Efficient organocatalysts derived from simple chiral acyclic amino acids in asymmetric catalysis
Zhuo
Chai
and
Gang
Zhao
*
Key Laboratory of Synthetic Chemistry of Natural Substances, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, China. E-mail: zhaog@mail.sioc.ac.cn; Fax: +86 21-64161628; Tel: +86 21-54925182
First published on 10th November 2011
Abstract
Chiral amino acids have played a key role in the development of organocatalysis from a biomimetic concept to an independent methodology, together with metal catalysis and enzyme catalysis, comprising the three major catalytic methodologies in modern organic synthesis. As an excellent pool for the design of organocatalysts, chiral amino acids have two obvious advantages: ready availability with usually affordable costs and modular structures allowing facile tuning of the catalytic efficiency. Recently, bifunctional/multifunctional primary–secondary amines, tertiary amine–thioureas, aminophosphines easily prepared from simple acyclic amino acids have been developed as efficient organocatalysts for various asymmetric reactions leading to a variety of useful chiral compounds. In this perspective, we present a personal overview of some of these recent advances in this field based on our own research experience.
1. Introduction
Asymmetric organocatalysis has seen tremendous development since the end of the 1990s, which has led to various new methods enabling organic synthesis to proceed in milder, more efficient and environmentally benign ways in many cases.1,2 To meet the high standards of modern organic synthesis, the discovery and development of efficient and cost-effective organocatalysts have both been one of the core tasks and added momentum to the fast-growing field of asymmetric organocatalysis. Due to their ready availability, inexpensiveness and modular chiral skeletons allowing structure fine-tuning,3 chiral α-amino acids have been one of the most valuable organocatalyst pools since the seminal work of List, Lerner and Barbas using the free amino acid proline to catalyze the direct asymmetric aldol reaction.4In general, the utilization of chiral α-amino acids in the development of organocatalysts has two options: the use of free amino acids or their derivatives. Although the direct use of free α-amino acids as catalysts is more preferable in terms of commercial availability and cheapness, they usually suffer from a rather narrow applicable scope of reactions, unsatisfactory catalytic activities and the lack of flexibility for structure fine-tuning. To overcome these limitations, simple modifications of free amino acids have become an expedient choice. Since the great success of Macmillan's imidazolidinone catalysts,5 a variety of chiral α-amino acid derivatives have been successfully developed as efficient and versatile catalysts for various kinds of reactions. The strategies used for the modification of free amino acids mainly focused on varying the electronic and/or steric properties of the amino and carboxylic groups.
In 2005, Ishihara and co-workers reported the preparation of novel chiral di- and triamine catalysts from simple acyclic amino acids and their applications in the asymmetric Diels–Alder reaction of α-substituted acroleins with 1,3-dienes.6 Thereafter, a great many new organocatalysts have been developed based on simple acyclic amino acids and applied in various reactions. Despite the diverse differences in their structures, the stereocontrol mechanisms of these amino acids-derived catalysts can still mainly be divided into the following modes: iminium, enamine, and hydrogen bonding (Fig. 1). In addition, with its unique good nucleophilic properties, the recent introduction of the phosphine atom to amino acid-derived catalysts has further greatly expanded their application scope. More importantly, the catalytic activities of these bifunctional or multifunctional catalysts are highly versatile and flexible in that a single catalyst can function through several activation modes in a single reaction or different activation modes in different reactions, even in a one-pot fashion, providing an excellent platform for the development of new cascade reactions.7 In this perspective, a personal overview of some recent developments in this field is provided, which are based on the recent works from our labs as well as others. Since the functioning of these organocatalysts in a single reaction was usually realized via the cooperation of several stereocontrolling elements (iminium, enamine, etc.), it should be mentioned here that the following rough classification was mainly made according to the presumed key stereocontrolling element in the transition state.
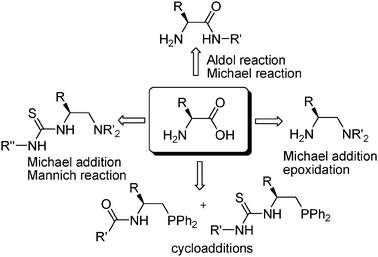 | ||
| Fig. 1 Representative organocatalysts derived from simple acyclic α-amino acids discussed in this perspective. | ||
2. Iminium catalysis mediated reactions
2.1 Michael addition to α,β-unsaturated ketones
Despite their role as one of the most important and major classes of Michael acceptors, α,β-unsaturated ketones have given not-so-satisfactory results (mostly below 95% ee and long reaction times of 2–6 days) with previously developed secondary-amine organocatalysts,8 probably due to the increased steric hindrance and reduced reactivity of the carbonyl group in these ketone substrates, compared to their aldehyde counterparts. Given that primary amines are less sterically demanding when they form iminium ions with carbonyl groups, we then presumed that certain chiral primary amine catalysts would be able to activate α,β-unsaturated ketones for Michael addition via efficient iminium catalysis to achieve good stereocontrol.Starting from commercially available amino acids, a series of novel bifunctional secondary–primary amine catalysts were then easily prepared in three steps: esterification of the carboxylic acid, amidation with different amines and reduction with LiAlH4.9 We first tested them in the Michael additions of malonates to various α,β-unsaturated ketones (Scheme 1).9 The highly modular structures of these catalysts proved to be very valuable in the catalyst screening process, which allowed easy fine-tuning of the catalytic efficiency from three aspects: the amino-acid skeleton, the two amine moieties and the acid additives. It should be noted that the addition of an acid additive could improve the catalytic efficiency remarkably and give better yields and ees (enantiomeric excess). The role of the acid additive was presumed to foster the formation of the iminium intermediate and the regeneration of the catalyst (see the catalytic cycle in Scheme 1).
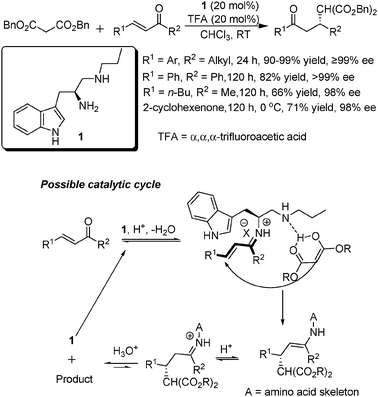 | ||
| Scheme 1 Michael additions of malonates to α,β-unsaturated ketones and a proposed possible catalytic cycle. | ||
After extensive screening, the best catalyst 1 derived from L-tryptophan and the acid additive TFA was identified as the best combination for this reaction. For most of the enones examined (R1 = Ar, R2 = alkyl), the desired Michael adducts could be obtained in excellent yields and ee values (mostly ≥99%). When both R1 and R2 of the enone (chalcone) were phenyl groups, the reaction was sluggish, but still with high yield and excellent ee. The reduced reactivity of the carbonyl group to form the iminium intermediate due to the conjugation with the phenyl group as well as the steric hindrance might be part of the reason. Reduced reactivity was also observed for aliphatic substrates (R1 and R2 were alkyl groups). In addition, the reaction of the cyclic 2-cyclohexenone also required a low temperature and long reaction time to give good results. As we would see from the ensuing examples, these points seemed rather common for the amino-acid based primary–secondary amine-catalyzed Michael additions to α,β-unsaturated ketones, which may occur via a similar activation mechanism as illustrated in Scheme 1.
Based on our above catalyst system, Wang and co-workers later realized performing the same reaction in water, the most environmentally-benign solvent, by using a phenylalanine-derived catalyst 2 (Scheme 2).10 It was found that the long alkyl chain in 2 was crucial for the reaction to proceed to give good chemical yields in water, which is in accord with previous findings from the organocatalyzed reactions performed in water. Also noteworthy is the almost complete ineffectiveness of some of the other secondary amine catalysts that the authors screened for this reaction, though these catalysts have found successful applications in other Michael reactions. This system also showed a broad range of substrate scope of α,β-unsaturated ketones. Compared to our homogeneous system, although this aqueous system gave somewhat decreased yields and ees and required an elevated reaction temperature of 50 °C, this example showed the possibility of developing more environmentally-benign procedures with this kind of catalyst system.
