Recent advances in organocatalytic asymmetric Michael reactions
Received
22nd August 2011
, Accepted 16th September 2011
First published on 25th October 2011
Abstract
The Michael addition reaction represents one of the most powerful methods for the formation of carbon–carbon bonds in organic synthesis. Thanks to the rapid development of asymmetric organocatalysis, significant progress has been made during the past years in achieving organocatalytic asymmetric Michael reactions with a diverse combination of Michael donors and acceptors. Many new substrates have been accordingly applied in this reaction, together with the new approaches developed for the purpose of target- and diversity-oriented asymmetric synthesis. This review surveys the advances in target- and diversity-oriented asymmetric organocatalytic Michael reactions developed between 2009 and early 2011.

Yong Zhang
| Yong Zhang was born in 1985 in Jiangxi, P. R. China. He received his BSc in 2007 from Gannan Normal University. Then, he joined the research group of Professor Wei Wang at the State Key Laboratory of Applied Organic Chemistry (SKLAOC) in Lanzhou University. Currently, he is completing his PhD on the development of organocatalytic asymmetric methodologies and their applications in the total synthesis of natural products and biologically active targets. |

Wei Wang
| Wei Wang was born in 1972 in Xinjiang, P. R. China. He received his PhD in physical organic chemistry from Lanzhou University in 1998. After his postdoctoral research at University of Stuttgart (2000–2001) and at University of Southern California (2001–2002), he worked at the Institute of Chemical Technology, University of Stuttgart until 2006. He then moved back to Lanzhou University and was appointed as the Cheung Kong Professor from the Ministry of Education of China. Currently he serves as the Deputy Director of the State Key Laboratory of Applied Organic Chemistry (SKLAOC). The research of his laboratory focuses on homogeneous and heterogeneous organocatalysis, organic porous materials, and solid-state NMR spectroscopy. |
1. Introduction
The development of new chemical transformations for efficient and practical synthesis of complex structures has emerged as the main objective in synthetic organic chemistry. For this purpose, numerous organic reactions have been explored, allowing the (stereocontrolled) synthesis of a diversity of important scaffolds. Among all the methods developed, the (asymmetric) Michael reaction proves to be one of the most versatile tools for the formation of carbon–carbon bonds, and has been widely used to generate valuable building blocks in organic synthesis. On the other hand, asymmetric organocatalysis1 has emerged as an attractive research area for the synthesis of optically active compounds with inexpensive and environmentally benign organocatalysts under mild reaction conditions. During the past decade, a number of elegant organocatalysts have been developed for the enantiocontrol in a large variety of reactions. Accordingly, the organocatalytic asymmetric Michael reaction has received much attention, the previous progress of which has also been subject to some excellent reviews.2 In contrast to the early research on testing the concept of asymmetric organocatalysis with simple reagents, recent investigations on organocatalytic asymmetric Michael reaction have been directed more towards the target- and diversity-oriented synthesis,3 in which many new substrates and approaches have been attempted. In this review, we summarise the progress made in organocatalytic asymmetric Michael reactions between 2009 and early 2011, with the focus on the target- and diversity-oriented synthesis via asymmetric organocatalysis. We first survey the new substrates applied in the reactions, such as protected 2-nitro-ethenamines, oxindoles, benzofuran-2(3H)-ones, β-carbonyl heteroaryl sulfones, oxazolones, nitrophenylacetonitriles, and 1-acetylindolin-3-ones. This is then followed by the description of new approaches applied in the reactions, such as vinylogous and intramolecular Michael reactions. The diverse strategies for applying the organocatalytic asymmetric Michael reactions in the synthesis of natural products (or their core structures) are emphasised. Other developments related to the asymmetric organocatalytic Michael reactions, such as organocatalytic cascade reaction and hetero-Michael addition are also briefly mentioned.
2. New substrates
2.1 Protected 2-nitro-ethenamines
The 1,2-diamino structural motifs as well as the substituted 3-aminopyrrolidines are encountered in a large variety of pharmaceutical molecules. Recently, Ma and co-workers4 developed an organocatalytic Michael addition of protected 2-nitro-ethenamine 1 to aldehydes 2 as an efficient way to provide the synthetically useful 1,2-diamino precursors 3 (Scheme 1). In the presence of diarylprolinol silylether catalysts 4, the Michael adducts 3 were obtained in excellent yields and enantioselectivities. Interestingly, the diastereoselectivity of 3 is controlled by the Z- or E-form of the functionalised nitroolefins. The phthaloyl-protected 2-nitroethenamine 1 with E-form gives the syn adducts 3 as the major products, while acyl-protected 2-nitroethenamine 8a with Z-form affords the anti adducts. The optically active adducts 3 could be further derivatised into the corresponding aminopyrrolidines 6 by a Zn/HOAc-mediated reductive/amination process. Moreover, via a similar strategy developed by Hayashi and co-workers,5 a more efficient and practical procedure4 was provided for the enantioselective synthesis of (−)-oseltamivir 13 from 7 and 8a (Scheme 1).
