C in 0.2 M PBS after use. The variation of the response current at the GOD/AgNPs/PANINFs/GCE decreased to about 96% of its initial response current on the 2nd day and about 90% on the 7th day, as shown in Fig. 6. This slight decrease of the response current may be ascribed to the decrease of the enzyme activity as well as leaching of the immobilized enzyme from the electrode surface.22 A comparison of the performance of our designed sensor with those already reported results regarding the performance of glucose detection is shown in Table S1 (ESI†), which indicates that our sensor exhibits relatively lower detection limit and wider linear range than most of them.
 |
| | Fig. 6 The variation in the response current of 4 mM glucose in PBS solution (pH: 7.4) at the GOD/AgNPs/PANINFs/GCE for 7 days (scan rate: 50 mV s−1). | |
4. Conclusions
Electrochemical polymerization of aniline monomers in acidic media has been proven to be an effective method for synthesizing PANINFs in the absence of any templates and surfactants. Such nanofibers have been further used as both supports and reductants for the preparation of AgNPs decorated PANINFs. As-formed nanocomposites exhibit excellent catalytic activity toward both reduction of 4-NP by NaBH4 and electrochemical reduction of H2O2. The use of these nanocomposites for enzymeless H2O2 detection and glucose detection has also been demonstrated successfully. Our present study is important because it provides us a facile templateless and surfactantless strategy toward PANI nanofibers for applications.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21175129) and the National Basic Research Program of China (No. 2011CB935800).
Notes and references
- J. F. Hulvat and S. I. Stupp, Adv. Mater., 2004, 16, 589 CrossRef CAS.
-
(a) M. J. Sailor and C. L. Curtis, Adv. Mater., 1994, 6, 688 CrossRef CAS;
(b) D. Normile, Science, 1999, 286, 2056 CrossRef CAS;
(c) Z. Yao, H. W. C. Postma, L. Balents and C. Dekker, Nature, 1999, 402, 273 CrossRef CAS.
-
(a) X. Li, Y. Zhoa, T. Zhuang, G. Wang and Q. Gu, Colloids Surf., A, 2007, 295, 146 CrossRef CAS;
(b) Z. Wei, Z. Zhang and M. Wan, Langmuir, 2002, 18, 917 CrossRef CAS.
-
(a)
T. A. Skoheim, R. L. Elsenbaumer and J. R. Reynolds, Handbook of Conducting Polymers, Marcel Dekker, New York, 2nd edn, 1997 Search PubMed;
(b) M. Lahav, C. Durkan, R. Gabai, E. Katz, I. Willner and M. E. Welland, Angew. Chem., Int. Ed., 2001, 40, 4095 CrossRef CAS.
- G. Li and Z. Zhang, Macromolecules, 2004, 37, 2683 CrossRef CAS.
-
(a) C. R. Martin, Acc. Chem. Res., 1995, 28, 61 CrossRef CAS;
(b) C. R. Martin, Science, 1994, 266, 1961 CAS;
(c) V. M. Cepak and C. R. Martin, Chem. Mater., 1999, 11, 1363 CrossRef CAS.
-
(a) J. Han, G. Song and R. Guo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4229 CrossRef CAS;
(b) K. Huang and M. Wan, Chem. Mater., 2002, 14, 3486 CrossRef CAS;
(c) X. Zhang and S. K. Manohar, Chem. Commun., 2004, 2360 RSC;
(d) L. Huang, Z. Wang, H. Wang, X. Cheng, A. Mitra and Y. Yan, J. Mater. Chem., 2002, 12, 388 RSC.