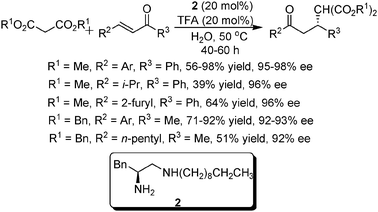 | ||
| Scheme 2 Michael additions of malonates to α,β-unsaturated ketones in water. | ||
Given the generality of the iminium activation mechanism of these primary–secondary amine catalysts proposed in Scheme 1, we later became interested in the utilization of other Michael donors instead of malonates in the Michael addition to α,β-unsaturated ketones. It was found that, using a phenylalanine-derived catalyst 3, a highly enantioselective tandem Michael addition–aldol-dehydration (Robinson-annulation-type) of benzoylacetates to α,β-unsaturated ketones could be realized to give synthetically useful densely substituted chiral cyclohexenones (Scheme 3).11 Although the dr values obtained for most substrates were not satisfactory, the enantioselectivity for each of the diastereoisomers was equally excellent. A simple decarboxylation procedure for the product was also developed to provide a synthetically useful chiral cyclohexenone, which previously had only been acquired via multistep synthesis. In this reaction, D-N-Boc-phenylglycine was identified as the acid additive of choice to give the best results while the use of L-N-Boc-phenylglycine gave somewhat decreased ee, but with comparable yields and dr values.
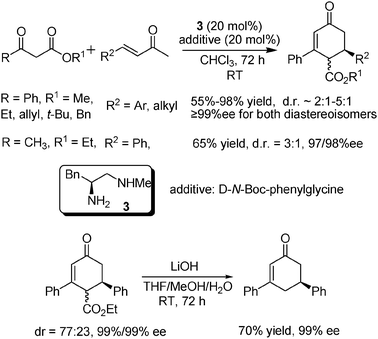 | ||
| Scheme 3 Tandem Michael addition–aldol-dehydration of benzoylacetates to α,β-unsaturated ketones. | ||
Later, it was found that α-fluoro-β-ketoesters were also suitable Michael donors in this system. Fluorine-containing compounds have found extensive applications in various kinds of fields such as materials, medicinal, pharmaceutical and agrochemical science due to the unique properties of the fluorine atom. Here, 4-nitrobenzoic acid (PNBA) was identified as the best acid additive, whose combination with catalyst 1 could catalyze the asymmetric Robinson annulation to give multiply substituted fluorinated cyclohexenones in excellent ee and dr values (Scheme 4).1219F NMR monitoring of the reaction process was done to get some insight into the reaction mechanism, which is supportive of a Michael–aldol-dehydration process similar to the addition of benzoylacetates described above. Actually, simple fluorinated acetylacetates and benzoylacetates were more reactive than their non-fluorinated counterparts for this transformation (see Scheme 3), for which only half loading-amount of the corresponding catalyst was required (Scheme 4). This might be ascribed to an enhanced acidity of the hydrogen atom between the two carbonyl groups after the introduction of the fluorine atom, which may in turn facilitate the deprotonation step to form the actual nucleophile, namely the corresponding enolate.
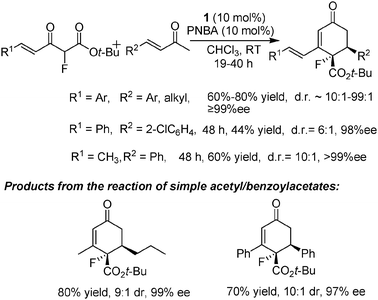 | ||
| Scheme 4 Asymmetric Robinson annulation of α-fluoro-β-ketoesters with α,β-unsaturated ketones. | ||
The Michael addition of simple nitroalkanes to α,β-unsaturated ketones has been one of the most important methods to construct very useful organic nitro group-containing compounds, which could be easily converted to many other useful structures.13 At the moment of our work (Scheme 5),14 successful organocatalytic systems for this kind of reaction were very limited for acyclic α,β-unsaturated ketone substrates and mostly the relatively costly chiral thiourea catalysts were utilized. When the primary–secondary amine catalysts were used to promote the additions of 2-nitropropane to enones, it was found that a new catalyst with a bulky t-Bu group on the secondary amine moiety was the one of choice for this reaction. Moreover, a new acid additive 4-nitrophenol was also identified. Compared to previous systems, the reaction times were significantly reduced from 2–5 days to generally 24 h in our system. In addition, using nitromethane as the nucleophile also gave comparably good results (Scheme 5). Although the system worked well for acyclic enones (around 90% ee), one limitation is that only 60% ee was obtained for the cyclic substrate 2-cyclohexenone. Also noteworthy here is that, as we mentioned above, although the enone (R1 = R2 = phenyl) could also participate in the reaction with the same good yield and ee, the reaction was much slower.
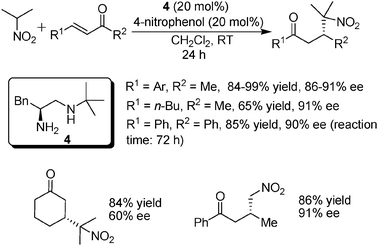 | ||
| Scheme 5 Michael additions of nitroalkanes to α,β-unsaturated ketones. | ||
Very recently, Wang and co-workers further extended the scope of this catalyst system to the Michael addition of N-Ac-indolin-3-one to α,β-unsaturated ketones (Scheme 6).15 This method provided an efficient access to a series of chiral indolin-3-ones, which are important structural motifs present in many biologically active natural products. With a catalyst combination of 5 and TFA, a broad range of α,β-unsaturated ketones worked well in the reaction to give the desired products in high yields, excellent dr and ee values. However, two heterocyclic substrates gave poor dr values while still maintaining excellent ee values. Similar to the above findings of some common features of this kind of catalytic system, aliphatic substrates (R1 and R2 were both alkyl groups) also displayed relatively lower reactivity in this system, for which 30 mol% of the catalyst was required with moderate yields and poor dr values. Moreover, even chalcones did not undergo the reaction while the cyclic 2-cyclohexenone also gave a moderate yield.
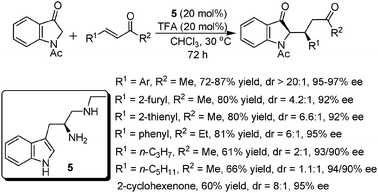 | ||
| Scheme 6 Michael additions of N-Ac-indolin-3-one to α,β-unsaturated ketones. | ||
2.2 Epoxidation of α,β-unsaturated ketones
In light of the above excellent results obtained with these amino-acid-derived primary–secondary amines in catalyzing the asymmetric Michael additions of various nucleophiles to α,β-unsaturated ketones, we next became interested in applying them to another important reaction: the asymmetric epoxidation of enones. Previously, Lattanzi pioneered the use of α,α-diaryl prolinols for the epoxidation of chalcones, in which the catalyst was believed to function through hydrogen bonding (Fig. 2).16a Our group have also contributed to this reaction.16b However, this secondary-amine catalyst system's success was mostly restricted to chalcones: when the two substituents on the enones were alkyl groups, there was often no reaction or only moderate yields and ee, which might be partly attributed to the reduced electrophilicity of these substrates. The latter problem has been solved by List and Deng, both using Cinchona alkaloids-derived primary amine catalysts through iminium catalysis.17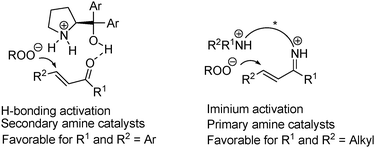 | ||
| Fig. 2 Two proposed activation models for the epoxidation of α,β-unsaturated ketones. | ||
Then we examined the efficiency of our primary–secondary amine catalysts for the asymmetric epoxidation of alkyl-substituted α,β-unsaturated ketones.18 After extensive screening of various parameters including the catalyst, acid additive, solvent and oxidant, the optimum catalyst combination was tested in a broad range of alkyl-substituted enone substrates (Scheme 7). Functional groups on the alkyl substituent R1 such as benzyl and silyl ethers and the ester group were well tolerated. A higher reaction temperature (40 °C) was required to obtain good yields when the bulk of the R2 substituent was increased. In addition, a significant drop in enantioselectivity was observed in the case of 2-cyclohexenone. The alkene moiety in the α,β-unsaturated ester moiety existing in the same molecule of the α,β-unsaturated ketone could remain intact in the reaction. It should be noted here that chalcones or conjugated enones with a trisubstituted or terminal alkene moiety were unreactive in this system. The mechanism of the reaction was presumed to be similar to that of previously proposed iminium catalysis for Cinchona alkaloids-derived primary amine catalysts (Fig. 2).