 |
| | Scheme 1 | |
Most recently, Yuan and co-workers6 successfully applied the protected 2-amino-1-nitroethenes 8 in the asymmetric Michael addition with 3-substituted oxindoles 14 for the construction of 3,3′-disubstituted oxindoles 15 bearing α,β-diamino functionality. The amino-indanol derivative 16 was found as the optimal catalyst to afford the reaction products 15 in very high yields with excellent diastereoselectivities and good enantioselectivities (Scheme 2). Other bifunctional catalysts, which proved to be efficient for normal nitroalkenes, showed, however, poor enantiocontrol in this reaction due to the strong intramolecular hydrogen bonding in acyl-protected 2-nitroethenamine.
 |
| | Scheme 2 | |
The first asymmetric conjugate addition of unprotected 3-alkyloxindoles 14 to α,β-unsaturated aldehydes 17a was reported by Melchiorre and co-workers.8 In the presence of a new bifunctional chiral primary amine thiourea catalyst 19, the adducts 18 with adjacent quaternary and tertiary stereocentres were obtained in good yields with high diastereoselectivities and enantioselectivities (Scheme 3).
 |
| | Scheme 3 | |
Later, Maruoka and co-workers9 developed a chiral quaternary phosphonium salt 22 as an efficient phase-transfer catalyst for this reaction. A variety of N-Boc-3-aryloxindoles 14 could be successfully reacted with enones 20 (or acrolein), giving rise to the adducts 21 with excellent yields and enantioselectivities. The optically active Michael adducts 21a can be readily derivatised into valuable natural products and their analogues as shown in Scheme 4.
 |
| | Scheme 4 | |
In 2010, Luo and Cheng et al.10 realised the Michael addition of N-Boc-oxindoles 14 to activated terminal alkenes 25 with a bifunctional tertiary-amine thiourea catalyst 27 (Scheme 5). Both vinyl ketones and vinyl sulfones are applicable, giving the chiral oxindole compounds 26 with moderate to high yields and up to high enantioselectivities.
 |
| | Scheme 5 | |
The asymmetric conjugate addition of nitroalkenes is an important synthetic process due to the wide possibilities for subsequent transformations of the nitro group. In 2009, Barbas III and co-workers11 found that catalyst 30 could efficiently promote the addition of N-Boc-3-alkyloxindoles 14 to nitroolefins 28. The desired products 29 with a quaternary stereocentre at the C3 position were produced in good yields, together with good to excellent diastereoselectivities and enantioselectivities. The synthetic utility of this reaction was shown by derivatising the product 29a for the formal synthesis of (+)-physostigmine 32 (Scheme 6).
 |
| | Scheme 6 | |
Enantioselective conjugate addition of N-Boc-3-aryloxindoles 14 with nitroolefins 28 was realised later by Maruoka and co-workers.12 In the presence of chiral phase-transfer catalyst 33, the reaction proceeded well in water-rich solvents, affording the desired products 29 in excellent yields, together with up to excellent diastereoselectivities and high to excellent enantioselectivities (Scheme 7). In contrast to the general conditions for phase-transfer catalysis, this reaction does not need additional base. The enantioenriched adduct 29b could be reduced to the corresponding amine, followed by deprotection and cyclisation to give product 34, which has a core structure similar to the natural products (Scheme 7).
 |
| | Scheme 7 | |
In 2010, Luo and Cheng et al.13 presented the Michael addition of N-phenyl-3-methyloxindoles 35 to nitroolefins 28 with a simple bifunctional alkyl-thiourea organocatalyst 37. The desired products 36 were obtained in excellent yields with moderate diastereoselectivities and good enantioselectivities (Scheme 8).
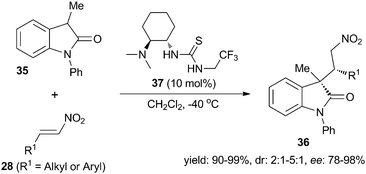 |
| | Scheme 8 | |
Chiral α-succinimides are synthetically and biologically important motifs in asymmetric synthesis. The asymmetric Michael addition of oxindoles to maleimides provides a practical route for the preparation of chiral α-succinimides. In 2010, Yuan and co-workers14 reported the enantioselective Michael addition of N-Boc-3-substituted oxindole 14 to maleimides 38 utilising a bifunctional thiourea-tertiary amine catalyst 30c (Scheme 9). A variety of oxindoles and N-protected maleimides (as well as maleic anhydride) could be used to provide the multifunctional products 39 in high yields and with high to excellent diastereo- and enantioselectivities.
 |
| | Scheme 9 | |
The enantioselective construction of 1,3-nonadjacent stereocentres remains a synthetic challenge in asymmetric catalysis. The use of α-branched Michael acceptors (such as 2-chloroacrylonitrile) is a conceivable strategy toward this issue. In 2010, Luo and Cheng et al.15 developed a highly enantioselective Michael addition of N-Boc-3-substituted oxindoles 14 to 2-chloroacrylonitrile 40 using a bifunctional tertiary-amine thiourea catalyst 37. Both 3-aryl and 3-alkyl oxindoles could be applied, providing the desired products 41 in high yields and high to excellent diastereo- and enantioselectivities. The utility of this methodology was exemplified by the synthesis of pyrroloindole derivatives 44 (Scheme 10).