-
(a) A. G. MacDiarmid, W. E. Jones, I. D. Norns, J. Gao, A. T. Johnson, N. J. Pinto, J. Hone, B. Han, F. K. Ko, H. Okuzaki and M. Llaguno, Synth. Met., 2001, 119, 27 CrossRef CAS;
(b) H. He, C. Li and N. Tao, Appl. Phys. Lett., 2001, 78, 811 CrossRef CAS;
(c) J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314 CrossRef CAS;
(d) J. Huang and R. B. Kaner, J. Am. Chem. Soc., 2004, 126, 851 CrossRef CAS;
(e) N. R. Chiou and A. J. Epstein, Adv. Mater., 2005, 17, 1679 CrossRef CAS;
(f) S. K. Pillalamarri, F. D. Blum, A. T. Tokuhiro, J. G. Story and M. F. Bertino, Chem. Mater., 2005, 17, 227 CrossRef CAS;
(g) L. K. Werake, J. G. Story, M. F. Bertino, S. K. Pillalamarri and F. D. Blum, Nanotechnology, 2005, 16, 2833 CrossRef CAS;
(h) X. Jing, Y. Wang, D. Wu, L. She and Y. Guo, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1014 CrossRef CAS;
(i) X. Lu, H. Mao, D. Chao, W. Zhang and Y. Wei, Macromol. Chem. Phys., 2006, 207, 2142 CrossRef CAS;
(j) X. Zhang, W. Goux and S. K. Manohar, J. Am. Chem. Soc., 2004, 126, 4502 CrossRef CAS;
(k) G. Chang, Y. Luo, F. Liao, W. Lu and X. Sun, J. Nanopart. Res., 2011, 13, 471 CrossRef CAS.
-
(a) Y. Zhao, L. Fan, H. Zhong, Y. Li and S. Yang, Adv. Funct. Mater., 2007, 17, 1537 CrossRef CAS;
(b) Y. Hsin, K. Hwang and C. Yeh, J. Am. Chem. Soc., 2007, 129, 9999 CrossRef CAS;
(c) Y. Ma, S. Jiang, G. Jian, H. Tao, L. Yu, X. Wang, J. Zhu, Z. Hu and Y. Chen, Energy Environ. Sci., 2009, 2, 224 RSC.
- Y. Gao, D. Shan, F. Cao, J. Gong, X. Li, H. Ma, Z. Su and L. Qu, J. Phys. Chem. C, 2009, 113, 15175 CAS.
- M. Hosseini and M. Momeni, J. Mater. Sci., 2010, 45, 3304 CrossRef CAS.
-
(a) M. Wan and J. Yang, J. Appl. Polym. Sci., 1995, 55, 399 CrossRef CAS;
(b) M. Trueba, A. L. Montero and J. Rieumont, Electrochim. Acta, 2004, 49, 4341 CrossRef CAS.
- Comments of one referee.
- X. Li, X. Ma, J. Sun and M. Huang, Langmuir, 2009, 25, 1675 CrossRef CAS.
- X. Li, R. Liu and M. Huang, Chem. Mater., 2005, 17, 5411 CrossRef CAS.
-
(a) J. Tian, S. Liu and X. Sun, Langmuir, 2010, 26, 15112 CrossRef CAS;
(b) J. Tian, H. Li, W. Lu, Y. Luo, L. Wang and X. Sun, Analyst, 2011, 136, 1806 RSC;
(c) J. Tian, Y. Luo, H. Li, W. Lu, G. Chang, X. Qin and X. Sun, Catal. Sci. Technol., 2011, 1, 1393 RSC.
-
(a) Y. Du, H. Chen, R. Chen and N. Xu, Appl. Catal., A, 2004, 277, 259 CrossRef CAS;
(b) J. F. Corbett, Dyes Pigm., 1999, 41, 127 CrossRef CAS;
(c) C. V. Rode, M. J. Vaidya and R. V. Chaudhari, Org. Process Res. Dev., 1999, 3, 465 CrossRef CAS;
(d) Z. Zhang, C. Shao, P. Zou, P. Zhang, M. Zhang, J. Mu, Z. Guo, X. Li, C. Wang and Y. Liu, Chem. Commun., 2011, 47, 3906 RSC.
-
(a) R. Leary and A. Westwood, Carbon, 2011, 49, 741 CrossRef CAS;
(b) Y. Zhang, S. Liu, W. Lu, L. Wang, J. Tian and X. Sun, Catal. Sci. Technol., 2011, 1, 1142 RSC.
- K. E. Toghill and R. G. Compton, Int. J. Electrochem. Sci., 2010, 5, 1246 CAS.
- W. Lu, Y. Luo, G. Chang and X. Sun, Biosens. Bioelectron., 2011, 26, 4791 CrossRef CAS.
- C. Guo, Z. Sheng, Y. Shen, Z. Dong and C. Li, ACS Appl. Mater. Interfaces, 2010, 2, 2481 CAS.
- P. He, G. Greenway and S. J. Haswell, Microfluid. Nanofluid., 2010, 8, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy00454b |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.