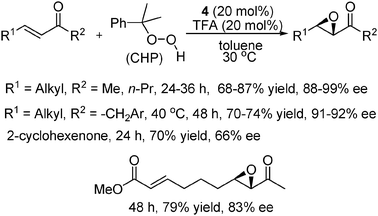 | ||
| Scheme 7 Asymmetric epoxidation of alkyl-substituted α,β-unsaturated ketones. | ||
3. Hydrogen bonding catalysis mediated reactions
3.1 Michael-type reactions
Thioureas bound to chiral 1,2-cyclohexanediamine and Cinchona alkaloids skeletons are good hydrogen bonding donors and have proved to be efficient organocatalysts in a myriad of reactions.19 Consequently, there is no surprise in people getting interested in installing the thiourea functionality in chiral amino acid skeletons. Generally, these organocatalysts are bifunctional: the thiourea moiety commonly activates electrophiles via hydrogen bonding while the tertiary amine moiety activates nucleophiles as a general base (Fig. 3).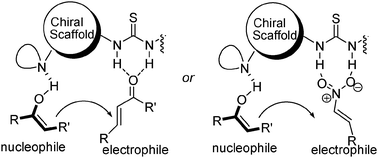 | ||
| Fig. 3 General plausible mechanisms for amino acid-based chiral ureas-catalyzed Michael additions to enones or nitroalkenes. | ||
In general, starting from commercially available N-Boc amino acids, these amino acids-based thioureas could be obtained in four steps with high yields. Pedrosa and co-workers first used these new chiral thioureas to mediate the Michael addition of malonates and diketones to nitroalkenes (Scheme 8).20 The identified optimum catalyst 6 derived from L-valine worked well in the addition of various malonates to nitroalkenes bearing aromatic substituents to give high yields and ees. In the case of an alkyl substituted nitroalkene, both the yield and ee decreased with a prolonged reaction time. In addition, while sterically demanding di-tert-butyl malonate was unreactive, the malonate with an electron-withdrawing chloride atom gave a very fast reaction and excellent results. The authors also found that an exchange of the positions of the thiourea and tertiary amine moieties in 6's structure led to much lower yield and ee.
 | ||
| Scheme 8 Asymmetric Michael additions of malonates and 1,3-diketones to nitroalkenes. | ||
Our group used catalyst 7 to promote the Michael additions of α-substituted cyanoketones to β,γ-unsaturated α-ketoesters (Scheme 9).21 The reaction worked well with a range of ketoesters with different aryl substituents (R1) and ester substituents (R2) with a catalyst loading of 2 mol%, irrespective of the substitution type of the aryl groups. When R1 was an alkyl group (PhCH2CH2), somewhat reduced yield and ee were obtained over a longer reaction time. A similar trend was also observed with the other reaction partner, namely cyanoketones, when R3 was also an aryl group. However, when R3 were groups such as t-Bu and EtO, the reaction failed to occur. The highly enantiomerically enriched dihydropyran products obtained by this method could be easily converted in a single step into chiral dihydropyridines (DHPs), which have been recognized as an important class of organic calcium-channel modulators as well as other pharmaceutical activities (Scheme 9).
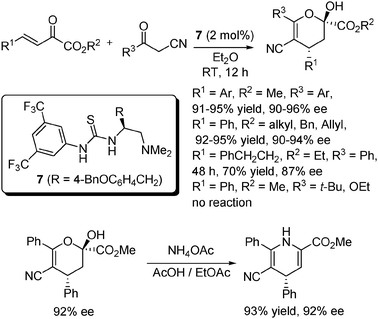 | ||
| Scheme 9 Michael addition to β,γ-unsaturated α-ketoesters. | ||
Trifluoromethyl-containing molecules are an important class of compounds in drug discovery as well as in the syntheses of pharmaceutical and agrochemical products. Similar to the above reaction, we also realized the asymmetric addition of α-substituted cyanoketones to α,β-unsaturated trifluoromethyl ketones to provide a series of chiral α-trifluoromethyldihydropyrans (Scheme 10).22 Under the catalysis of the novel thiourea 8 derived from L-phenylalanine, the reaction proceeded very well when R1 and R2 were aryl groups, except for R1 = 2-OMeC6H4, with which a poor dr value was obtained but still with excellent yield and ee. Unlike that in β,γ-unsaturated α-ketoesters, no appreciable reduction in reactivity was observed when R1 was an alkyl group, albeit with a slight drop in ee. When R2 was an alkyl group (t-Bu), excellent dr and ee values could still be obtained with diminished yield and a longer reaction time. However, the reaction failed to take place when R2 was an alkoxyl group (OEt). With similar conditions to those shown in Scheme 9, the α-trifluoromethyldihydropyran products could also be converted to chiral trifluoromethyldihydropyridines in one step without the loss of enantioselectivity.
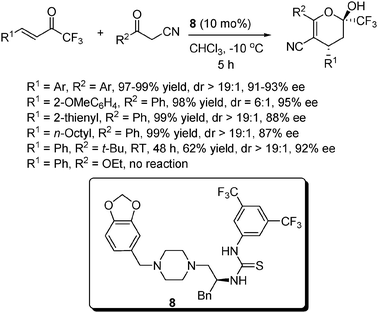 | ||
| Scheme 10 Michael addition to α,β-unsaturated trifluoromethyl ketones. | ||
Trichloromethyl ketones are also synthetically useful structures because of their easy elaboration to other functionalities such as carboxylic acid, ester and amide, owing to the good leaving ability of the trichloromethyl group. We later found that thiourea 8 was also the optimal catalyst for the asymmetric additions of α-substituted cyanoketones to α,β-unsaturated trichloromethyl ketones (Scheme 11).23 It is worthy of note that a series of Cinchona alkaloid-derived catalysts as well as Cinchona alkaloid-based thioureas all gave inferior results in this reaction. For the trichloromethyl ketone substrates, both aryl and alkyl substituents (R1) were well tolerated, though a lower ee was obtained in the case of an alkyl group (R1 = n-Pr). For α-substituted cyanoketones, when an electron-donating group was on the aryl group (R2) or an alkyl group (R2 = n-Pr), the reaction proceeded much slower and gave reduced yields, but still with comparably good ees. However, when R2 was a bulky t-Bu, the reaction hardly took place. Similar to the above two cases, the resulting products could also be transformed to chiral trichloromethyldihydropyridines in one step with almost complete retention of the enantioselectivity.
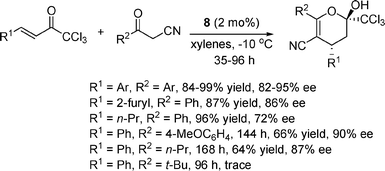 | ||
| Scheme 11 Michael addition to α,β-unsaturated trichloromethyl ketones. | ||
Interestingly, thiourea 8 was also found to be the catalyst of choice for the asymmetric Michael addition of α-fluoro-α-phenylsulfonyl ketones to nitroolefins (Scheme 12).24 This reaction provided an easy access to chiral fluorine-containing multifunctional molecules containing adjoining chiral fluorine-substituted quaternary and tertiary centers. Substrates with two aryl substituents (R1 and R2) performed well in the reaction, irrespective of the substitution type of the aryl groups. When R1 on the Michael donor ketone was an alkyl group like Me, the reaction still gave excellent yield and ee value, albeit with a drop in the dr value. Moreover, comparably good results were also obtained when alkyl-substituted nitroalkenes (R2 = n-Bu, i-Pr) were used, except for a decreased dr value in the case of the less sterically hindered n-Bu substrate. The resulting product could be converted to useful chiral cyclic nitrones via a single hydrogenation step without the loss of diastereoselectivity and enantioselectivity.