 |
| | Scheme 10 | |
2.3 Benzofuran-2(3H)-ones
Benzofuranones with an all-carbon quaternary centre at the C3 position widely exist in a number of bioactive compounds. The first application of 3-substituted benzofuranones in an organocatalytic asymmetric Michael addition was recently reported by Luo and Cheng.16 In the presence of alkyl-substituted bifunctional thiourea catalyst 37, the addition of 3-arylbenzofuran-2(3H)-ones 45 to chalcones 46 proceeded smoothly to afford the desired products 47 with high yields and excellent enantioselectivities, but with poor diastereoselectivities (Scheme 11).
 |
| | Scheme 11 | |
In 2010, Luo and Cheng et al.17 extended their methodology to include maleimides 38 as the substrates. Utilising Takemoto's catalyst 30a, the Michael addition of 38 with a broad scope of substrates 45 affords the corresponding adducts 48 in excellent yields with high to excellent diastereoselectivities and enantioselectivities (Scheme 12).
 |
| | Scheme 12 | |
2.4
β-Carbonyl heteroaryl sulfones
In most cases, the Michael reactions are applicable only for the bond formation between two sp3–hybridised carbon atoms. The asymmetric bond formation between sp–sp3 or sp2–sp3 centres remains a challenging task. Recent developments have demonstrated that the use of β-carbonyl heteroaryl sulfones as necliphiles could easily tackle this synthetic challenge.18 Jørgensen and co-workers19 report the first application of β-carbonyl heteroaryl sulfones 49a in the Michael reaction with α,β-unsaturated aldehydes 17b. The reaction was catalysed by diarylprolinol silylether 5c, affording the privileged addition intermediates 50 with high yields and excellent enantioselectivities. The intermediates 50 could then be transformed into highly valuable alkynes 51 and alkenes 52via the Smiles rearrangement reaction. This also represents the first example for the asymmetric formation of β-alkyne-substituted aldehydes (Scheme 13).
 |
| | Scheme 13 | |
Further extension of this strategy to the asymmetric functionalisation of cyclic α,β-unsaturated ketones 53 was later achieved by the same group.20 In the presence of 9-epi-amino cinchona alkaloid salt 55 as the catalyst, the reaction proceeded well to afford the key intermediates 54, which are useful precursors for a series of important building blocks. When R1 are aryl substituents, alkynes 56 and alkenes 57 were obtained in moderate to high yields with moderate to excellent enantioselectivities. An organomediated desulfonylation further led to 1,5-diketo products 58 in moderate yields with excellent enantioselectivities. Interestingly, when R1 are alkyl substituents,21 bicyclic products 59 were obtained via the sequential pathway of intramolecular aldol reaction, Smiles rearrangement, and elimination (Scheme 14).
 |
| | Scheme 14 | |
2.5 Oxazolones
The asymmetric synthesis of non-natural amino acids and their derivatives is an area of great importance. Oxazolones have proven to be excellent reagents for the synthesis of α,α-disubstituted (quaternary) α-amino acids.22
In 2008, Jørgensen and co-workers23 reported (followed by Hayashi et al.24) the first nucleophilic addition of oxazolones 60 with α,β-unsaturated aldehydes 17 catalysed by diarylprolinol silylether 5c. Despite using oxazolones 60 with different nucleophilic sites, only C4-addition products 61 were observed. Having been obtained with good diastereoselectivities and excellent enantioselectivities, the Michael adducts 61 could then be derivatised to form different amino acid derivatives (Scheme 15).
 |
| | Scheme 15 | |
Soon after this, the Michael addition of oxazolones 60 to nitrostyrenes 28 was reported by the same group (Scheme 16).25 With a broad range of nitrostyrenes and oxazolones as the substrates, the reaction was catalysed by cinchona alkaloid thiourea catalyst 67 with good yields, excellent diastereoselectivities, and moderate to good enantioselectivities. The substituents on the oxazolone ring were shown to have a great impact on the regioselectivity. As illustrated in Scheme 16, the aryl substituents in the C2-position of oxazolone lead to the regioselectivity on C2 while aliphatic substituents (i.e. tert-butyl) give only the C4-substituted adducts.
 |
| | Scheme 16 | |
In 2009, Ooi and co-workers26 developed a supramolecular catalyst 70 for the conjugate addition of oxazolone 60a to α,β-unsaturated acylbenzotriazoles 68 (Scheme 17). The C2 addition products were obtained with excellent yields and enantioselectivities. To show the synthetic utility of this protocol, optically active methylsuccinic acid 72 was synthesised in four steps from 69 without loss of the enantiomeric excess.
 |
| | Scheme 17 | |
Enantioselective nucleophilic addition of simple conjugated esters or amides remains a challenging task in organocatalysis due to the low reactivities of substrates. In 2010, Jørgensen and co-workers27 demonstrated that the use of acyl phosphonates 73 as surrogates can effectively solve this problem through a double nucleophilic reaction. By employing a bifunctional thiourea 67 as catalyst, the conjugate addition of α,β-unsaturated acylphosphonates 73 with oxazolones 60 proceeded well to afford the N,O-aminal products 74 in moderate to high yields and excellent enantioselectivities (Scheme 18). Other nucleophiles such as indoles and 1,3-dicarbonyl compounds are also tolerable in this reaction.
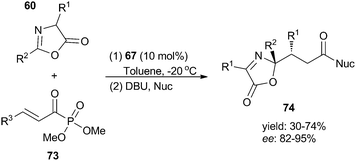 |
| | Scheme 18 | |
By using Takemoto thiourea catalyst 30a, Rios et al. reported the Michael addition of oxazolones 60 to 1,1-bis(phenylsulfonyl)ethylene 7528 and to maleimides 38,29 respectively. These two types of electrophiles showed distinct regioselectivities toward the conjugate addition with oxazolones. In the case of 75, the Michael addition took place exclusively at the C4 position, giving the precursors of quaternary amino acids with high yields and enantioselectivities. However, when maleimides 38 were used as electrophiles, the regioselectivity was reversed to give only C2 adducts 77 with excellent yields, diastereo- and enantioselectivities (Scheme 19).
 |
| | Scheme 19 | |
2.6 Nitrophenylacetonitriles
In 2010, Cid and Ruano30 found that arylacetonitriles bearing an electron-withdrawing group (such as a nitro group) at the ortho- or para-position could serve as suitable nucleophiles in organocatalytic processes. In the presence of prolinol ethers 5c, the enantioselective Michael addition of p-nitrophenylacetionitrile 78 to β-alkyl unsaturated aldehydes 17 could proceed in a highly enantioselective manner (Scheme 20). The reaction was performed in a sequential procedure as Michael addition/NaBH4 reduction/lactonisation, allowing the synthesis of diastereomerically pure lactones 79 in very high yields and enantioselectivities.
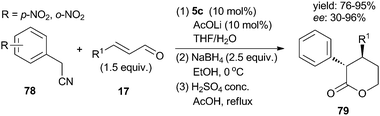 |
| | Scheme 20 | |
2.7 1-Acetylindolin-3-ones
Indole motifs are widely distributed in nature (especially in bioactive natural products) and pharmaceutical compounds. Due to the inherent electron characteristics of indole, the selective functionalisation of indole at the C2 position has met with limited success.31 Very recently, Xu and co-workers32 addressed a new methodology for this synthetic challenge. The asymmetric Michael addition of 1-acetylindolin-3-ones 80 to nitrostyrenes 28 was catalysed by a bifunctional thiourea-tertiary amine catalyst 67b, affording the desired products 81 with excellent yields and high diastereo- and enantioselectivities. These optically active products could be reduced to the corresponding alcohols, followed by elimination to give the 2-functionalised indoles 82 without loss of enantioselectivities (Scheme 21).
 |
| | Scheme 21 | |
Soon after this, Wang et al.33 reported a highly enantioselective Michael addition of 1-acetylindolin-3-one 80 to enones 83 by using a primary-secondary amine 85 as catalyst (Scheme 22). A variety of enones could be applied, giving the corresponding adducts 84 in good yields, moderate to excellent diastereoselectivities, and excellent enantioselectivities.
 |
| | Scheme 22 | |
3. Vinylogous Michael reaction
In spite of the substantial progress made in asymmetric Michael reactions, most of the catalytic methodologies are applicable only for the functionalisation of electron-deficient olefins at the β position. The enantioselective construction of C–C bonds at the γ position of olefins has met with limited success. Recently, with the exploitation of the vinylogy concept,34 the asymmetric vinylogous Michael reaction has emerged as an effective strategy to address this synthetic issue.35
3.1 α,β-Unsaturated γ-butenolides
γ-Butenolides and their derivatives are common motifs found in natural products and biologically active molecules. In 2003, MacMillan and co-workers36 realised the enantioselective γ-functionalisation of butenolides through an organocatalysed Mukaiyama-Michael addition of silyloxyfuran to enals. This reaction is synthetically useful for constructing the enantioenriched γ-butenolides architecture. However, the use of silyloxyfuran is unfavourable from the standpoint of atom economy. Thus, development of the direct vinylogous additions of γ-butenolides (as pronucleophiles) has received much attention.
Li and co-workers37 disclosed the first enantioselective organocatalytic direct vinylogous Michael addition of γ-butenolides 86 to chalcones 47. The reaction was catalysed by the vicinal primary-diamine salt 88, giving rise to highly valuable chiral γ-butenolides 87 with good yields, high diastereo- and enantioselectivities. However, the substrate scope was limited to the substituted γ-butenolides and chalcones. For the unsubstituted γ-butenolide, the enantioselectivity is low and no diastereoselectivity could be obtained. The use of simple 2(5H)-furanone 86a in the same reaction in the presence of Takemoto thiourea catalyst was almost simultaneously reported by Wang and co-workers38 (Scheme 23).
 |
| | Scheme 23 | |
Further development with much improved substrate scope and efficiency was later reported by Ye and co-workers39 by using a new catalyst 91 (Scheme 24). A variety of enones 83 including benzalacetone, chalcones, and alkyl substituted enones are applicable, affording the synthetically versatile γ-substituted butenolides 90 with satisfactory yields, diastereo- and enantioselectivities.
 |
| | Scheme 24 | |
Utilising the aminal-pyrrolidine catalyst 94, Angelica lactones 92 were successfully applied in the direct vinylogous Michael addition with enals by Alexakis and co-workers.40 Various aldehydes 17 could be used, affording the γ-tetrasubstitued butenolides 93 with excellent stereoselectivities. However, 2(5H)-furanone and γ-substituted butenolide proved to be unsuitable for this reaction, because the enol equilibrium of Angelica lactone occurs independently in the absence of the catalyst (Scheme 25).