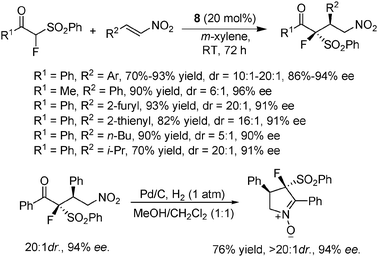 | ||
| Scheme 12 Addition of α-fluoro-α-phenylsulfonyl ketones to nitroolefins. | ||
As another important class of Michael donors, cyclic 1,3-dicarbonyl compounds were also investigated in our system (Scheme 13).25 Their additions to α,β-unsaturated carbonyl systems could produce many chiral compounds with biological and pharmaceutical activities such as neoflavonoids and coumarins. Using the thiourea catalyst 9, most 1,3-dicarbonyl compounds added to various β,γ-unsaturated α-ketoesters with excellent yields and ee values. Similar to the above cases, there were also inferior results when it came to alkyl-substituted ketoesters (R1 = n-Bu and PhCH2CH2), the former gave a complicated system while the latter gave reduced yield and enantioselectivity. It should be pointed out that two acyclic 1,3-dicarbonyl compounds were also tested as Michael donors in this system: acetyl acetone gave much less satisfactory results while ethyl acetoacetate hardly reacted. Several α,β-unsaturated trifluoromethyl ketones were also examined as the Michael acceptors in the same system, which provided excellent results but with significantly reduced enantioselectivities.
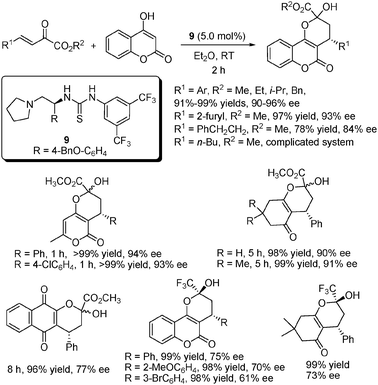 | ||
| Scheme 13 Michael addition of cyclic 1,3-carbonyl compounds to β,γ-unsaturated α-ketoesters and α,β-unsaturated trifluoromethyl ketones. | ||
3.2 Mannich reaction
In addition to the above Michael addition examples, Lu and co-workers realized a highly enantioselective Mannich reaction of α-fluoro-β-ketoesters to N-Boc imines using a L-tryptophan-derived thiourea catalyst 10 (Scheme 14).26 Notably, some of the screened Cinchona alkaloids-based organocatalysts gave much poorer results in this reaction. When both R1 and R2 substituents of the two reactants were aryl groups, excellent yields and ees, and moderate to excellent dr values were generally obtained, irrespective of the substitution type of the aryl groups. Poor dr values were observed when R1 was an alkyl group, with the exception of the bulky t-Bu group, while excellent yields and ees were still obtained. As for the more challenging alkyl imine substrates, this system gave a better result than previously reported systems for this type of reaction using Cinchona alkaloids-based organocatalysts,27 although 100 mol% loading of the catalyst was still required. Moreover, when alkyl N-tosylimines were used, the loading amount of the catalyst could still be reduced to 10 mol% with good results. Notably, in addition to α-fluoro-β-ketoesters, three other β-ketoesters and two malonates were also tested as nucleophiles in this system and comparably good results were obtained.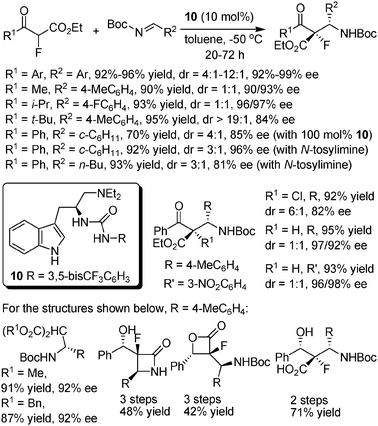 | ||
| Scheme 14 Mannich reaction of α-fluoro-β-ketoesters to N-Boc imines. | ||
As for the mechanism, the authors proposed a plausible explanation for the stereochemical outcome of the reaction, which was built on density functional theory (DFT) calculations. It was proposed that the protonated tertiary amine in the catalyst activated the imine, while the indole NH and the thiourea directed the ketoenolate (formed by deprotonation by the tertiary amine), all through hydrogen-bonding interactions. In addition, the utility of this reaction was also illustrated by facile conversions of one of the typical products in two or three steps to three other compounds such as α-fluoro-β-lactam, α-fluoro-β-amino acid and α-fluoro-β-lactone.
3.3 Vinylogous aldol reaction
The asymmetric vinylogous aldol reaction has received much attention in the past few decades. Most previous works in this field used 2-silyloxyfurans as nucleophiles, and the direct use of 2-furanones has mainly been restricted to the reaction with aldehydes.28 Very recently, using the same catalyst 10, Lu and co-workers also realized the highly diastereo- and enantioselective vinylogous aldol reactions between 2-furanone derivatives and α-ketoesters (Scheme 15).29 This reaction enabled a ready access in an organocatalytic way to chiral 2-substituted glycerol derivatives, which are both important structural motifs in many pharmaceuticals and versatile synthetic intermediates.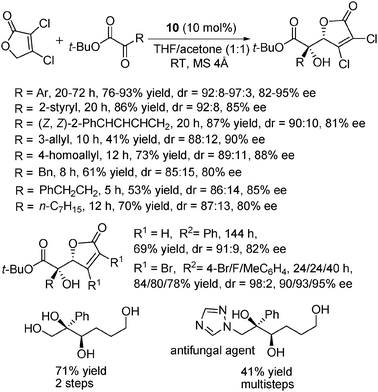 | ||
| Scheme 15 Vinylogous aldol reaction of furanones with α-ketoesters. | ||
With 3,4-dichlorofuran-2(5H)-one, the authors examined the scope of ketoesters. Aryl, alkenyl and alkyl-substituted ketoesters were well-tolerated in the reaction, though some reduced yields were observed for alkyl-substituted substrates. While the use of 3,4-dibromofuran-2(5H)-one gave comparable results, the use of the simple furan-2(5H)-one itself gave a very sluggish reaction. This is probably due to the reduced acidity of the 5-hydrogen atoms. Elaboration of a typical product to other useful compounds was also done: one is a chiral 2-substituted glycerol derivative and the other is an antifungal agent. Notably, several examined Cinchona alkaloids-based organocatalysts gave inferior results for this reaction.
A plausible transition model was proposed to explain the stereochemical outcome of this reaction system. Similar to the working model for this type of catalysts in the Michael reactions, hydrogen-bonding interaction between the ketoester and the thiourea moiety (activation of the electrophile) was supposed to be crucial for the stereocontrol while the tertiary amine moiety was assumed to activate the nucleophile (furanones) via deprotonation.