 |
| | Scheme 25 | |
Although the direct vinylogous Michael addition of 2(5H)-furanone to nitroalkenes was reported in 2009 by Trost using a chiral metal complex,41 the use of organocatalysts for this reaction was not realised until recently.42 In this context, α-tert-butylthio-substituted furanones 86b were utilised as the vinylogous pronucleophiles. The Michael reaction with nitroalkenes 28 was catalysed by axially chiral guanidine 96, leading to densely functionalised γ-butenolides 95 in high syn-diastereo- and enantioselectivities. To show the synthetic utility of this reaction, the α,γ-functionalised γ-butenolide product 95a was then derivatised into β,γ-disubstituted butenolide 98via standard chemical transformations (Scheme 26).
 |
| | Scheme 26 | |
3.2 α,β-Unsaturated γ-butyrolactams
5-Substituted 3-pyrrolidin-2-ones motifs are privileged heterocyclic structures, which widely exist in synthetic bioactive molecules and natural products. Recently, α,β-unsaturated γ-butyrolactams 99 have emerged as one of the most efficient precursors for the synthesis of 5-substituted 3-pyrrolidin-2-one derivatives. Shibasaki and co-workers43 reported the first asymmetric Mannich and Michael reaction of α,β-unsaturated γ-butyrolactams catalysed by a chiral metal complex. Thereafter, three publications of asymmetric Michael reaction of α,β-unsaturated γ-butyrolactams involving organocatalysis appeared.
In 2010, Chen and co-workers44 presented an organocatalytic asymmetric conjugate addition of α,β-unsaturated aldehydes 17via iminium catalysis. Catalysed by diarylprolinol silylether 5a, the reaction proceeded well with the addition of o-fluorobenzoic acid (OFBA), affording the highly valued adducts 100 in excellent enantioselectivities and with up to outstanding diastereoselectivities. To show the synthetic utility, a number of natural-product-like or drug-like molecules with versatile skeletons have been efficiently constructed from the Michael adducts 100 (Scheme 27).
 |
| | Scheme 27 | |
The first organocatalytic conjugate addition to enones was reported in 2011 by our group,45 utilising the cinchona alkaloid-based thiourea catalyst 67a (Scheme 28). Various chalcones 47 could be applied, affording the synthetically versatile γ-substituted butyrolactams 107 with excellent diastereo- and enantioselectivities. However, the substrate scope was limited to chalcones only. When R was an aliphatic moiety (e.g.CH3 or t-Bu), no reaction could be observed. Almost simultaneously, the group of Ye realised the asymmetric addition to benzalacetone or alkyl substituted enones 83 using a multifunctional primary amine salt 91.46 The corresponding Michael adducts 108 could be obtained in high yields, together with excellent diastereo- and enantioselectivities. These adducts were derivatised into certain important fragments under standard chemical transformations (Scheme 29).
 |
| | Scheme 28 | |
 |
| | Scheme 29 | |
3.3 5-Styrylisoxazoles
Adamo et al.47 have previously developed styrylisoxazoles as new vingylogous electrophiles in the Michael addition with a variety of soft nucleophiles. In 2009, Bernardi, Adamo and co-workers fulfilled the successful application of styrylisoxazoles 111 in the asymmetric vinylogous conjugate addition with nitroalkanes.48 Using phase-transfer catalyst 114, the corresponding adducts 113 were formed in moderate to high yields and with good to excellent enantiomeric excesses. These enantiomerically enriched adducts were then transformed to γ-nitroesters 115 and γ-amino acids 117 without any loss of enantiomeric excess (Scheme 30).
 |
| | Scheme 30 | |
3.4 β-Substituted cyclohexenone derivatives
Inspired with the dienamine catalysis49 developed by Jørgensen and co-workers, Melchiorre et al.50 discovered that the chiral primary amine salts 120 could selectively activate the γ-position of unmodified cyclic α,β-unsaturated ketones 118 through dienamine catalysis. The direct vinylogous Michael addition of β-substituted cyclohexenones 118 to nitroalkenes 28 proceeded smoothly, affording the γ-site-selective adducts 119 with high yields and excellent enantioselectivities. However, the substrate scope was limited to arylphatic nitroalkenes. Aliphatic nitroalkenes did not react under the optimal conditions. The five-membered ring nucleophilic component (i.e. 3-methy 2-cyclopenten-1-one) was inactive, which suggested that the geometry of the cyclic scaffold strongly influenced the selective formation of the dienamine intermediate. Other β,β-disubstituted nitrostyrene 121 and α,α-disubstituted nitrostyrene 122 could also be applied, leading to the compounds 123 and 124 with an all-carbon quaternary stereogenic centre, respectively (Scheme 31).
 |
| | Scheme 31 | |
4. Intramolecular Michael reaction
The enantioselective intramolecular Michael reaction provides an important method for the construction of chiral, cyclic carbon skeletons, which are common motifs in natural products. Early in 2004, List and Fonseca51 presented the first organocatalytic asymmetric intramolecular Michael reaction of aldehydes for the synthesis of chiral trans-disubstituted cyclopentanes and pyrrolidine derivatives. Despite of the synthetic utility of this method in organic synthesis, little progress has been made in this area.52
Recently, Cobb and co-workers53 successfully used this methodology for the synthesis of cyclic γ-amino acids. The catalytic asymmetric intramolecular Michael addition of nitronates to conjugated esters was firstly realised by using a bifunctional tertiary-amine thiourea catalyst 67. A variety of substrates except Z-esters could be applied, affording the disubstituted cyclohexane derivatives 126 in poor to high yields, with up to excellent diastereoselectivities and enantioselectivities. Notably, up to three contiguous stereocentres can be constructed in one step with this strategy. To illustrate the synthetic utility of these products in the synthesis of peptides, N- and C-terminal derivatisations were performed (Scheme 32).
 |
| | Scheme 32 | |
In 2009, Christmann et al.54 disclosed a Ranhut-Currier type intramolecular Michael reaction viadienamine activation for the construction of an iridoid framework. In the presence of Jørgensen-Hayashi catalyst 5a, the reaction proceeded well to afford the cyclopentene derivatives 130 in moderate to good yields with good enantioselectivities. This also provides a simple operational procedure for the enantioselective synthesis of (+)-rotundial 130a (Scheme 33).
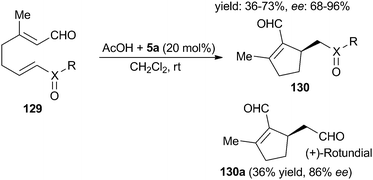 |
| | Scheme 33 | |
Further expansion of the scope of Michael acceptors to vinyl sulfones was reported by Alexakis and co-workers recently.55Trans-4-hydroxyprolylamide 133 was found to be the optimal catalyst to promote the intramolecular Michael addition of aldehydes to vinyl sulfone, affording the desired products 132 in good yields, together with good diastereoselectivities and enantioselectivities (Scheme 34).
 |
| | Scheme 34 | |
5. Miscellaneous
Some elegant examples have appeared for applying asymmetric organocatalytic Michael reactions in the total synthesis of natural products.56 Most recently, Fan and co-workers57 reported an unprecedented Michael addition of α-cyanoketones 134 and acrylates 135 as the key step for the diversity-oriented asymmetric synthesis of bioactive hydrodibenzofuran alkaloids. By using bifunctional tertiary amine-thiourea catalyst 30a or 67c, highly functionalised building blocks 136 with a sterically-congested aryl-substituted quaternary carbon atom were obtained in high yields and excellent enantioselectivities after one recrystallisation. The building blocks 136 were then transformed to 137 with the cis-hydrodibenzofuran core structure under an intramolecular ketone-ester condensation and oxa-Michael sequence. Three natural hydrodibenzofuran alkaloids, (−)-lycoramine, (−)-galanthamine and (+)-lunarine, were constructed on the basis of this method (Scheme 35).
 |
| | Scheme 35 | |
The organocatalytic cascade reactions involving Michael addition have emerged as powerful strategies for the synthesis of complex natural products and bioactive compounds. Since the cascade reactions offer advantages such as protecting-group-free, atom and step economy, several elegant examples have been produced in organocatalysis and the progress has been summarised recently.58 Besides, the conjugate additions of non-carbon nucleophiles such as amines,59alcohols,60thiols61 and phosphorus62 leading to the formation of a carbon–heteroatom bond via hetero-Michael addition have been achieved, the description of which have, however, not been included in this review.
6. Conclusion
In summary, considerable progress in the area of asymmetric Michael reactions has been made recently through the combination of various Michael donors and acceptors. Organocatalysis proved to be one of the most efficient methods for the target- and diversity-oriented synthesis, and many successful examples in the construction of important building blocks for natural products synthesis have emerged. Further extension of the concepts to vinylogous as well as intramolecular reactions gives vitality in this area. Hence, future developments in this field will probably focus on broadening the scope, exploiting new approaches and developing new concepts of the organocatalytic asymmetric Michael reaction, especially towards the target- and diversity-oriented synthesis.
Acknowledgements
The National Natural Foundation of China (no. 20972064 and 21172103) is gratefully acknowledged for financial support.
Notes and references
-
(a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS
 ;
(b)
A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c)
P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed ;
(d) B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS ;
(e) Special issue on organocatalysis, Chem. Rev., 2007, 107, 5413;
(f) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS ;
(g) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323 CrossRef CAS .
;
(b)
A. Berkessel and H. Gröger, Asymmetric Organocatalysis: From Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed;
(c)
P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed ;
(d) B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS ;
(e) Special issue on organocatalysis, Chem. Rev., 2007, 107, 5413;
(f) D. W. C. MacMillan, Nature, 2008, 455, 304 CrossRef CAS ;
(g) J. T. Mohr, M. R. Krout and B. M. Stoltz, Nature, 2008, 455, 323 CrossRef CAS .
-
(a) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS ;
(b) D. Almasi, D. A. Alonso and C. Najera, Tetrahedron: Asymmetry, 2007, 18, 299 CrossRef CAS ;
(c) J. L. Vicario, D. Badia and L. Carrillo, Synthesis, 2007, 2065 CrossRef CAS ;
(d) M. Thirumalaikumar, Org. Prep. Proced. Int., 2011, 43, 67 CrossRef CAS .