4. Reactions mediated by amino acids-derived chiral phosphines
4.1 Asymmetric [3+2] cycloadditions
The phosphine-catalyzed cycloadditions of allenoates have become an efficient way to access various synthetically useful carbo- and heterocycles due to intensive research efforts after the pioneering works of Lu and co-workers in 1995.30 Thereafter, most efforts in developing enantioselective variants for this kind of reaction had been done for a long time with chiral mono/biphosphine catalysts since the first example reported by Zhang.31 Despite the early use of phosphines derived from amino acids in metal-catalyzed reactions,32 it was until 2007 that an ingenious work from Miller and co-workers appeared, using phosphine-incorporated amino acid derivatives to catalyze the asymmetric [3+2] annulation of allenoates and α,β-unsaturated ketones (Scheme 16).33 In principle, a synergistic co-operation of the nucleophilic phosphine and a hydrogen bonding donor is considered to be crucial for the success of this kind of multifunctional phosphine catalysts.![Asymmetric [3+2] cycloaddition of allenoates with α,β-unsaturated ketones.](/image/article/2012/CY/c1cy00347j/c1cy00347j-s16.gif) | ||
| Scheme 16 Asymmetric [3+2] cycloaddition of allenoates with α,β-unsaturated ketones. | ||
After a survey of catalysts with different NH functionalities, catalyst 11 was identified as the best one, with which excellent selectivities of α-addition to cyclic enones with an exo-alkene moiety were obtained, together with moderate to good enantioselectivities. It should be pointed out that the problem of regioselectivity (α or γ-addition) is common in [3+2] cycloadditions of allenoates with simple electron-deficient olefins.30 Notably, the replacement of the NH-Boc group in 11 with NH-Ts or MeN-Boc led to a racemic product, which may suggest the importance of H-bonding interaction in the stereodifferentiating process. For acyclic enones, a significant drop in the α-selectivity was observed, but still with comparable yields and ee values. When a racemic γ-substituted allenoate was subjected to the reaction with chalcones, a unique deracemization of this allenoate was observed, together with exclusive α-selectivity. This deracemization can be easily understood by referring to the key zwitterion intermediate proposed for the reaction, in which the planar chirality of the allenoate is erased. However, this reaction required a very high catalyst loading amount (100 mol%) to maintain a high yield. It should be pointed out that in the proposed transition state, H-bonding interaction and P–O interaction were held as important controlling elements of the stereochemistry.34
In order to circumvent the regioselectivity problem associated with the cycloadditions of allenoates with simple electron-deficient olefins, Lu and co-workers used dual-activated olefins for the reaction.35 In our efforts to develop an enantioselective variant for the reaction, a small library consisting of over 16 bifunctional N-acyl aminophosphine catalysts with different chiral amino acid skeletons and NH protecting groups was prepared and screened, inspired by Miller's work (Scheme 17).36 These catalysts were generally prepared in four steps from the corresponding N-Boc-protected amino alcohols. During the catalysts screening process, the benefit of their modular structures was again manifested by enabling facile fine-tuning of their catalytic activity. It was found that both the chiral amino acid skeletons and the protecting group of the NH functionality had pronounced influence on the catalytic activity. Unlike Miller's case, the aminophosphine 12 derived from L-isoleucine with a 3,5-bistrifluoromethyl benzoyl group on the NH was identified as the optimal catalyst for the reaction.
![Asymmetric [3+2] cycloaddition of allenoates with dual-activated olefins.](/image/article/2012/CY/c1cy00347j/c1cy00347j-s17.gif) | ||
| Scheme 17 Asymmetric [3+2] cycloaddition of allenoates with dual-activated olefins. | ||
With malonitrile-derived alkenes with an aromatic group, the reaction generally performed well to achieve completion in one hour, while substrates bearing electron-withdrawing substituents in general gave higher ee values than those with electron-donating substituents. Substrates with two different electron-withdrawing functions (CN and CO2Et), with which poor regioselectivities and diastereoselectivities were observed when PPh3 was employed as the catalyst for accessing the corresponding racemic cyclopentenes, were also well tolerated in the system, though a longer reaction time was required. The diastereoselectivities observed in these substrates also suggested that the cycloaddition step might have proceeded via a stepwise way. One limitation was that alkyl substituted substrates failed in the reaction. A racemic γ-substituted allenoate was also tested in the reaction. Unlike Miller's case, the reaction could still proceed rather fast to give excellent yields with a catalytic loading amount of 12, albeit with moderate diastereoselectivities and somewhat reduced enantioselectivities. A transition state similar to Miller's was proposed to account for the stereochemical results of the reaction.
Substituted acrylates are commonly problematic substrates for phosphine-catalyzed cycloadditions with rare examples reported in the literature, probably due to their reduced reactivities (less electrophilic) compared to enones. Lu and co-workers very recently realized the enantioselective [3+2] cycloaddition of allenoates with α-substituted acrylates (Scheme 18).37 The optimal catalyst 13 that they identified for the reaction was based on a simple dipeptide derived from D-threonine and L-tert-leucine. The assorted combinations of different amino acids for the dipeptide skeleton offered additional benefits for the fine-tuning of the catalysts to acquire better results.
![Asymmetric [3+2] cycloaddition of allenoates with α-substituted acrylates.](/image/article/2012/CY/c1cy00347j/c1cy00347j-s18.gif) | ||
| Scheme 18 Asymmetric [3+2] cycloaddition of allenoates with α-substituted acrylates. | ||
A significant electronic effect of the α-substituted acrylates was observed: aryl substituents (R) bearing electron-withdrawing groups underwent the reaction fast within half an hour to deliver the products in high yields and ee values while those bearing electron-donating ones gave sluggish reactions. This is in accord with the expected electrophilicity (reactivity) of these substrates towards nucleophilic attacks. A remarkable drop in the enantioselectivity was also observed when R was an alkyl group.
Different from Miller's as well as our mechanistic proposals involving a chair-like six-membered ring, the authors came up with an open-chain transition state to explain the stereochemical outcome of this reaction. In this model, the dipeptide skeleton was assumed to take on conformation favoring its hydrogen-bonding interaction with the acrylates, which was attacked by the phosphonium from the Re face. The steric repulsion between the bulky t-Bu group of the allenoate and the 9-phenanthryl of the acrylate was thought to be responsible for the exclusive α-selectivity of the allenoate.
All the above three examples used allenoates as the “3C” unit. The use of the MBH (Morita–Baylis–Hillman) adducts as the “3C” unit in phosphine-catalyzed cycloadditions has also been developed by Lu in 2003.38 Very recently, the asymmetric versions of this type of reactions have also gained advances.39 Lu and co-workers realized an asymmetric [3+2] addition of MBH adducts with isatin-derived α,α-dicyanoalkenes (Scheme 19).39a Different from previously developed amino acids-based phosphines, the best catalyst 14 for this reaction featured a thiourea moiety as the hydrogen-bonding donor. It should be noted that the problem of regioselectivity (α or γ-selectivity) remains in this type of reaction (see the proposed transition state in Scheme 19).
![Asymmetric [3+2] addition of MBH adducts with isatin-derived α,α-dicyanoalkenes.](/image/article/2012/CY/c1cy00347j/c1cy00347j-s19.gif) | ||
| Scheme 19 Asymmetric [3+2] addition of MBH adducts with isatin-derived α,α-dicyanoalkenes. | ||
When the R groups in the MBH adducts were aryl groups with substituents on the 3 or 4-position of the benzene ring, excellent γ-selectivities, yields and ee values were obtained. Poor γ-selectivities were observed for R = heterocyclic groups, an alkyl group and an ester group. Also notable is that the α-addition isomers were usually obtained in much lower ee values than the corresponding γ-products. Substituted isatins with substituents on the indole ring were well-tolerated in the reaction. The authors also developed a three-component one-pot reaction using an N-PMB (PMB = para-methoxylbenzyl) protected isatin, malonitrile and a MBH adduct to give comparably excellent results.
An open-chain transition state similar to the above reaction was also proposed to explain the stereochemical results. The phosphonium salt was assumed to be formed by an initial SN2′ attack of the phosphine catalyst to the MBH adduct followed by deprotonation by the in situ formed t-BuO− anion. Hydrogen-interactions between the thiourea NH and the carbonyl group in the isatin were presumed to play a key role in the stereocontrol process.