-
(a) D. R. Spring, Org. Biomol. Chem., 2003, 1, 3867 RSC ;
(b) T. E. Nielsen and S. L. Schreiber, Angew. Chem., Int. Ed., 2008, 47, 48 CrossRef CAS ;
(c) S. L. Schreiber, Nature, 2009, 457, 153 CrossRef CAS ;
(d) W. Galloway, A. Isidro-Llobet and D. R. Spring, Nat. Commun., 2010, 1 CrossRef .
- S. L. Zhu, S. Y. Yu, Y. Wang and D. W. Ma, Angew. Chem., Int. Ed., 2010, 49, 4656 CrossRef CAS .
- H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., Int. Ed., 2009, 48, 1304 CrossRef CAS .
- X.-L. Liu, Z.-J. Wu, X.-L. Du, X.-M. Zhang and W.-C. Yuan, J. Org. Chem., 2011, 76, 4008 CrossRef CAS .
-
(a) F. Zhou, Y. L. Liu and J. Zhou, Adv. Synth. Catal., 2010, 352, 1381 CrossRef CAS ;
(b) J. J. Badillo, N. V. Hanhan and A. K. Franz, Curr. Opin. Drug Discovery Dev., 2010, 13, 758 CAS .
- P. Galzerano, G. Bencivenni, F. Pesciaioli, A. Mazzanti, B. Giannichi, L. Sambri, G. Bartoli and P. Melchiorre, Chem.–Eur. J., 2009, 15, 7846 CrossRef CAS .
- R. J. He, C. H. Ding and K. Maruoka, Angew. Chem., Int. Ed., 2009, 48, 4559 CrossRef CAS .
- X. Li, Z. G. Xi, S. Z. Luo and J. P. Cheng, Org. Biomol. Chem., 2010, 8, 77 CAS .
- T. Bui, S. Syed and C. F. Barbas, J. Am. Chem. Soc., 2009, 131, 8758 CrossRef CAS .
- R. J. He, S. Shirakawa and K. Maruoka, J. Am. Chem. Soc., 2009, 131, 16620 CrossRef CAS .
- X. Li, B. Zhang, Z. G. Xi, S. Z. Luo and J. P. Cheng, Adv. Synth. Catal., 2010, 352, 416 CrossRef CAS .
- Y. H. Liao, X. L. Liu, Z. J. Wu, L. F. Cun, X. M. Zhang and W. C. Yuan, Org. Lett., 2010, 12, 2896 CrossRef CAS .
- X. Li, S. Z. Luo and J. P. Cheng, Chem.–Eur. J., 2010, 16, 14290 CrossRef CAS .
- X. Li, Z. G. Xi, S. Z. Luo and J. P. Cheng, Adv. Synth. Catal., 2010, 352, 1097 CrossRef CAS .
- X. Li, S. S. Hu, Z. G. Xi, L. Zhang, S. Z. Luo and J. P. Cheng, J. Org. Chem., 2010, 75, 8697 CrossRef CAS .
- M. Nielsen, C. B. Jacobsen, N. Holub, M. W. Paixão and K. A. Jørgensen, Angew. Chem., Int. Ed., 2010, 49, 2668 CAS .
- M. Nielsen, C. B. Jacobsen, M. W. Paixão, N. Holub and K. A. Jørgensen, J. Am. Chem. Soc., 2009, 131, 10581 CrossRef CAS .
- M. W. Paixão, N. Holub, C. Vila, M. Nielsen and K. A. Jørgensen, Angew. Chem., Int. Ed., 2009, 48, 7338 CrossRef .
- N. Holub, H. Jiang, M. W. Paixão, C. Tiberi and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 4337 CrossRef CAS .
- A. N. R. Alba and R. Rios, Chem.–Asian. J., 2011, 6, 720 CrossRef CAS .
- S. Cabrera, E. Reyes, J. Alemán, A. Milelli, S. Kobbelgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2008, 130, 12031 CrossRef CAS .
- Y. Hayashi, K. Obi, Y. Ohta, D. Okamura and H. Ishikawa, Chem.–Asian. J., 2009, 4, 246 CrossRef CAS .
- J. Alemán, A. Milelli, S. Cabrera, E. Reyes and K. A. Jørgensen, Chem.–Eur. J., 2008, 14, 10958 CrossRef .
- D. Uraguchi, Y. Ueki and T. Ooi, Science, 2009, 326, 120 CrossRef CAS .
- H. Jiang, M. W. Paixão, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775 CrossRef CAS .
- A. N. R. Alba, X. Companyó, G. Valero, A. Moyano and R. Rios, Chem.–Eur. J., 2010, 16, 5354 CrossRef CAS .
- A. N. R. Alba, G. Valero, T. Calbet, M. Font-Bardía, A. Moyano and R. Rios, Chem.–Eur. J., 2010, 16, 9884 CrossRef CAS .
- M. B. Cid, S. Duce, S. Morales, E. Rodrigo and J. L. G. Ruano, Org. Lett., 2010, 12, 3586 CrossRef CAS .
-
(a) S. L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC ;
(b) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CAS .
- Y. Z. Liu, R. L. Cheng and P. F. Xu, J. Org. Chem., 2011, 76, 2884 CrossRef CAS .
- W. Sun, L. Hong and R. Wang, Chem.–Eur. J., 2011, 17, 6030 CrossRef CAS .