4.2 Asymmetric [4+2] reaction
As we could see from the above examples, most enantioselective annulations in phosphine-catalyzed cycloadditions were achieved only with asymmetric [3+2] cycloadditions. In 2003, Kwon and co-workers developed α-substituted allenoate–imine [4+2] cycloadditions as an efficient way to afford tetrahydropyridines.40 Two years later, only Fu and co-workers reported an enantioselective variant of this reaction using a chiral monodentate phosphine catalyst.41 Very recently, we found that our bifunctional aminophosphine 12 was also an efficient catalyst for this reaction (Scheme 20).42![Asymmetric [4+2] addition of allenoates with N-tosyl imines.](/image/article/2012/CY/c1cy00347j/c1cy00347j-s20.gif) | ||
| Scheme 20 Asymmetric [4+2] addition of allenoates with N-tosyl imines. | ||
Catalyst screening for this reaction showed that varying the amino acid skeletons and the NH protecting groups of these phosphines brought a negligible effect on the diastereoselectivity, which was more influenced by the choice of solvent. Generally, N-tosylaldimines derived from aromatic aldehydes worked well in the reaction. Similar to previous reports, N-tosylaldimines derived from enolizable aliphatic aldehydes proved to be unreactive in this system. While some similar phenomena to Fu's monophosphine system were observed, such as the relatively poor diastereoselectivity for ortho-substituted imines, some improvements for the reactions of substrates such as 4-anisyl imine and 2-chlorobenzene aldimine were also obtained. Notably, the reactivity of the allenoates was affected by the anion-stabilizing ability of the substituents on the α carbon atom: changing the ester group to an aryl group led to decreased yield and enantioselectivity, but improved diastereoselectivity (Scheme 20). The steric hindrance of these α-substituents may be also responsible for the change of the regioselectivity to γ-selectivity from the α-selectivity observed for unsubstituted allenoates in the above examples. To demonstrate the potential utility of the present [4+2] cycloaddition reaction, a simple two-step procedure for the conversion of the cycloadduct to a highly functionalized chiral piperidine product was also developed.
Both a six-membered ring and an open-chain transition state may be invoked to explain the observed stereochemical outcome of this reaction. As mentioned in previous examples, both hydrogen bonding interaction and P–O interaction were considered crucial for the stereocontrol process.
4.3 Asymmetric aza-Morita–Baylis–Hillman reaction
The Morita–Baylis–Hillman (MBH) reaction and its aza counterpart are one of the most efficient atom-economical ways to construct densely functionalized molecules.43 In addition to the above cycloaddition examples, Lu, Huang et al. have also developed an asymmetric aza-Morita–Baylis–Hillman reaction using the amino acid-based phosphine catalysts (Scheme 21).44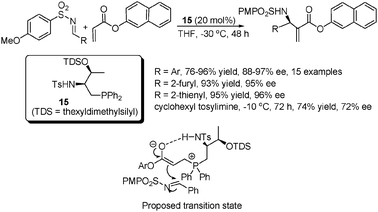 | ||
| Scheme 21 Asymmetric aza-MBH reaction of acrylate and imine. | ||
With a novel L-threonine-derived phosphine–sulfonamide catalyst 15, a range of imines derived from aromatic aldehydes worked very well in the reaction, irrespective of the electronic nature or positions of the substituents on the benzene ring. Heteroaryl substrates were also well tolerated. A less successful result was obtained with an alkyl-substituted substrate for which significantly lower yield and enantioselectivity were obtained.
A plausible mechanism involving the transition state for the addition step (shown in Scheme 21) was also proposed for the reaction. The hydrogen bonding interaction within the phosphoenolate and the bulky OTDS group were believed to be of key importance for the stereocontrol as supported by experimental observations.
5. Enamine catalysis mediated reactions
5.1 Michael addition
Córdova and co-workers developed a simple L-alanine-derived primary amine–amide catalyst 16 for the Michael addition of ketone to nitroolefin (Scheme 22).45 This system worked well for six-membered cyclic ketones to give excellent dr and ee values, though a decreased yield was observed in one case. However, much poorer ees were obtained for the two non-symmetric acyclic ketone substrates tested while excellent regioselectivities were obtained (more reactive at the more substituted site). Notably, the free L-alanine itself was found to give much inferior results in this reaction. Water or Brønsted acid additives were found to be able to both enhance the enantioselectivity and accelerate the reaction, which was explained by their synergistic stabilization of the transition state and facilitation of the inter-conversion of the different intermediates in the catalytic enamine cycle, respectively.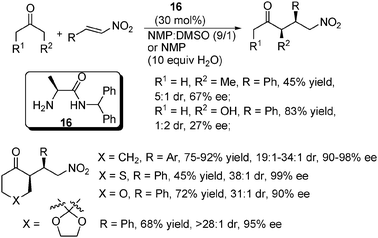 | ||
| Scheme 22 Asymmetric Michael addition of ketone to nitroolefin. | ||
5.2 Aldol reaction
The asymmetric aldol reaction is one of the most studied reactions in organocatalysis. In this field, the use of 1,3-dihydroxyacetone and its protected variants as donors has attracted particular interest, which is partly because this strategy is adopted by Nature for the metabolism and catabolism of carbohydrates.46b While the initially used L-proline and its derivatives catalyzed the direct aldol reaction of 1,3-dihydroxyacetone and its protected variants with aldehydes to give the anti 1,2-diol products, primary amine amides and carboxylic acids derived from simple acyclic amino acids were found to catalyze the reaction to give the syn 1,2-diol products.3fBarbas and coworkers developed a O-tBu-L-Thr based primary amine–amide catalyst 17 for this reaction (Scheme 23).46a When an organic solvent like DMF was used and in the presence of an acid cocatalyst 5-methyl-1H-tetrazole (5-Me-tet), the catalyst promoted the reaction of both aromatic and non-aromatic aldehydes quite well, albeit with moderate to good diastereoselectivities favoring the syn diol products. It is worth mentioning that the results obtained with non-aromatic aldehydes were generally superior to those obtained in the system of O-tBu-L-Thr,46c which is the parent catalyst of amide 17. An additional advantage of 17 is its water compatibility: using the TBS-protected 1,3-dihydroxyacetone in brine, the system gave much improved yields and dr values while maintaining the same excellent ee values in many cases, when compared to the O-tBu-L-Thr catalysis performed in NMP (containing 3% vol of water).46b
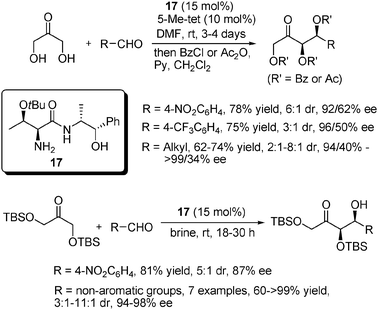 | ||
| Scheme 23 Asymmetric syn-aldol reaction of 1,3-dihydroxyacetone and aldehyde. | ||
After screening a series of amino acid-based organocatalysts, our group later identified O-tBu-L-Thr based amide 18 as a privileged catalyst for the direct syn-aldol reaction of hydroxyacetone (Scheme 24).47 Compared to previous works, this system required a low catalyst loading amount and no acidic cocatalyst was needed. For alkyl aldehydes, it was found that 2 mol% of catalyst provided improved results when a higher catalyst loading amount (8 mol%) was used. When applied to aromatic aldehydes, this system obtained less successful results with reduced dr and ee values. Notably, a chiral aldehyde substrate was also subjected to the reaction to give the desired product in acceptable yield and diastereoselectivity, which is a useful building block in the total synthesis of the natural product amphidinolide B1 (Scheme 24).
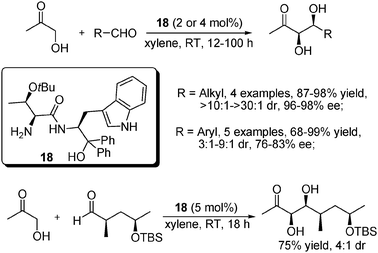 | ||
| Scheme 24 Asymmetric syn-aldol reaction of hydroxyacetone and aldehydes. | ||
In addition to amides 17 and 18 with an additional alcohol moiety, as shown in Fig. 4, amides 19a and 19b with a chiral thiourea structure were also synthesized and screened in the aldol reaction between acetone and 4-nitrobenzaldehyde by Kokotos and co-workers, though they were found to be less effective than the corresponding amide derived from L-proline.48 Also mentionable is amide 20 with a sulfonamide moiety and 21 with a tertiary amine moiety developed by Barbas and coworkers, which in some cases gave comparable results to that of catalyst 17.46 These different components in these amide catalysts have diverse reactivities, which may allow applications to other reactions.
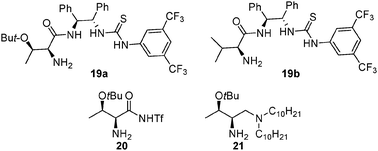 | ||
| Fig. 4 representative amide organocatalysts derived from simple acyclic chiral amino acids. | ||
6. Conclusions and outlook
Chiral amino acids created by Nature as one of the most important substances are an ideal foundation and toolbox for organic chemists to utilize in the creation of new catalysts to make organic reactions more efficient and environmentally-benign. The several recently developed organocatalysts derived from simple acyclic amino acids introduced in this perspective are only a small portion of the contribution of chiral amino acids to the development of organocatalysts. Some of the other recent important advances in the utilization of simple acyclic amino acids such as primary amine–carboxylic acid catalysts for asymmetric aldol, Mannich reactions3f,g and small peptide catalysts3e,49 are also excellent evidence for the great to-be-explored potential of amino acids in organocatalysis, despite the great previous achievements in this field.As shown in this paper, several organocatalysts based on chiral acyclic amino acids have accomplished excellent results in some asymmetric reactions surpassing previously developed other catalyst systems. However, there are still many important points to be addressed. For example, the reaction types applicable for these types of catalysts are still rather limited: mainly for Michael-type reactions and phosphine-catalyzed cycloadditions. The recent elegant application of a small peptide catalyst to realize asymmetric bromination reported by Miller et al.49 indicates that extension of the utility of these catalysts could be attained through judicious design of reactions.50 Moreover, compared to some of the other more mature catalyst systems, the application of these catalysts to more complex systems such as total syntheses of natural products is also very much underdeveloped. Most mechanisms proposed are still very preliminary, lacking solid and thorough experimental and theoretical studies. We believe that deeper understanding of the working mechanisms of these catalysts would lead to more rational designs of new catalysts and their applications, and thus would be extremely beneficial for the future development in this field.
Acknowledgements
Research supports from National Basic Research Program of China (973 Program, 2010CB833300), the National Natural Science Foundation of China (No. 20172064, 203900502, 21032006), Shanghai Natural Science Council, and Excellent Young Scholars Foundation of National Natural Science Foundation of China (20525208) are greatly acknowledged.Notes and references
- For monographs and special issues on asymmetric organocatalysis: (a) A. Berkessel and H. Gröger, Asymmetric Organocatalysis from Biomimetic Concepts to Applications in Asymmetric Synthesis, Wiley-VCH, Weinheim, 2005 Search PubMed; (b) P. I. Dalko, Enantioselective Organocatalysis, Wiley-VCH, Weinheim, 2007 Search PubMed; (c) K. N. Houk and B. List, Acc. Chem. Res., 2004, 37, 487 CrossRef CAS; (d) B. List, Chem. Rev., 2007, 107, 5413 CrossRef CAS; (e) J. B. Brazier and N. C. O. Tomkinson, Top. Curr. Chem., 2010, 291, 281 CrossRef CAS.
- For general reviews: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (b) H. Pellissier, Tetrahedron, 2007, 63, 9267 CrossRef CAS; (c) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS; (d) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS; (e) M. Ueda, T. Kano and K. Mamoka, Org. Biomol. Chem., 2009, 7, 2005 RSC; (f) M. Bella and T. Gasperi, Synthesis, 2009, 1583 CrossRef CAS; (g) S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 RSC; (h) B. List, Angew. Chem., Int. Ed., 2010, 49, 1730 CrossRef CAS.
- For reviews on the isolation and synthesis of α-amino acids: (a) C. Nájera and J. M. Sansano, Chem. Rev., 2007, 107, 4584 CrossRef; (b) K. Ditrich, T. Habicher, B. Hauer, M. Keβeler, R. Stürmer and T. Zelinski, Angew. Chem., Int. Ed., 2004, 43, 788 CrossRef; (c) R. O. Duthaler, Tetrahedron, 1994, 50, 1539 CrossRef CAS; (d) T. Hermann, J. Biotechnol., 2003, 104, 155 CrossRef CAS . For reviews on the use of amino acids and their derivatives as organocatalysts: ; (e) E. A. C. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Chem. Rev., 2007, 107, 5759 CrossRef; (f) L.-W. Xu, J. Luo and Y. Lu, Chem. Commun., 2009, 1807 RSC; (g) L.-W. Xu and Y. Lu, Org. Biomol. Chem., 2008, 6, 2047 RSC; (h) L.-W. Xu, L. Li and Z.-H. Shi, Adv. Synth. Catal., 2010, 352, 243 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 2000, 122, 2395 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. Macmillan, J. Am. Chem. Soc., 2000, 122, 4243 CrossRef CAS.
- K. Ishihara and K. Nakano, J. Am. Chem. Soc., 2005, 127, 10504 CrossRef CAS.
- For recent reviews on multiple catalysis and organocatalytic cascade reactions: (a) S. Poivesana, D. M. Scarpino Schietroma and M. Bella, Angew. Chem., Int. Ed., 2011, 50, 6216 CrossRef; (b) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS; (c) Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef.
- (a) N. Halland, P. S. Abured and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 661 CrossRef CAS; (b) K. R. Knudsen, C. E. T. Mitchell and S. V. Ley, Chem. Commun., 2006, 66 RSC; (c) V. Wascholowski, K. R. Knudsen, C. E. T. Mitchell and S. V. Ley, Chem.–Eur. J., 2008, 14, 6155 CrossRef CAS.
- Y.-Q. Yang and G. Zhao, Chem.–Eur. J., 2008, 14, 10888 CrossRef CAS.
- Z. Mao, Y. Jia, W. Li and R. Wang, J. Org. Chem., 2010, 75, 7428 CrossRef CAS.
- Y.-Q. Yang, Z. Chai, H.-F. Wang, X.-K. Chen, H.-F. Cui, C.-W. Zheng, H. Xiao, P. Li and G. Zhao, Chem.–Eur. J., 2009, 15, 13295 CrossRef CAS.
- H.-F. Cui, Y.-Q. Yang, Z. Chai, P. Li, C.-W. Zheng, S.-Z. Zhu and G. Zhao, J. Org. Chem., 2009, 74, 117 Search PubMed.
- For a monography on the chemistry of the nitro group: N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, Weinheim, 2001 Search PubMed.
- Y.-Q. Yang, X.-K. Chen, H. Xiao, W. Liu and G. Zhao, Chem. Commun., 2010, 46, 4130 RSC.
- W. Sun, L. Hong and R. Wang, Chem.–Eur. J., 2011, 17, 6030 CrossRef CAS.
- For reviews, see: (a) A. Lattanzi, Curr. Org. Synth., 2008, 5, 117 CrossRef CAS; (b) C.-W. Zheng and G. Zhao, Chin. Sci. Bull., 2010, 55, 1712 CrossRef CAS.
- (a) X. Wang, C. M. Reisinger and B. List, J. Am. Chem. Soc., 2008, 130, 6070 CrossRef CAS; (b) C. M. Reisinger, X. Wang and B. List, Angew. Chem., Int. Ed., 2008, 47, 8112 CrossRef CAS; (c) X. Lu, Y. Liu, B. Sun, B. Cindric and L. Deng, J. Am. Chem. Soc., 2008, 130, 8134 CrossRef CAS.
- Y. Lu, C. Zheng, Y. Yang, G. Zhao and G. Zou, Adv. Synth. Catal., 2011 DOI:10.1002/adsc.201100230.
- For recent reviews on chiral thiourea catalysts: (a) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) Y. Takemoto, Chem. Pharm. Bull., 2010, 58, 593 CrossRef CAS.
- J. M. Andrés, R. Manzano and R. Pedrosa, Chem.–Eur. J., 2008, 14, 5116 CrossRef.
- S.-L. Zhao, C.-W. Zheng, H.-F. Wang and G. Zhao, Adv. Synth. Catal., 2009, 351, 2811 CrossRef CAS.