- R. C. FusoN, Chem. Rev., 1935, 16, 1 CrossRef CAS .
-
(a) G. Casiraghi, L. Battistini, C. Curti, G. Rassu and F. Zanardi, Chem. Rev., 2011, 111, 3076 CrossRef CAS ;
(b) H.-L. Cui and Y.-C. Chen, Chem. Commun., 2009, 4479 RSC .
- S. P. Brown, N. C. Goodwin and D. W. C. MacMillan, J. Am. Chem. Soc., 2003, 125, 1192 CrossRef CAS .
- J. F. Wang, C. Qi, Z. M. Ge, T. M. Cheng and R. T. Li, Chem. Commun., 2010, 46, 2124 RSC .
- Y. A. Zhang, C. G. Yu, Y. F. Ji and W. Wang, Chem.–Asian. J., 2010, 5, 1303 CAS .
- H. C. Huang, F. Yu, Z. C. Jin, W. J. Li, W. B. Wu, X. M. Liang and J. X. Ye, Chem. Commun., 2010, 46, 5957 RSC .
- A. Quintard, A. Lefranc and A. Alexakis, Org. Lett., 2011, 13, 1540 CrossRef CAS .
- B. M. Trost and J. Hitce, J. Am. Chem. Soc., 2009, 131, 4572 CrossRef CAS .
- M. Terada and K. Ando, Org. Lett., 2011, 13, 2026 CrossRef CAS .
- N. E. Shepherd, H. Tanabe, Y. J. Xu, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2010, 132, 3666 CrossRef CAS .
- X. Feng, H. L. Cui, S. Xu, L. Wu and Y. C. Chen, Chem.–Eur. J., 2010, 16, 10309 CrossRef CAS .
- Y. Zhang, Y. L. Shao, H. S. Xu and W. Wang, J. Org. Chem., 2011, 76, 1472 CrossRef CAS .
- H. C. Huang, Z. C. Jin, K. L. Zhu, X. M. Liang and J. X. Ye, Angew. Chem., Int. Ed., 2011, 50, 3232 CrossRef CAS .
-
(a) M. F. A. Adamo, E. F. Duffy, D. Donati and P. Sarti-Fantoni, Tetrahedron, 2007, 63, 2684 CrossRef CAS ;
(b) M. F. A. Adamo and V. R. Konda, Org. Lett., 2007, 9, 303 CrossRef CAS ;
(c) M. F. A. Adamo, D. Donati, E. F. Duffy and P. Sarti-Fantoni, J. Org. Chem., 2005, 70, 8395 CrossRef CAS .
- A. Baschieri, L. Bernardi, A. Ricci, S. Suresh and M. F. A. Adamo, Angew. Chem., Int. Ed., 2009, 48, 9342 CrossRef CAS .
- S. Bertelsen, M. Marigo, S. Brandes, P. Dinér and K. A. Jørgensen, J. Am. Chem. Soc., 2006, 128, 12973 CrossRef CAS .
- G. Bencivenni, P. Galzerano, A. Mazzanti, G. Bartoli and P. Melchiorre, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20642 CrossRef CAS .
- M. T. Hechavarria Fonseca and B. List, Angew. Chem., Int. Ed., 2004, 43, 3958 CrossRef CAS .
-
(a) Y. Hayashi, H. Gotoh, T. Tamura, H. Yamaguchi, R. Masui and M. Shoji, J. Am. Chem. Soc., 2005, 127, 16028 CrossRef CAS ;
(b) M. Kikuchi, T. Inagaki and H. Nishiyama, Synlett, 2007, 1075 CAS .
- W. J. Nodes, D. R. Nutt, A. M. Chippindale and A. J. A. Cobb, J. Am. Chem. Soc., 2009, 131, 16016 CrossRef CAS .
- E. Marqués-López, R. P. Herrera, T. Marks, W. C. Jacobs, D. Könning, R. M. de Figueiredo and M. Christmann, Org. Lett., 2009, 11, 4116 CrossRef .
- C. Bournaud, E. Marchal, A. Quintard, S. Sulzer-Mossé and A. Alexakis, Tetrahedron: Asymmetry, 2010, 21, 1666 CrossRef CAS .
- E. Marqués, R. P. Herrera and M. Christmann, Nat. Prod. Rep., 2010, 27, 1138 RSC .
- P. Chen, X. Bao, L.-F. Zhang, M. Ding, X.-J. Han, J. Li, G.-B. Zhang, Y.-Q. Tu and C.-A. Fan, Angew. Chem., Int. Ed., 2011, 50, 8161 CrossRef CAS .
-
(a) D. Enders, C. Grondal and M. R. M. Huettl, Angew. Chem., Int. Ed., 2007, 46, 1570 CrossRef CAS ;
(b) X. Yua and W. Wang, Org. Biomol. Chem., 2008, 6, 2037 RSC ;
(c) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS .
- D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS .
- C. F. Nising and S. Bräese, Chem. Soc. Rev., 2008, 37, 1218 RSC .
- D. Enders, K. Luettgen and A. A. Narine, Synthesis, 2007, 959 CrossRef CAS .
- D. Enders, A. Saint-Dizier, M. I. Lannou and A. Lenzen, Eur. J. Org. Chem., 2006, 29 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.