- P. Li, Z. Chai, S.-L. Zhao, Y.-Q. Yang, H.-F. Wang, C.-W. Zheng, Y.-P. Cai, G. Zhao and S.-Z. Zhu, Chem. Commun., 2009, 7369 RSC.
- H.-F. Wang, P. Li, H.-F. Cui, X.-W. Wang, J.-K. Zhang, W. Liu and G. Zhao, Tetrahedron, 2011, 67, 1774 CrossRef CAS.
- H.-F. Cui, P. Li, X.-W. Wang, S.-Z. Zhu and G. Zhao, J. Fluorine Chem., 2011 DOI:10.1016/j.jfluchem.2011.05.029.
- X.-K. Chen, C.-W. Zheng, S.-L. Zhao, Z. Chai, Y.-Q. Yang, G. Zhao and W.-G. Cao, Adv. Synth. Catal., 2010, 352, 1648 CrossRef CAS.
- X. Han, J. Kwiatkowski, F. Xue, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2009, 48, 7604 CrossRef CAS.
- J. Song, Y. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 6048 CrossRef CAS and references therein.
- For reviews on vinylogous aldol reaction: (a) G. Casiraghi, F. Zanardi, G. Appendino and G. Rassn, Chem. Rev., 2000, 100, 5713 CrossRef; (b) M. Kalesse, Top. Curr. Chem., 2005, 244, 43 CAS; (c) S. E. Denmark, J. R. Heemstra, Jr. and G. L. Beutner, Angew. Chem., Int. Ed., 2005, 44, 4682 CrossRef CAS . For examples of reactions between furanones and aldehydes: ; (d) K. D. Sarma, J. Zhang and T. T. Curran, J. Org. Chem., 2007, 72, 3311 CrossRef; (e) Y. Yang, K. Zheng, J. Zhao, J. Shi, L. Lin, X. Liu and X. Feng, J. Org. Chem., 2010, 75, 5382 CrossRef CAS; (f) H. Ube, N. Shimada and M. Terada, Angew. Chem., Int. Ed., 2010, 49, 1858 CrossRef CAS.
- J. Luo, H. Wang, X. Han, L.-W. Xu, J. Kwiatkowski, K.-W. Huang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 1861 CrossRef CAS.
- (a) C. Zhang and X. Lu, J. Org. Chem., 1995, 60, 2906 CrossRef CAS . For reviews on phosphine-catalyzed reactions: ; (b) X. Lu, C. Zhang and Z. Xu, Acc. Chem. Res., 2001, 34, 535 CrossRef CAS; (c) J. L. Methot and W. R. Roush, Adv. Synth. Catal., 2004, 346, 1035 CrossRef CAS; (d) L.-W. Ye, J. Zhou and Y. Tang, Chem. Soc. Rev., 2008, 37, 1140 RSC; (e) B. J. Cowen and S. J. Miller, Chem. Soc. Rev., 2009, 38, 3102 RSC; (f) A. Marinetti and A. Voituriez, Synlett, 2010, 174 CrossRef CAS.
- G. Zhu, Z. Chen, Q. Jiang, D. Xiao, P. Cao and X. Zhang, J. Am. Chem. Soc., 1997, 119, 3836 CrossRef CAS.
- S. R. Gilbertson, S. E. Collibee and A. Agarkov, J. Am. Chem. Soc., 2000, 122, 6522 CrossRef CAS.
- (a) B. J. Cowen and S. J. Miller, J. Am. Chem. Soc., 2007, 129, 10988 CrossRef CAS . Jacobsen et al. later reported a new chiral 1,2-cyclohexanediamine-based phosphine catalyst: ; (b) Y.-Q. Fang and E. N. Jacobsen, J. Am. Chem. Soc., 2008, 130, 5660 CrossRef CAS.
- (a) Y. Liang, S. Liu, Y. Xia, Y. Li and Z.-X. Yu, Chem.–Eur. J., 2008, 14, 4361 CrossRef CAS; (b) Y. Xia, Y. Liang, Y. Chen, M. Wang, L. Jiao, F. Huang, S. Liu, Y. Li and Z.-X. Yu, J. Am. Chem. Soc., 2007, 129, 3470 CrossRef CAS; (c) X.-F. Zhu, C.-E. Henry and O. Kwon, J. Am. Chem. Soc., 2007, 129, 6722 CrossRef CAS.
- X. Lu, Z. Lu and X. Zhang, Tetrahedron, 2006, 62, 457 CrossRef CAS.
- H. Xiao, Z. Chai, C.-W. Zheng, Y.-Q. Yang, W. Liu, J.-K. Zhang and G. Zhao, Angew. Chem., Int. Ed., 2010, 49, 4467 CrossRef CAS.
- X. Han, Y. Wang, F. Zhang and Y. Lu, J. Am. Chem. Soc., 2011, 133, 1726 CrossRef CAS.
- (a) Y. Du, X. Lu and C. Zhang, Angew. Chem., Int. Ed., 2003, 42, 1035 CrossRef CAS ; For a review on contributions from the Baylis–Hillman reaction to organic synthesis: ; (b) D. Basavaiah, B. S. Reddy and S. S. Badsara, Chem. Rev., 2010, 110, 5447 CrossRef CAS.
- (a) F. Zhong, X. Han, Y. Wang and Y. Lu, Angew. Chem., Int. Ed., 2011, 50, 7837 CrossRef CAS ; For a report on a similar reaction using biphosphine-catalysts: ; (b) B. Tan, N. R. Candeias and C. F. Barbas III, J. Am. Chem. Soc., 2011, 133, 4672 CrossRef CAS.
- X.-F. Zhu, J. Lan and O. Kwon, J. Am. Chem. Soc., 2003, 125, 4716 CrossRef CAS.
- R. P. Wurz and G. C. Fu, J. Am. Chem. Soc., 2005, 127, 12234 CrossRef CAS.
- H. Xiao, Z. Chai, H.-F. Wang, X.-W. Wang, D.-D. Cao, W. Liu, Y.-P. Lu, Y.-Q. Yang and G. Zhao, Chem.–Eur. J., 2011, 17, 10562 CrossRef CAS.
- For recent comprehensive reviews on MBH reaction: (a) D. Basavaiah, K. V. Rao and R. J. Reddy, Chem. Soc. Rev., 2007, 36, 1581 RSC; (b) V. Declerck, J. Martinez and F. Lamaty, Chem. Rev., 2009, 109, 1 CrossRef CAS and ref. 38b.
- F. Zhang, Y. Wang, X. Han, K.-W. Huang and Y. Lu, Org. Lett., 2011, 13, 1310 CrossRef.
- Y. Xu and A. Córdova, Chem. Commun., 2006, 460 RSC.
- (a) S. S. V. Ramasastry, K. Albertshofer, N. Utsumi and C. F. Barbas III, Org. Lett., 2008, 10, 1621 CrossRef CAS; (b) N. Utsumi, M. Imai, F. Tanaka, S. S. V. Ramasastry and C. F. Barbas III, Org. Lett., 2007, 9, 3445 CrossRef CAS; (c) S. S. V. Ramasastry, K. Albertshofer, N. Utsumi, F. Tanaka and C. F. Barbas III, Angew. Chem., Int. Ed., 2007, 46, 5572 CrossRef CAS.
- X. Wu, Z. Ma, Z. Ye, S. Qian and G. Zhao, Adv. Synth. Catal., 2009, 351, 158 CrossRef CAS.
- S. Fotaras, C. G. Kokotos, E. Tsandi and G. Kokotos, Eur. J. Org. Chem., 2011, 1310 CrossRef CAS.
- J. L. Gustafson, D. Lim and S. J. Miller, Science, 2010, 328, 1251 CrossRef CAS.
- For example, a very recent report utilized free primary amino acids to catalyze the asymmetric α-amination of branched aldehydes with azadicarboxylates: J.-Y. Fu, Q.-C. Yang, Q.-L. Wang, J.-N. Ming, F.-Y. Wang, X.-Y. Xu and L.-X. Wang, J. Org. Chem., 2011, 76, 4661 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |